

Abstract
In Escherichia coli, cell division is performed by a multimolecular machinery called the divisome, made of 10 essential proteins and more than 20 accessory proteins. Through a bacterial two-hybrid library screen, we identified the E. coli β-lactam resistance protein Blr, a short membrane polypeptide of 41 residues, as an interacting partner of the essential cell division protein FtsL. In addition to FtsL, Blr was found to associate with several other divisomal proteins, including FtsI, FtsK, FtsN, FtsQ, FtsW, and YmgF. Using fluorescently tagged Blr, we showed that this peptide localizes to the division septum and that its colocalization requires the presence of the late division protein FtsN. Although Blr is not essential, previous studies have shown that the inactivation of the blr gene increased the sensitivity of bacteria to β-lactam antibiotics or their resistance to cell envelope stress. Here, we found that Blr, when overproduced, restores the viability of E. coli ftsQ1(Ts) cells, carrying a thermosensitive allele of the ftsQ gene, during growth under low-osmotic-strength conditions (e.g., in synthetic media or in Luria-Bertani broth without NaCl). In contrast, the inactivation of blr increases the osmosensitivity of ftsQ1(Ts) cells, and blr ftsQ1 double mutants exhibit filamentous growth in LB broth even at a moderate salt concentration (0.5% NaCl) compared to parental ftsQ1(Ts) cells. Altogether, our results suggest that the small membrane polypeptide Blr is a novel component of the E. coli cell division apparatus involved in the stabilization of the divisome under certain stress conditions.
INTRODUCTION
Cell division is one of the most fundamental processes in biology. In bacteria, it is mediated by a group of proteins called the divisome or septosome (for reviews, see references 11, 19, 29, and 37) In Gram-negative bacteria, these proteins drive the coordinated invagination of the inner and outer membranes along with septal peptidoglycan (PG) synthesis. To effect this, the divisomal proteins form a multicomponent ringlike structure at the potential cell division site. In Escherichia coli, 10 essential and more than 20 nonessential proteins are involved in divisome assembly (19, 21, 28, 29). This assembly occurs in an ordered manner, starting with the polymerization of the central cell division protein FtsZ, a bacterial tubulin homolog, at midcell to form a cytoskeletal scaffold known as the Z ring. FtsA, ZipA, ZapA, ZapB, and ZapC subsequently localize at midcell independently of each other, most probably to stabilize and to tether the Z-ring structure to the inner membrane. Several integral membrane proteins, FtsK, FtsQ, FtsL/FtsB, FtsW, FtsI, and FtsN, are then recruited to the Z ring in a nearly sequential order, allowing the maturation of the core divisome (for reviews, see references 19 and 45). Interestingly, in the past years, different experimental approaches have revealed a more complex interaction network among the E. coli divisome components than originally thought (3, 20, 24, 32). The precise functions of many of these proteins remain to be characterized. Several of them, such as FtsZ, FtsI, FtsW, and FtsN, are required for septal PG synthesis (for a review, see reference 19). The FtsQ/FtsL/FtsB heterocomplex has been hypothesized to connect the Z-ring components and the proteins involved in PG synthesis (25). Another group of proteins includes the periplasmic amidases AmiA, AmiB, and AmiC and their activators, EnvC and NlpD, which are responsible for proper PG splitting during cell constriction (41, 50). The multifunctional protein FtsK, which coordinates chromosomal segregation, is also involved in septum constriction during cell division (17). Finally, the Tol-Pal complex together with the PBP1 and LpoB proteins are crucial for outer membrane invagination during the late stages of E. coli cell division (19, 40, 49).
In recent years, FtsE, FtsX, FtsP, ZapB, ZapC, and YmgF as well as SPOR-domain proteins such as DamX, DedD, and RplA were shown to associate with the E. coli division machinery (19). These proteins, although not essential for cell viability (in rich medium and/or under standard growth conditions), might become critical for the cell division process under specific conditions. One hypothesis is that these accessory proteins (often with overlapping functions) may confer some robustness to the divisome assembly process, especially under stress conditions (19).
In this study, we identified Blr (β-lactam resistance protein), a 41-amino-acid inner membrane polypeptide, as a novel component of the E. coli cell division machinery. Blr was previously reported by Wong and colleagues to be involved in the resistance of E. coli cells to certain classes of antibiotics that inhibit PG synthesis (52). Here, Blr was isolated through a bacterial two-hybrid (BACTH) screen (33) as a binding partner of the essential membrane-bound cell division protein FtsL, which forms a trimeric complex with FtsQ and FtsB (13). In addition to FtsL, Blr is able to interact with multiple components from the E. coli divisome, including FtsI, FtsK, FtsN, FtsQ, FtsW, and YmgF. We showed that the Blr peptide localizes to the E. coli division septum in an FtsQ- and FtsN-dependent manner. We found that the inactivation of blr exacerbates the osmosensitivity of E. coli cells carrying a thermosensitive allele of the ftsQ gene, ftsQ1(Ts). Conversely, the overproduction of Blr allows E. coli ftsQ1(Ts) cells to overcome this osmosensitivity.
Taken together, our results suggest that the small membrane polypeptide Blr might be a novel component of the bacterial cell division apparatus possibly involved in the stabilization of the E. coli divisome under low-osmotic-stress conditions.
MATERIALS AND METHODS
General methods.
Bacteria were routinely grown at 30°C in LB broth containing 0.5% NaCl (38). When necessary, LB broth with no NaCl added (LB0) and LB broth containing 2% NaCl (LB2) were used. M63 synthetic medium was used as a minimal medium (38). When necessary, M63 medium was supplemented with Casamino Acids (at a concentration of 50 μg/ml). Plates contained 15 g agar per liter. Unless stated otherwise, the following antibiotics were added: ampicillin at 100 μg/ml, chloramphenicol at 30 μg/ml, kanamycin at 50 μg/ml, and tetracycline at 30 μg/ml. d-Glucose and l-arabinose were used at a concentration of 0.2% to modulate the expression of genes cloned under the control of the PBAD promoter (27). Standard protocols for molecular cloning, PCR, DNA analysis, and transformation were used (47). Regular PCRs were performed with DyNAzyme EXT polymerase (Thermo Scientific Finnzymes), while Pfu Turbo DNA polymerase (Agilent Technologies-Stratagene) was used for site-directed in vitro mutagenesis. Unless otherwise indicated, genomic DNA from E. coli K-12 MG1665 (a wild-type strain) was used as a template in the PCR procedures. PCR primer synthesis and DNA sequencing were carried out by the Eurofins MWG Operon Company (Ebersberg, Germany).
Strains and plasmids.
Bacterial strains and plasmids used and constructed in this study are listed in Tables 1 and 2, respectively. For all routine cloning experiments, E. coli strain XL1-Blue (Agilent Technologies-Stratagene) was used. BACTH complementation assays and BACTH library screenings were performed by using E. coli cya strain DHM1 (35). E. coli cya strain AK2000 (34) was used as a source of genomic DNA for the construction of a BACTH DNA library, which was then propagated in strain DH10B (Life Technologies-Invitrogen).
Table 1.
Bacterial strains used and constructed in this study
| Strain | Genotypea | Source or reference |
|---|---|---|
| XL1-Blue | F′::Tn10 proAB+ lacIq Δ(lacZ)M15 endA1 glnV44(AS) gyrA96 hsdR17 recA1 thi-1 lac | Agilent Technologies-Stratagene |
| DH5α | F− Δ(argF-lac)U169 ϕ80Δ(lacZ)M15 deoR endA1 glnV44(AS) gyrA96 hsdR17 recA1 relA1 thi-1 | Laboratory collection |
| MG1655 | F− ilvG rfb-50 rph-1 | Laboratory collection |
| MC4100 | F− araD139 Δ(argF-lac)U169 rspL150 relA1 flbB5301 fruA25 deoC1 ptsF25 | Laboratory collection |
| MCQ1 | MC4100 ftsQ1(Ts) leu260::Tn10 | Coli Genetic Stock Center |
| JOE309 | MC4100 ara+ | 15 |
| JOE417 | JOE309 ftsQE14::kan (pJC10) | 15 |
| JOE565 | JOE309 ftsN::kan (pJC83) | 14 |
| JW5963 | Keio Collection strain BW25113 Δblr::kan | 7 |
| JW1156 | Keio Collection strain BW25113 ΔymgF::kan | 7 |
| DHM1 | F− glnV44(AS) recA1 endA1 gyrA96 thi-1 hsdR17 spoT1 rfbD1 cya-854 | 32 |
| AK2000 | MG1655 cya-854 | 34 |
| GK1503 | MG1655 Δblr::kan | This work |
| GK1504 | MC4100 Δblr::kan | This work |
| GK1505 | GK1503 Δ(λattL-lom)::bla lacIq gfp-blr | This work |
| GK1506 | GK1505 Δblr::ftr | This work |
| GK1507 | GK1506, ftsQE14::kan (pJC10) | This work |
| GK1508 | GK1506 ftsN::kan (pJC83) | This work |
| GK1510 | GK1504 ftsQ1(Ts) leu260::Tn10 | This work |
| GK1511 | GK1503 Δblr::ftr | This work |
| GK1512 | GK1511 ftsQ1(Ts) leu260::Tn10 | This work |
| GK1513 | GK1511 ΔymgF::kan | This work |
| GK1514 | GK1512 ΔymgF::kan | This work |
AS, amber suppressor.
Table 2.
Plasmids used and constructed in this study
| Plasmid | Description | Reference |
|---|---|---|
| pKT25 | BACTH vector expressing a given polypeptide fused in frame at its N-terminal end with T25; p15 ori Kmr | 35 |
| pUT18C | BACTH vector expressing a given polypeptide fused in frame at its N-terminal end with T18; ColE1 ori Apr | 35 |
| pDSW207 | Vector expressing GFP fused at the N terminus of a given polypeptide; ColE1 ori Apr | 51 |
| pDSW913 | Vector expressing RFP fused at the C terminus of a given polypeptide; ColE1 ori Apr | 6 |
| pCP20 | Vector expressing the yeast flp recombinase gene; pSC101 ori RepA(Ts) Apr Cmr | 16 |
| pBAD33 | Arabinose-inducible protein expression vector; p15 ori Cmr | 27 |
| pKTop | Vector expressing dual reporter PhoA22–472/LacZ4–60; p15 ori Kmr | 34 |
| pKT25-ftsA | The full-length ftsA ORF was cloned into pKT25 | 32 |
| pKT25-ftsI | The full-length ftsI ORF was cloned into pKT25 | 32 |
| pKT25-ftsL | The full-length ftsL ORF was cloned into pKT25 | 32 |
| pKT25-ftsB | The full-length ftsB ORF was cloned into pKT25 | 32 |
| pKT25-ftsQ | The full-length ftsQ ORF was cloned into pKT25 | 32 |
| pKT25-ftsN | The full-length ftsN ORF was cloned into pKT25 | 32 |
| pKT25-ftsW | The full-length ftsW ORF was cloned into pKT25 | 32 |
| pKT25-ftsX | The full-length ftsX ORF was cloned into pKT25 | 32 |
| pKNT25-ftsZ | The full-length ftsZ ORF was cloned into pKNT25 | 32 |
| pKNT25-ymgF | The full-length ymgF ORF cloned into pKNT25 | 34 |
| pUT18C-ftsI | The full-length ftsI ORF was cloned into pUT18C | 32 |
| pUT18C-ftsL | The full-length ftsL ORF was cloned into pUT18C | 32 |
| pUT18C-ftsB | The full-length ftsB ORF was cloned into pUT18C | 32 |
| pUT18C-ftsQ | The full-length ftsQ ORF was cloned into pUT18C | 32 |
| pUT18C-ftsN | The full-length ftsN ORF was cloned into pUT18C | 32 |
| pUT18C-ftsX | The full-length ftsX ORF was cloned into pUT18C | 32 |
| pUT18C-ftsA | The full-length ftsA ORF was cloned into pUT18C | 32 |
| pUT18C-ftsW | The full-length ftsW ORF was cloned into pUT18C | 32 |
| pUT18-ymgF | The full-length ymgF ORF was cloned into pUT18 | 34 |
| pKT25-ftsK | pKT25 derivative expressing T25-FtsK1–271 | This work |
| pUT18C-ftsK | pUT18C derivative expressing T18-FtsK1–271 | This work |
| pKT25-blr | The full-length blr ORF was cloned into pKT25 | This work |
| pUT18C-blr | The full-length blr ORF was cloned into pUT18C | This work |
| pUT18C-blrmut1 | pUT18C derivative expressing T18-Blrmut1 | This work |
| pUT18C-blrmut2 | pUT18C derivative expressing T18-Blrmut2 | This work |
| pUT18C-blrmut3 | pUT18C derivative expressing T18-Blrmut3 | This work |
| pDSW207-blr | pDSW207 derivative expressing GFP-Blr | This work |
| pDSW207-blrmut1 | pDSW207 derivative expressing GFP-Blr Blrmut1 | This work |
| pDSW207-blrmut2 | pDSW207 derivative expressing GFP-Blr Blrmut2 | This work |
| pDSW207-blrmut3 | pDSW207 derivative expressing GFP-Blr Blrmut3 | This work |
| pDSW913-blr | pDSW913 derivative expressing Blr-RFP | This work |
| pBAD33-blr | pBAD33 derivative expressing wild-type Blr | This work |
| pBAD33-ymgF | pBAD33 derivative expressing wild-type YmgF | This work |
| pKTOP-blr | pKTop derivative expressing tripartite Blr/PhoA22–472/LacZ4–60 | This work |
| pKTOP-blrmut1 | pKTop derivative expressing tripartite Blrmut1/PhoA22–472/LacZ4–60 | This work |
| pKTOP-blrmut2 | pKTop derivative expressing tripartite Blrmut2/PhoA22–472/LacZ4–60 | This work |
| pKTOP-blrmut3 | pKTop derivative expressing tripartite Blrmut3/PhoA22–472/LacZ4–60 | This work |
Strain constructions.
GK1503 is a derivative of E. coli wild-type strain K-12 MG1655 in which the blr gene has been inactivated. For this, the cassette, which encodes the blr open reading frame (ORF) replaced by the kanamycin resistance marker (Δblr::kan) from Keio Collection strain JW5963 (7), was introduced into MG1655 through P1 transduction (38) by selection for kanamycin-resistant (Kmr) colonies.
Strain GK1504 was constructed similarly by the P1 transduction of the same Δblr::kan cassette into strain MC4100.
GK1505 is a derivative of GK1503 that contains a translational gfp-blr fusion integrated at the λ attachment site of its chromosome (λattB). The strain was created by integrating plasmid pDSW207-blr (see below) onto the chromosome of GK1503 as described previously (10).
GK1506 was constructed from GK1505 by removing the kanamycin resistance marker of the Δblr::kan cassette. In Δblr::kan, the antibiotic resistance marker is flanked by ftr repeats, which are the recognition site of the FLP nuclease (7). Temperature-sensitive plasmid pCP20 expressing FLP was used to eliminate the kanamycin resistance marker from the GK1505 chromosome (16). Briefly, GK1505 cells were transformed with pCP20, and chloramphenicol-resistant (Cmr) colonies were selected at 30°C. After overnight growth in antibiotic-free LB broth at 42°C, several of these clones were no longer Cmr and Kmr, indicating a simultaneous loss of pCP20 and the kanamycin resistance marker from the bacterial chromosome. This FLP-catalyzed excision created an in-frame deletion of the blr ORF, leaving behind a 102-bp scar sequence (Δblr::ftr) (7). One clone was chosen and named GK1506.
Strain GK1507 is derivative of GK1506 that contains a null allele of ftsQ. It was constructed in two steps. First, GK1506 was transformed with plasmid pJC10, which carries a wild-type copy of ftsQ under the control of an arabinose-dependent PBAD promoter and a chloramphenicol resistance gene (14). Next, the ftsQE14::kan locus from strain JOE417 was introduced into GK1506(pJC10) via P1 transduction by selecting for Cmr and Kmr clones on LB agar supplemented with antibiotics and arabinose (0.2%) to induce the expression of the plasmid complementing the ftsQ allele. Several clones were then tested for FtsQ depletion by growth in M63 synthetic medium in the absence of arabinose to repress expression from the PBAD promoter (with 0.2% glucose). The cellular filaments were visualized by light microscopy. One clone was chosen and called GK1507.
Similarly, GK1508, a derivative of GK1506 containing a null allele of ftsN, was constructed by using plasmid pJC83, which harbors a wild copy of ftsN under the control of the PBAD promoter (15) and a P1 lysate made with strain JOE565 (ftsN::kan).
Strain GK1510 is a derivative of GK1504 carrying a thermosensitive allele of ftsQ (ftsQ1). To construct this strain, a P1 phage lysate was first prepared on strain MCQ1 (ftsQ1 leu260::Tn10). Tetracycline-resistant (Tcr) clones of the GK1504 recipient cells were isolated by direct selection on LB2 agar plates containing tetracycline and kanamycin after 20 h of incubation at 30°C. Out of the 16 isolated Tcr Kmr candidates, two clones grew as filaments at 42°C; i.e., they harbored the ftsQ1(Ts) allele of the ftsQ gene together with the leu260::Tn10 marker. One of these clones was designated GK1510.
GK1511 is a derivative of GK1503 in which the drug resistance marker of the Δblr::kan cassette was eliminated, as described above for strain GK1506.
GK1512 is a derivative of GK1511 that carries the ftsQ1(Ts) allele of the ftsQ gene together with the leu260::Tn10 marker. It was constructed similarly to strain GK1510, except that candidates were isolated on LB2 agar plates containing tetracycline.
To obtain double mutant strain GK1513, the ΔymgF::kan cassette from Keio Collection strain JW1156 (7) was brought into GK1511 by selecting transductants on LB agar plates supplemented with kanamycin.
Strain GK1514 was constructed similarly by the P1 transduction of the same ΔymgF::kan cassette into the GK1512 strain.
Plasmid constructions.
To construct the recombinant plasmids expressing blr fusions for the BACTH complementation assays, the ORF sequence of blr was PCR amplified by using primers blr1 and blr2 (Table 3) and genomic DNA of MG1655 as a template. The 145-bp amplified blr DNA fragment was digested with BamHI and Acc65I (the restriction sites were included in PCR primers) and subcloned into the corresponding sites of the pKT25 and pUT18C vectors (35). The resulting recombinant plasmids, pKT25-blr and pUT18C-blr, expressed hybrid proteins in which the Blr polypeptide was fused to the C terminus of the T25 or T18 fragment.
Table 3.
Oligonucleotides used in this study
| Oligonucleotide | Sequence (5′–3′)a |
|---|---|
| blr1 | CGCGGATCCCATGAATCGTCTTATTGAATTAACAGGTTGG |
| blr2 | GCGGGTACCTTACTTGTGTTGTACCGAAGC |
| blr3 | CCCGGTACCATGAATCGTCTTATTGAATTAACAGGTTGG |
| blr4 | CCCGGATCCTTACTTGTGTTGTACCGAAGC |
| blr5 | GCGGAGCTCAGGAGGCTGTAATGAATCGTCTTATTG> |
| blr6 | CAGGTTGGATCGTTGTACTAGTCGTTTCAGTCATTCTTCTTG |
| blr7 | CAAGAAGAATGACTGAAACGACTAGTACAACGATCCAACCTG> |
| blr8 | CAGGTTGGATCGTTTGGCCACTTGTCGTTTCAGTCATTCTTC |
| blr9 | GAAGAATGACTGAAACGACAAGTGGCCAAACGATCCAACCTG> |
| blr10 | GAATTAACAGGTTGGATCGTTTGGCCAGTACTAGTCGTTTCAGTCATTCTTCTTGG |
| blr11 | CCAAGAAGAATGACTGAAACGACTAGTACTGGCCAAACGATCCAACCTGTTAATTC> |
| blrrfprev | CCCGGATCCCTTGTGTTGTACCGAAGC |
| ymgF10 | GGGGAGCTCTTGACAGGATTGCCATTAGTAGC> |
| ymgF11 | GGGTCTAGATTATTGCTGATCCCTGTGGTTGACG |
| ftsK1 | CCCGGATCCCATGAGCCAGGAATACATTGAAGAC> |
| ftsK271 | CCCGGTACCCGCTTACCGGAGAACAACGC |
Restriction sites are in boldface type; mismatched bases generating mutations are underlined.
To create recombinant plasmids pKT25-ftsK(1-271) and pUT18C-ftsK(1-271), a fragment DNA encoding a variant of FtsK that contains the first 271 residues of the protein was generated by PCR using primers ftsK1 and ftsK271. After digestion with BamHI and Acc65I, the DNA fragment was ligated with the pKT25 or pUT18C vector, which was cut with the corresponding enzymes.
Plasmid pDSW207-blr was used for the integration of a gfp-blr fusion at the λattB site of the E. coli chromosome. To construct this plasmid, a DNA fragment encoding the ORF of blr was PCR amplified by using primers blr3 and blr4 (Table 3), cut with Acc65I and BamHI, and subcloned into the corresponding sites of plasmid pDSW207 (51). In the resulting plasmid, the expression of the fused green fluorescent protein (GFP)-Blr protein was under the control of an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible modified trc promoter (51).
To create plasmid pDSW913-blr, which codes for the Blr peptide labeled at its C terminus with the mCherry variant of the red fluorescent protein (RFP) (Blr-RFP), a 141-bp DNA encoding the blr ORF without the stop codon was generated from chromosomal DNA with primers blr3 and blr12. The resulting DNA fragment was digested with Acc65I and BamHI and ligated into pDSW913 (6), which had been linearized with the same restriction enzymes.
Plasmid pBAD33-blr contains the blr gene, whose expression is controlled by an arabinose-inducible PBAD promoter (27). To create this plasmid, a 149-bp DNA fragment containing the potential ribosome-binding (RB) site for blr and its ORF was generated by PCR using primers blr5 and blr2 (Table 2). After cleavage with SacI and Acc65I, the DNA fragment was ligated into the corresponding sites of pBAD33.
To construct plasmids expressing the Blr variants with one, two, or three additional amino acids that were inserted into the transmembrane (TM) segment of the Blr polypeptide, we performed site-directed mutagenesis using Pfu Turbo DNA polymerase (Agilent Technologies-Stratagene). The pairs of oligonucleotide primers containing the desired mutations, blr6/blr7 (for the insertion of the amino acid valine), blr8/blr9 (for the insertion of the dipeptide tryptophan-proline), and blr10/blr11 (for the insertion of the tripeptide tryptophan-proline-valine), are shown in Table 3. In brief, the pairs of oligonucleotide primers, each complementary to opposite strands of the mutagenized plasmids (pUT18C-blr or pDSW207-blr), were extended during temperature cycling by Pfu Turbo DNA polymerase. The PCR protocol involved a denaturation step at 95°C (5 min) followed by 18 cycles of 1 min at 95°C, 1 min at 55°C, and 5 min at 68°C. After DpnI treatment, the PCR mixtures were transformed into XL1-Blue cells. The presence of each mutation was further confirmed by DNA sequencing. The resulting plasmids, pUT18C-blrmut1 and pDSW207-blrmut1, code for the hybrid proteins T18-Blrmut1 and GFP-Blrmut1, respectively, which harbor an additional amino acid within the TM segment of Blr; pUT18C-blrmut2 and pDSW207-blrmut2 encode T18-Blrmut2 and GFP-Blrmut2, respectively, with two additional amino acids within the TM segment of Blr; and pUT18C-blrmut3 and pDSW207-blrmut3 encode T18-Blrmut3 and GFP-Blrmut3, respectively, which have three additional amino acids within the TM segment of Blr.
To construct plasmid pBAD33-ymgF, which expresses the E. coli ymgF gene under the control of the PBAD promoter, a 242-bp DNA fragment comprising the RB site and the ORF of ymgF was created by PCR amplification using primers ymgF10 and ymgF11. The fragment was digested with SacI and XbaI and cloned into pBAD33.
All PCR-generated DNA sequences used to construct recombinant plasmids were verified by sequencing.
E. coli BACTH library construction and screening.
A library of E. coli genomic DNA fragments fused to the 3′ end of the T25 ORF was constructed in the pKT25 vector as follows.
Genomic DNA of AK2000 (a Δcya derivative of MG1665) was prepared from a 100-ml culture grown overnight by SDS (sodium dodecyl sulfate)-mediated cell lysis, proteinase K treatment, and phenol extraction, as described previously (18). About 50 μg of the purified genomic DNA in 200 μl of Tris-EDTA (TE) buffer (pH 8.0) was randomly sheared by sonication (to yield fragments ranging from 300 to 1,000 bp). The DNA fragments were end repaired with mung bean nuclease (90 units) in 400 μl of T4 DNA polymerase buffer for 45 min at 30°C, extracted with phenol-chloroform, and recovered by standard ethanol precipitation. Next, the DNA fragments were treated with a mixture of T4 DNA polymerase (90 units) and Klenow fragment (20 units) in the presence of deoxynucleoside triphosphate (dNTP) (200 μM) at 16°C for 1 h. In parallel, the pKT25 vector (10 μg of DNA) was digested with SmaI (100 units with 3 h of incubation at 37°C in the appropriate buffer) and dephosphorylated with shrimp alkaline phosphatase (10 units with 1 h of incubation at 37°C in the same buffer). The linearized vector was gel purified by using a QIAquick gel extraction kit (Qiagen). The blunt-ended DNA fragments were ligated with 5 μg of the SmaI-digested pKT25 vector for 20 h at 16°C using T4 DNA ligase (12 Weiss units). The ligation mixture was recovered by standard ethanol precipitation, and aliquots corresponding to about 100 ng of the ligated pKT25 vector were transformed into 150 μl of electrocompetent ElectroMAX DH10B (Life Technologies-Invitrogen) cells: 20 independent transformations were carried out, each culture was plated onto LB agar-kanamycin (50 μg/ml), and colonies were allowed to grow at 30°C for 36 h. The overall transformation process yielded ∼5 × 105 independent clones. The sizes of the DNA fragments inserted into the pKT25 vector were analyzed in 20 randomly selected clones by PCR amplification: the mean size distribution of the inserts ranged from 200 to 1,500 bp. All the colonies were then pooled, and the plasmid DNA was purified (Plasmid Midi kit; Qiagen) from the mixture and used as a stock for the BACTH plasmid DNA library.
For library screen experiments, the E. coli cell division protein FtsL was used as bait. Plasmid pUT18C-ftsL (which encodes the T18-FtsL hybrid protein) was first introduced into DHM1. The resultant strain, DHM1(pUT18C-ftsL), was rendered electrocompetent and transformed with an E. coli DNA library using 50 to 100 ng of DNA. After incubation in LB nutrient broth at 30°C for 90 min, cells were collected by centrifugation, washed several times with M63 synthetic medium, and then spread onto M63 agar plates supplemented with maltose (0.2%), 5-bromo-4-chloro-3-indolyl-β-d-galactoside (X-Gal) (40 μg/ml), IPTG (0.5 mM), kanamycin (25 μg/ml), and ampicillin (50 μg/ml) (∼5 × 105 transformants per plate). Plates were incubated at 30°C for 5 to 10 days until the appearance of blue Cya+ (Mal+ and Lac+) colonies, which were reisolated and further characterized by the sequencing of the DNA inserts from the corresponding plasmids.
BACTH complementation assays.
For BACTH complementation assays, Blr variants were genetically fused to T25 or T18 fragments of the catalytic domain of Bordetella pertussis adenylate cyclase and coexpressed with various E. coli divisomal proteins in DHM1 cells. After transformation, cells harboring a pair of the appropriate plasmids were plated onto LB agar containing X-Gal and IPTG plus antibiotics and incubated at 30°C for 24 to 36 h. The efficiency of the interaction between two tested hybrid proteins was quantified by measuring the β-galactosidase (β-Gal) activity in liquid cultures in a 96-well format based on an assay described previously (26, 46). For each set of transformations, the β-Gal assay was performed on eight cultures that were grown overnight at 30°C in 300 μl LB broth in the presence of 0.5 mM IPTG and the appropriate antibiotics in a 96-well microtiter plate (2.2-ml 96-well storage plate; Thermo Fisher Scientific). Before measurement, the cultures were diluted 1:5 into M63 medium in the same microplate, and 175 μl of the diluted culture from each well was transferred into a flat-bottom microtiter plate to record the optical density at 595 nm (OD595) absorbance data. To permeabilize cells, 7 μl of 0.05% SDS and 10 μl of chloroform were added to 200 μl of bacterial suspensions, which were previously transferred onto another plate (1.2-ml polypropylene 96-well storage block; Thermo Fisher Scientific). The solutions were vigorously intermixed and incubated at room temperature for 30 to 40 min (to evaporate the chloroform). For the enzymatic reaction, aliquots (20 μl) of the permeabilized cells were added to a microtiter plate, the wells of which contained 105 μl of a reaction mixture containing PM2 buffer (70 mM Na2HPO4 · 12H2O, 30 mM NaHPO4 · H2O, 1 mM MgSO4, and 0.2 mM MnSO4 [pH 7.0] plus 100 mM β-mercaptoethanol) and 0.1% o-nitrophenol-β-galactoside (ONPG). To develop the enzymatic reaction, the plate was incubated at room temperature for 20 to 30 min. The reaction was stopped by the addition of 50 μl of 1 M Na2CO3 to the mixture, and the OD405 absorbance data were recorded. All the absorbance readings were collected by using a GENios plate reader (Tecan Group Ltd.) and analyzed with Microsoft Excel software. The enzymatic activity, A (in relative units), was calculated according to the following equation: A = 1,000 × (OD405 − OD405 in control well)/(OD595 − OD595 in control well)/t (min) of incubation.
Microscopy analyses.
Fluorescence microscopy studies to localize GFP-tagged Blr were carried out mainly on E. coli cells, which contained a single copy of the gfp-blr fusion inserted at the λattB site of the bacterial chromosome and had no wild-type blr gene (GK1506 and its derivatives). In addition, strain GK1504(pDSW207-blr), harboring the same gfp-blr fusion expressed from plasmid pDSW207, was utilized. In the localization study with a blr-rfp fusion, strain GK1504(pDSW913-blr) was used.
For fluorescence microscopy, cells were prepared as described previously (34). Briefly, cultures grown overnight were diluted 1:100 or 1:200 in M63 minimal medium containing IPTG (5 to 50 μM), Casamino Acids (50 μg/ml), and glucose (0.2%) plus the appropriate antibiotics and incubated until the early exponential phase for 3 to 4 h at 30°C. To induce the expression of Blr-RFP, 1 mM IPTG was used.
For FtsQ depletion experiments using the conditional PBAD promoter, GK1507 was grown in LB broth supplemented with arabinose (0.2%), ampicillin (25 μg/ml), chloramphenicol (30 μg/ml), and kanamycin (50 μg/ml) at 30°C overnight. The next day, cells were diluted 1:200 into M63 medium containing 5 μM IPTG, 0.2% glucose, and the appropriate antibiotics and grown for 3 to 5 h until cell filamentation was visible. For FtsN depletion experiments, GK1508 cells were treated similarly.
To analyze the effect of Blr (or YmgF) overproduction on the MCQ1 phenotype, cultures of strains MCQ1(pBAD33), MCQ1(pBAD33-blr), and MCQ1(pBAD33-ymgF) grown overnight in LB2 broth plus arabinose (0.2%) and the corresponding antibiotics were diluted 1:200 in M63 medium supplemented with Casamino Acids, chloramphenicol (15 μg/ml), tetracycline (15 μg/ml), and either arabinose (0.2%) or glucose (0.2%) and incubated for 4 to 10 h at 30°C.
To visualize the effect of the Blr inactivation, GK1510 cells harboring a temperature-sensitive allele of ftsQ (ftsQ1) and a blr null mutation (Δblr::kan) were grown overnight in LB2 at 30°C. The next day, the cells were diluted 1:200 in LB broth and incubated for 4 h at 30°C. The control parental strains, MCQ1 (ftsQ1) and GK1504 (Δblr::kan), were treated similarly to GK1510.
For complementation assays, the strains were grown in LB2 plus appropriate antibiotics at 30°C overnight. The next day, they were diluted 1:200 in LB broth supplemented with ampicillin and IPTG and incubated for 4 h at 30°C.
Images of living, nonfixed cells were acquired with a Nikon Eclipse 80i epifluorescence microscope equipped with a 100× Plan-Apo oil immersion objective and a 100-W mercury lamp. The mCherry variant of RFP was visualized with a TE/ZP MCherry filter (Nikon). Images were captured with a 5-megapixel color charge-coupled-device (CCD) DS-5Mc camera and processed by using Adobe Photoshop or ImageJ software (1).
Protein localization analysis.
In order to examine the subcellular localizations of Blr and its modified variants Blrmut1, Blrmut2, and Blrmut3, we used plasmid pKTop, which encodes a dual pho-lac reporter and allows the determination of protein localization in vivo (34). DNA fragments corresponding to the coding region of Blr or of the different variants were generated by PCR from the corresponding pUT18C-derived plasmids by using oligonucleotide pair blr1 and blr2 (Table 3) and cloned between the BamHI and Acc65I sites of pKTop. For cellular localization assays in vivo, the resulting plasmids were transformed into E. coli DH5α cells. A 10-μl aliquot from each transformation was spotted onto a dual-indicator LB agar plate containing 6-chloro-3-indolyl-β-d-galactopyranoside (Red-Gal; Sigma) at 100 μg/ml, 5-bromo-4-chloro-3-indolyl phosphate disodium salt (X-Phos; Sigma) at 80 μg/ml, IPTG (1 mM), 50 mM phosphate buffer (pH 7.0), and 50 μg/ml kanamycin. The plates were scanned after 36 h of growth at 30°C.
Western blot analysis.
Western blot analysis was performed to determine the stability of the different Blr variants fused to the T18 fragment (T18-Blr, T18-Blrmut1, T18-Blrmut2, or T18-Blrmut3). For this, the plasmids encoding the different fusions were cotransformed with pKT25-ftsQ into DHM1 cells. The transformants were plated onto LB–X-Gal–IPTG agar and incubated at 30°C for 24 h. Cultures of transformed cells grown overnight were diluted 1:100 into fresh LB broth supplemented with kanamycin, ampicillin, and 2 mM cyclic AMP (cAMP) (to allow for equal conditions of expression of interacting and noninteracting partners, given that the T18 hybrids were expressed under a cAMP-dependent lac promoter) and incubated for 3 h at 30°C. IPTG (0.5 mM) was then added, and the cells were incubated for another 3 h at 30°C. The cells were harvested by centrifugation at 7,000 × g for 10 min at 4°C and resuspended in B-PER bacterial protein extraction buffer (Thermo Scientific Fisher) containing 8 M urea (the volumes were normalized to the optical density at 600 nm of each culture). Proteins were separated on a NuPage Novex 4 to 12% Bis-Tris SDS-PAGE gel (Life Technologies-Invitrogen) and electrotransferred onto a polyvinylidene difluoride (PVDF) membrane (Pall Life Sciences) by using the iBlot dry blotting system (Life Technologies-Invitrogen). The membrane was blocked in 5% milk with 0.05% Tween 20 for 1 h, washed, and incubated with anti-CyaA mouse monoclonal antibody (Ab) 3D1 (dilution, 1:1,000), which recognizes a sequence at the C terminus of the T18 fragment (Santa Cruz Biotechnology). After extensive washing, the membrane was incubated with horseradish peroxidase-labeled goat anti-mouse whole Ab (dilution, 1:5,000; GE Healthcare Life Sciences). Bound horseradish peroxidase-labeled Abs were detected with the ECL-Plus detection system (GE Healthcare Life Sciences), exposed to film, and developed. The image of the film was then scanned and processed with Adobe Photoshop software.
RESULTS
Isolation of Blr as an interacting partner of E. coli divisomal protein FtsL.
To isolate novel putative components of the E. coli cell division apparatus, we performed a bacterial two-hybrid (BACTH) screen (33) using FtsL, a core, bitopic membrane component of the E. coli divisome, as bait. In a previous study, this approach allowed us to identify YmgF, a 72-amino-acid integral membrane protein, as a new divisomal protein from E. coli (34). In the BACTH system, the proteins of interest are genetically fused to two fragments (T25 and T18) of the catalytic domain of B. pertussis adenylate cyclase and coexpressed in an E. coli cya strain (i.e., lacking its endogenous adenylate cyclase). Upon an interaction between the hybrid proteins, the enzymatic activity of the separately inactive T25 and T18 fragments is restored, leading to cAMP synthesis and, in turn, to the transcriptional activation of catabolic operons (such as the lac operon or the mal regulon) (33).
For two-hybrid screening assays, we first constructed a library of genomic E. coli chromosomal DNA fragments fused to the 3′ end of the T25 ORF in the BACTH vector pKT25. In this genomic library, hybrid proteins are expressed from low-copy-number plasmid pKT25, at variance with our previously reported E. coli libraries, which were made with a high-copy-number vector, pUT18C (34). We anticipated that, with this new library, a lower level of expression of the hybrid proteins could enlarge the repertoire of fusion proteins expressed in a functional form (e.g., integral membrane proteins with multiple transmembrane segments) and could also diminish the potential toxicity associated with the overproduction of certain hybrids. Genomic DNA of an E. coli Δcya strain (AK2000, a Δcya derivative of MG1655) was randomly fragmented by sonication (size range of 500 to 1,500 bp), end repaired, and cloned into the SmaI site of pKT25 (see Materials and Methods). After transformation into DH10B cells, ∼5 × 105 independent clones were obtained. All these colonies were pooled, and their plasmid DNA was purified and used as a stock for the BACTH DNA library.
The pooled plasmid DNA was used to transform competent E. coli DHM1 cells harboring plasmid pUT18C-ftsL, which expresses a T18-FtsL hybrid protein (32). The transformed cells were plated onto synthetic solid medium supplemented with maltose as the only carbon source, kanamycin, ampicillin, IPTG, and X-Gal and incubated during 5 to 10 days. Thirty-four cya+ clones were isolated out of a total of ∼2 × 106 transformants. The resulting cya+ clones were further analyzed by the sequencing of the DNA fragments inserted into pKT25 library plasmids. Among the 34 cya+ clones, 9 clones were found to harbor the same fragment of the FtsW protein fused in frame to T25 at residue Leu26 (before the first FtsW transmembrane segment) (36), and one clone expressed a fusion with FtsI (Table 4). Three other E. coli ORFs (Blr, YfgM, and RseA) were found to be fused in frame with the T25 moiety in at least three independent clones, and one (Flk) was found in two independent clones (Table 4). All other cya+ clones were disregarded, as they harbored (i) genes picked up only a single time, (ii) out-of-frame fragments, (iii) fragments inserted in the antisense orientation, or (iv) noncoding DNA.
Table 4.
FtsL-binding proteins identified by the BACTH screena
| Protein (size [aa]) | No. of isolated clones | Location of protein fragment fused to T25 (aa) | Localization | Function |
|---|---|---|---|---|
| FtsW (414) | 7 | 26–213b | IM | Cell division |
| FtsI (588) | 1 | 9–193b | IM | Cell division |
| Blr (41) | 3 | 3–41 | IM | β-Lactam resistance |
| YfgM (206) | 3 | 17–206 | IM | Unknown |
| RseA (216) | 3 | 68–216 | IM | Anti-sigma E factor |
| Flk (331) | 2 | 114–331 | IM | Predicted flagellar assembly protein |
aa, amino acids; IM, bacterial inner membrane.
The last codon correctly determined by DNA sequencing though the in-frame DNA insert may extend beyond that point.
In the present study, we further characterized the Blr polypeptide, as we had previously isolated (albeit as a single clone) this polypeptide in a two-hybrid screen of FtsL interactors using another E. coli genomic library made with the pUT18C vector (32). In addition, Wong and collaborators previously showed that a TnphoA insertion within the blr gene increased the susceptibility of E. coli cells to certain drugs that inhibit peptidoglycan synthesis (52). Consequently, we hypothesize that the Blr polypeptide might be involved in the E. coli cell division process. Blr is a 41-amino-acid peptide predicted to have a single transmembrane segment (extending from residues 5 to 25) with an amino-terminal end in the cytosol and a carboxy terminus exposed to the periplasm (Topcons [http://topcons.net/]), a topology that was confirmed by Wong et al. with a TnphoA fusion (52). The clones isolated in the present screen encode a T25 fusion with residues 3 to 41 of Blr (while the clone isolated in our prior screen encoded a T18 fusion with residues 4 to 41).
Blr interacts in vivo with E. coli divisome proteins.
We first examined the ability of Blr to interact with other known components of the E. coli cell division machinery by performing BACTH complementation assays. For these experiments, the full-length blr coding region was PCR amplified and cloned into the pKT25 or pUT18C vector to generate plasmids expressing the Blr peptide fused to the C terminus of the T25 fragment (hybrid protein T25-Blr) or the T18 fragment (hybrid protein T18-Blr). The Blr hybrid proteins were then coexpressed in DHM1 cells with various E. coli divisomal proteins fused to the complementary T25 or T18 fragments. The efficiency of complementation between pairs of hybrid proteins was quantified by measuring β-galactosidase activities, as described in Materials and Methods.
The results of the BACTH complementation assays, shown in Fig. 1, demonstrated that, in addition to FtsL, Blr could efficiently interact with several other E. coli cell division proteins. All of them are integral membrane proteins. These interactions were established in both BACTH configurations, that is, when Blr was fused to T25 (Fig. 1, filled bars) as well as when it was fused to T18 (Fig. 1, open bars), except for the interaction involving FtsW. In that case, an interaction was detected only with the T25-FtsW fusion, as we previously observed that the T18-FtsW hybrid protein was nonfunctional for unknown reasons (32).
Fig 1.

Blr is able to interact with the E. coli cell division proteins. Shown are data for BACTH analyses of associations between the indicated E. coli cell division proteins and Blr fused to T25 or T18. The efficiencies of functional complementation between the indicated hybrid proteins were quantified by measuring beta-galactosidase (β-Gal) activities in E. coli DHM1 cells carrying the corresponding plasmids, as described in Materials and Methods. Interactions are reported as relative units of β-Gal activity. FtsK* corresponds to the first 271 residues of the FtsK protein.
In sum, these data indicate that Blr, which could associate with several E. coli cell division proteins, might be a part of the bacterial divisome.
Blr localizes to the E. coli division septum.
To study the subcellular localization of Blr, we used a hybrid protein in which GFP (green fluorescent protein) was attached to the amino terminus of Blr. For this, we constructed strain GK1506, a derivative of wild-type strain MG1655 in which a gfp-blr translational fusion, driven by an IPTG-inducible modified trc promoter (51), was inserted into the bacterial chromosome at the lambda attachment site, while the wild-type blr gene was replaced by a kanamycin cassette.
For fluorescence microscopy analysis, the cells expressing the GFP-Blr hybrid protein were grown in M63 synthetic medium in the presence of 5 to 20 μM IPTG until the early exponential phase, as described in Materials and Methods. Under these conditions, the expression of GFP-Blr did not interfere with the division process, as the cells displayed wild-type morphology and normal growth curves (data not shown). As shown in Fig. 2, a distinct fluorescent signal could be detected at midcell, indicating that Blr tagged with GFP was localized at the septum of dividing E. coli cells. The accumulation of GFP-Blr at midcell was observed for ∼40% of the cells (Table 5), while the others showed mainly a membrane-associated fluorescence signal. Moreover, ∼63% of the cells with a GFP-Blr septal localization were clearly undergoing cell division, as indicated by the presence of a visible septal constriction. Interestingly, the septum localization of GFP-Blr could be detected only in the absence of the wild-type copy of the blr gene (Table 5), suggesting that the endogenous native Blr protein might efficiently compete with GFP-Blr for recruitment to the divisome.
Fig 2.

Localization pattern of GFP-Blr. GK1506 cells expressing the chromosomally localized GFP-Blr fusion were grown in minimal medium in the presence of 20 μM IPTG at 30°C and examined by fluorescence microscopy. The arrow points to the septal localization of GFP-Blr in one of the dividing cells. Scale bar, 5 μm.
Table 5.
Septal localization of the fluorescently labeled Blr in E. coli cells
| Strain | Characteristic | No. of scored cells | No. of cells with GFP (or RFP) signal at septum (% of total) | No. of cells with GFP (or RFP) signal at septum undergoing division (% of septal localization) |
|---|---|---|---|---|
| GK1506 | MG1665 Δblr::ftr gfp-blr fusion intergrated at λatt site | 458 | 189 (41) | 120 (63) |
| GK1504(pDSW207-blr) | MC4100 Δblr::kan gfp-blr fusion expressed from multicopy plasmid | 471 | 152 (30) | 108 (71) |
| GK1504 (pDSW913-blr) | MC4100 Δblr::kan blr-rfp fusion expressed from multicopy plasmid | 238 | 36 (15) | 25 (69) |
| MC4100(pDSW207-blr) | MC4100 gfp-blr fusion expressed from multicopy plasmid | 253 | 0 (0) | 0 |
A comparable midcell localization of GFP-Blr was found in another genetic background, namely, in strain GK1504, a blr::kan derivative of MC4100. In these cells, the GFP-Blr fusion protein was expressed from a multicopy plasmid. GK1504(pDSW207-blr) cells displayed the same pattern of fluorescent signals as that shown by strain GK1506 (Table 5). A septal localization was also observed when the Blr polypeptide was tagged at its carboxy terminus with the mCherry variant of RFP (Blr-RFP), although only ∼15% of the scored cells displayed a distinct midcell recruitment of the hybrid protein. It is possible that the Blr-RFP fusion was less stable than GFP-Blr or that, when modified at its carboxy terminus, Blr associated less efficiently with the E. coli divisome (Table 5). The septal localization pattern of Blr identified this protein as a late-recruited component of the E. coli divisome, such as FtsN, AmiC, YmgF, DedD, and RlpA (2, 9, 23, 34, 41).
Altogether, the present results indicate that Blr is able to localize at midcell.
Blr septal localization requires the assembly of the E. coli core divisome.
To test whether the recruitment or GFP-Blr to the cell division site was dependent on the presence of other E. coli Fts proteins, we analyzed the septal localization of the GFP-Blr fusion in strains that could be selectively depleted in the E. coli cell division proteins FtsQ and FtsN.
Therefore, we constructed a ΔftsQ strain (GK1507) containing a chromosomal insertion of ftsQE14::kan and plasmid pJC10, which harbors a wild-type copy of ftsQ under the control of an arabinose-dependent PBAD promoter. In the same way, strain GK1508, which contains the chromosomal insertion ftsN::kan and a plasmid encoding a wild-type copy of the ftsN gene under the control of the PBAD promoter (pJC83), was constructed. In both strains GK1507 and GK1508, the depletion of FtsQ or FtsN, respectively, could be obtained by replacing arabinose with glucose in the growth medium. As expected, in the presence of arabinose, cells of both strains displayed wild-type morphology, and the GFP-Blr fusion was found to localize at midcell (Fig. 3, insets). However, when the strains were grown in the presence of glucose, cells formed filaments, which is indicative of a significant depletion of the corresponding E. coli cell division proteins. In those filamentous cells, the GFP fluorescent signal was dispersed homogenously throughout the cells, meaning that under these conditions, GFP-Blr did not localize at midcell. We conclude that Blr requires the presence of the complete E. coli divisome to be targeted to the septum.
Fig 3.
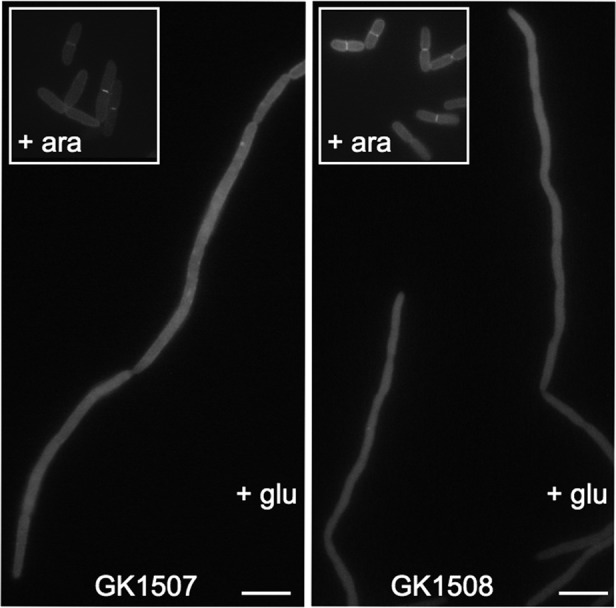
Blr localization in FtsQ- and FtsN-depleted strains. GFP fluorescence images of filaments built by GK1507 and GK1508 cells during growth in the presence of glucose are shown. The insets show the GFP-Blr septal localization in the same cells grown under permissive conditions (in the presence of 0.2% arabinose). For fluorescence microscopy, cells were prepared as described in Materials and Methods. Scale bars, 5 μm.
Blr is critical for E. coli ftsQ1(Ts) cells to grow under low-osmolarity conditions.
The Blr peptide is dispensable for E. coli cell viability under standard growth conditions (rich LB medium at 37°C) (7). We further examined the cells lacking Blr (i.e., GK1503, GK1504, and GK1506 harboring the disrupted blr gene) grown under various conditions (in rich or synthetic media at 30°C or 42°C) but could not detect any obvious division defects (data not shown). At variance with the original report of Wong and colleagues (52), in our hands, the Δblr strains did not exhibit any significant difference in susceptibility toward beta-lactam antibiotics (data not shown). Additionally, we could not confirm the results reported previously by Hobbs and coauthors, who showed that a Δblr mutant exhibited a reduced sensitivity to cell envelope stress compared to wild-type cells (30). When the Δblr (GK1506) cells were grown in one-to-one competition assays against otherwise wild-type cells in LB medium supplemented with SDS or EDTA, as described previously (30), no significant changes in the relative proportions of the Δblr or wild-type cells could be detected (data not shown). Also, when we checked whether the overproduction of Blr might affect bacterial growth and/or cell morphology, we did not notice any significant difference compared to wild-type cells.
To determine whether Blr might play some role in bacterial cell division, we examined the effect of the overproduction or inactivation of Blr on the growths of various conditional E. coli fts mutants, with the hope of finding mutant combinations that could be synthetically lethal or cause cell sickness.
Among the four different fts temperature-sensitive alleles tested, ftsZ84(Ts), ftsA12(Ts), ftsI23(Ts), or ftsQ1(Ts), significant results were obtained only with strains carrying a missense mutation within the ftsQ gene, ftsQ1(Ts), resulting from a change of Glu125 to Lys (48), which causes temperature-sensitive cell division in E. coli. Like a number of fts mutants, the bacteria carrying the ftsQ1(Ts) mutation also displayed an osmosensitive phenotype; i.e., they exhibited a filamentous phenotype when grown at a permissive temperature (30°C) in low-salt media (e.g., in minimal medium or in LB medium lacking NaCl), while normal growth was observed in LB medium containing at least 0.5% NaCl (8, 34, 42–44, 48).
We first examined whether the overproduction of the Blr peptide might rescue the osmosensitivity of MCQ1 cells carrying the ftsQ1(Ts) allele. Here, MCQ1 cells were transformed with plasmid pBAD33-blr, which expresses the blr gene under the control of an arabinose-inducible PBAD promoter, or, as a control, with empty plasmid pBAD33. As shown in Fig. 4, MCQ1(pBAD33) cells, harboring the empty vector, could not divide in minimal medium in the presence of arabinose or glucose and formed long filaments. In contrast, MCQ1(pBAD33-blr) cells exhibited wild-type morphology when Blr was expressed in the presence of arabinose but formed filaments in the presence of glucose. However, the overproduction of Blr could not rescue the thermosensitive defect of ftsQ1 cells. Interestingly, similar results were obtained previously with YmgF, another small membrane polypeptide that associates with the E. coli cell division machinery (34).
Fig 4.

Overproduction of Blr (or YmgF) rescues the osmosensitive phenotype but not the temperature-sensitive phenotype of ftsQ1(Ts) cells. Shown are phase-contrast micrographs of strains MCQ1(pBAD33), MCQ1(pBAD33-blr), and MCQ1(pBAD33-ymgF) grown in M63 minimal medium supplemented with Casamino Acids (and appropriate antibiotics). The left panels show cells growing in the presence of arabinose, which allows the expression of the genes controlled by the PBAD promoter. The middle panels show cells growing in the presence of glucose. The right panels show cells grown in LB medium in the presence of arabinose at 42°C (i.e., at a nonpermissive temperature). Scale bars, 5 μm.
Conversely, we investigated the effect of the disruption of the blr gene on the growth of ftsQ1(Ts) cells. Therefore, we constructed an ftsQ1(Ts) Δblr::kan double mutant strain (GK1510). Phenotypic analysis showed that the inactivation of blr rendered the ftsQ1(Ts) cells more sensitive to the osmolarity conditions. As shown in Fig. 5, the morphology of ftsQ1 Δblr cells grown at 30°C in LB medium containing 0.5% NaCl was strongly altered compared to the growth of the ftsQ1 or Δblr single mutant under the same conditions. After 4 h of incubation, the GK1510 cells were elongated, indicating severe problems in cell division, in contrast to the parental, single mutant strains, which grew robustly and exhibited normal morphology under these conditions. We conclude that the deletion of the blr gene increased the osmosensitivity of the ftsQ1(Ts) cells.
Fig 5.
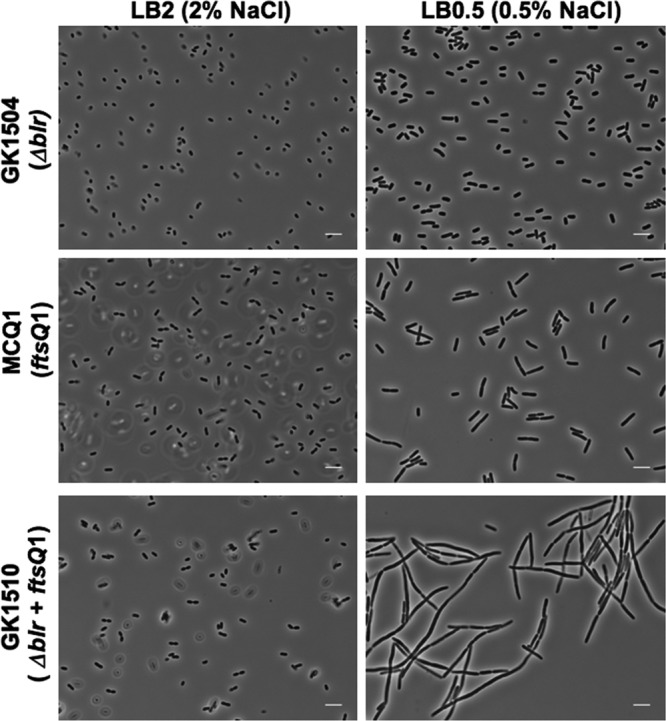
Effect of Blr inactivation on the growth of E. coli ftsQ1(Ts) cells. Shown are phase-contrast micrographs of cells grown in LB broth with different concentrations of salt (LB2 [2% NaCl] or LB [0.5% NaCl]). GK1510 and the two parental strains MCQ1 and GK1504 were grown as described in Materials and Methods. Scale bars, 5 μm.
Finally, to examine whether the two small membrane polypeptides Blr and YmgF (34) could have overlapping functions in cell division, we constructed a blr ymgF double mutant and found that it had no detectable phenotype. Furthermore, the blr ymgF ftsQ1(Ts) triple mutant did not exhibit a synergistic phenotype compared to that of the blr ftsQ1(Ts) or ymgF ftsQ1(Ts) double mutant (data not shown). Hence, Blr and YmgF might play a similar stabilization function toward FtsQ1(Ts) independently of each other.
Analysis of Blr variants modified in the transmembrane segment.
To determine whether the biological functionality of Blr was correlated with its ability to interact with the different E. coli cell division proteins, we constructed Blr variants by introducing one, two, or three amino acids (Blrmut1, Blrmut2, or Blrmut3, respectively) in the middle of the predicted helical transmembrane segment of the peptide. For this, template plasmids pUT18C-blr and pDSW207-blr were mutagenized as described in Materials and Methods. The modified Blr variants were then characterized for their ability to associate with the Fts components, to localize to the septum, and to rescue the increased osmosensitivity of the ftsQ1(Ts) Δblr double mutant cells.
As shown in Fig. 6a, the insertion of a single amino acid, Val, between residues Val12 and Leu13 (variant Blrmut1) reduced the association with four out of seven tested Fts partners (FtsI, FtsK, FtsN, and YmgF) but did not affect the interactions with FtsL, FtsQ, and FtsW. The introduction of two (Trp-Pro [Blrmut2]) or three (Trp-Pro-Val [Blrmut3]) (data not shown) residues at the same position diminished drastically the interaction of the Blr variants with all tested Fts components.
Fig 6.

Mutagenesis analysis of the Blr TM region. (a) BACTH analysis of interactions between the modified Blr variants and E. coli cell division proteins. The variants of Blr fused to T18 were coexpressed in DHM1 cells with the indicated E. coli cell division proteins fused to T25. The efficiency of functional complementation between the indicated hybrid proteins was quantified by measuring beta-galactosidase (β-Gal) activities as described in Materials and Methods. Interactions are reported as relative (rel) units of β-Gal. (b) Stabilities of the different Blr variants fused to the T18 fragment. Western blot analysis of total protein extracts from cells grown in the presence of 2 mM cAMP and 0.5 mM IPTG at 30°C was performed as described in Materials and Methods. Equivalent amounts of protein samples were loaded into each line. The blot was probed with an anti-T18 monoclonal Ab. Molecular mass standards in kilodaltons are indicated on the left (M). (c) Experimental determination of cellular localizations of the Blr variants. E. coli DH5α cells expressing the different Blr′-PhoLac fusions (“−” corresponds to the empty pKTop vector expressing a cytosolic PhoLac) were spotted onto a dual-indicator plate containing Red-Gal (for β-galactosidase activity) and X-Phos (for phosphatase activity) and incubated for 36 h at 30°C. The blue coloration (high phosphatase activity) of the colonies indicates a membrane or periplasmic localization of a tested protein. The red coloration of the colonies (high β-galactosidase activity) indicates a cytosolic localization.
Western blot analysis using an anti-T18 monoclonal antibody showed that all Blr variants were produced at the same level as that of the wild-type protein (Fig. 6b), indicating that the defects in two-hybrid associations were not due to a diminished stability of the modified variants of Blr. We also checked with an alkaline phosphatase (PhoA) fusion approach that the modifications introduced into the TM part of Blr did not impair the correct insertion of the corresponding polypeptides in the inner membrane. As shown in Fig. 6c, bacteria expressing PhoA fused to the C terminus of the different Blr variants showed high alkaline phosphatase activity, demonstrating a periplasmic localization of the PhoA reporter. We conclude that the mutations within the TM part of Blr affected only the protein interaction with its partners but neither its stability nor its proper insertion into the inner membrane.
In good agreement with the interaction properties of the Blr variants, GFP-Blrmut1, which maintained the capacity to associate with a number of Fts partners, could localize at midcell although with a reduced frequency compared to that of the wild-type GFP-Blr protein. The GFP-Blrmut2 and GFP-Blrmut3 fusions, which could not interact with any of the tested E. coli cell division proteins, could not be detected at midcell (Table 6).
Table 6.
Septal localizations of the different variants of Blra
| Strain | GFP-fused protein | Amino acid(s) inserted between Val12 and Leu13 | No. of scored cells | % of cells with GFP signal at septum |
|---|---|---|---|---|
| GK1504(pDSW207-blr) | Blr | None | 121 | 33 |
| GK1504(pDSW207-blrmut1) | Blrmut1 | Val | 112 | 16 |
| GK1504(pDSW207-blrmut2) | Blrmut2 | Trp-Pro | 153 | <1 |
| GK1504(pDSW207-blrmut3) | Blrmut3 | Trp-Pro-Val | 293 | <1 |
The results from a representative experiment out of three independent ones are shown.
Furthermore, when overproduced, the GFP-Blrmut1 fusion protein allowed GK1510 to overcome the osmosensitive defect: GK1510(pDSW207-blrmut1) cells grew normally in the presence of moderate salt concentrations (LB medium). In contrast, GK1510 cells expressing either GFP-Blrmut2 (Fig. 7) or GFP-Blrmut3 (data not show) displayed a filamentous phenotype when grown under the same conditions.
Fig 7.
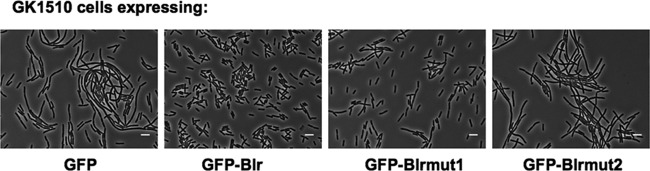
Functional assay of the modified Blr variants. Shown are phase-contrast micrographs of GK1510 cells expressing the modified variants of Blr fused to GFP. Cells were grown in LB medium (0.5% NaCl) for 4 h at 30°C.
Taken together, these results show that the capability of Blr to rescue the osmosensitive phenotype of the ftsQ1(Ts) allele is correlated directly with its ability to associate with the E. coli cell division machinery. These results also indicate that the transmembrane domain of Blr is a major determinant of its interaction with the different Fts partners.
DISCUSSION
In the present study, we identified the β-lactam resistance protein Blr, a short, 41-amino-acid, membrane-associated polypeptide, as a novel component of the cell division machinery of Escherichia coli. Blr was isolated by a two-hybrid screen as an interacting partner of FtsL and was shown to interact efficiently with several core components of the divisome apparatus. Fluorescence imaging revealed that Blr was indeed recruited to the division site in an FtsQ- and FtsN-dependent manner. The inactivation of Blr was shown to exacerbate the osmosensitivity of E. coli ftsQ1(Ts) cells harboring a thermosensitive variant of the essential cell division protein FtsQ. In contrast, when overproduced, Blr allowed E. coli ftsQ1(Ts) cells to overcome their intrinsic osmosensibility and to grow under low-salt growth conditions. Taken together, our findings indicate that the small membrane polypeptide Blr could be a novel component of the E. coli cell division machinery.
Blr was originally identified by Wong and colleagues, who reported that the inactivation of this peptide resulted in a 2- to 4-fold increase in the intrinsic susceptibility of the host strain to a wide spectrum of beta-lactam antibiotics or other drugs that inhibit PG synthesis (52). As Blr was shown to be an integral membrane peptide, those authors hypothesized that it could be part of an efflux pump or involved in murein metabolism, but they did not provide any clues about its possible molecular mode of action. More recently, Blr was independently found by Hobbs and colleagues during a large-scale screening of small proteins that might be involved in the resistance of E. coli to cell envelope stress and/or acid shock (30). Those authors reported that a blr deletion mutant displayed an increased resistance to cell envelope stress, triggered by exposure to SDS and EDTA, compared to an otherwise wild-type E coli cell. Again, no detailed molecular mechanism was proposed to explain the Blr-induced sensitivity to envelope stress. Unfortunately, we could not replicate these results with Δblr strains of various genetic backgrounds. This finding suggests that the potential phenotype associated with the deletion of blr may be highly dependent upon the specific genetic background of the bacteria. In our hands, the most pronounced effect of the blr deletion was observed for E. coli cells harboring a particular thermosensitive allele of the cell division protein FtsQ. This ftsQ1(Ts) mutation also conferred marked osmosensitivity, as the cells became highly filamentous when grown at a permissive temperature in low-salt medium (LB medium without NaCl or minimal medium) (8, 34, 48). We found that in the absence of Blr, the osmosensitive defect of ftsQ1(Ts) can be detected even in medium with a moderate salt concentration (0.5% NaCl).
As it was previously shown that the FtsQ1(Ts) variant protein displayed a reduced stability even at a permissive temperature (12), one could hypothesize that Blr could contribute to the stabilization of the modified FtsQ protein under low-salt “stress” conditions. Such a stabilization could be achieved through direct protein-protein interactions with FtsQ and/or with other components of the divisome. Indeed, we could show that 2- or 3-amino-acid insertions within the transmembrane segment of Blr abolished its associations with all the tested divisomal partners and also abrogated its ability to rescue the osmosensitivity of cells carrying the ftsQ1(Ts) allele.
Blr appears to play a role similar to that of YmgF, another small membrane protein that interacts with many Fts proteins and that can stabilize the FtsQ1(Ts) variant (34). One striking characteristic of the Blr peptide, also shared by YmgF, is its ability to interact with several divisomal proteins. This is quite remarkable given the small size of the polypeptide, made essentially of a single transmembrane segment (or two in the case of YmgF). How Blr (or YmgF) can associate selectively with the different components of the divisome and stabilize their assembly into a functional machinery that is able to complete septation is an intriguing question. One could hypothesize that these peptides may be important to “fill up” the membrane space in between the different components of the divisome and may allow them to adjust to the increased curvature of the inner membrane bilayer as it progressively invaginates to segregate into the two future daughter cells. These adaptor proteins could thus contribute to the stabilization of the divisome and might become critical in cells exposed to certain stress conditions, as evidenced here or in previous studies (30, 52).
Interestingly, Modell et al. recently identified a small inner membrane protein of 29 residues in Caulobacter crescentus, SidA, that is induced in response to DNA damage and that can inhibit cell division by binding directly to the FtsW protein to prevent the final constriction of the Z ring (39). A number of recent studies also highlighted the role of small proteins in membrane-associated macromolecular machineries (for reviews, see references 4 and 31). Several small conserved polypeptides are intrinsic constituents of photosystems in plants and cyanobacteria and contribute to the stabilization of these complex machineries (for a review, see reference 31). The KdpABC potassium transporter of E. coli is stabilized in vitro by a 29-residue-long polypeptide, KdpF (22), while in Salmonella enterica serovar Typhimurium, the degradation of MgtC, a virulence factor important for intramacrophage survival, is regulated by a 30-residue-long membrane-associated peptide, MgtR (5). Other integral membrane peptides were recently described as a new class of regulators of two-component systems, which interact with membrane-associated sensor kinases and modulate their activity (for a review, see reference 31). How such small membrane-associated peptides can modulate the stability and/or the activity of cognate membrane protein complexes at the molecular level remains largely unknown. Further studies will be needed to clarify the precise role and mechanisms of action of the small membrane polypeptides Blr and YmgF in the regulation of the bacterial divisome.
ACKNOWLEDGMENTS
We thank David Weiss, the Coli Genetic Stock Center, and the National Bio-Resource Project (NIG, Japan) for providing us with various E. coli strains and plasmids. We are greatly indebted to Agnes Ullmann and Scot Oullette for helpful discussions, insightful comments on the manuscript, and encouragement, and we thank other members of the Ladant laboratory for their constant interest.
This work was supported by the Institut Pasteur and the Centre National de la Recherche Scientifique (CNRS UMR 3528, Biologie Structurale et Agents Infectieux).
Footnotes
Published ahead of print 10 August 2012
REFERENCES
- 1.Abràmoff MD, Magalhães PJ, Ram SJ. 2004. Image processing with ImageJ. Biophotonics Int. 11:36–42 [Google Scholar]
- 2.Addinall SG, Cao C, Lutkenhaus J. 1997. FtsN, a late recruit to the septum in Escherichia coli. Mol. Microbiol. 25:303–309 [DOI] [PubMed] [Google Scholar]
- 3.Alexeeva S, Gadella TWJ, Verheul J, Verhoeven GS, den Blaauwen T. 2010. Direct interactions of early and late assembling division proteins in Escherichia coli cells resolved by FRET. Mol. Microbiol. 77:384–398 [DOI] [PubMed] [Google Scholar]
- 4.Alix E, Blanc-Potard AB. 2009. Hydrophobic peptides: novel regulators within bacterial membrane. Mol. Microbiol. 72:5–11 [DOI] [PubMed] [Google Scholar]
- 5.Alix E, Blanc-Potard AB. 2008. Peptide-assisted degradation of the Salmonella MgtC virulence factor. EMBO J. 27:546–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arends SJ, et al. 2010. Discovery and characterization of three new Escherichia coli septal ring proteins that contain a SPOR domain: DamX, DedD, and RlpA. J. Bacteriol. 192:242–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baba T, et al. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008 doi: 10.1038/msb41000050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Begg KJ, Hatfull GF, Donachie WD. 1980. Identification of new genes in a cell envelope-cell division gene cluster of Escherichia coli: cell division gene ftsQ. J. Bacteriol. 144:435–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernhardt TG, de Boer PA. 2004. Screening for synthetic lethal mutants in Escherichia coli and identification of EnvC (YibP) as a periplasmic septal ring factor with murein hydrolase activity. Mol. Microbiol. 52:1255–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyd D, Weiss DS, Chen JC, Beckwith J. 2000. Towards single-copy gene expression systems making gene cloning physiologically relevant: lambda InCh, a simple Escherichia coli plasmid-chromosome shuttle system. J. Bacteriol. 182:842–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bramkamp M, van Baarle S. 2009. Division site selection in rod-shaped bacteria. Curr. Opin. Microbiol. 12:683–688 [DOI] [PubMed] [Google Scholar]
- 12.Buddelmeijer N, Aarsman ME, Kolk AH, Vicente M, Nanninga N. 1998. Localization of cell division protein FtsQ by immunofluorescence microscopy in dividing and nondividing cells of Escherichia coli. J. Bacteriol. 180:6107–6116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buddelmeijer N, Beckwith J. 2004. A complex of the Escherichia coli cell division proteins FtsL, FtsB and FtsQ forms independently of its localization to the septal region. Mol. Microbiol. 52:1315–1327 [DOI] [PubMed] [Google Scholar]
- 14.Chen JC, Beckwith J. 2001. FtsQ, FtsL and FtsI require FtsK, but not FtsN, for co-localization with FtsZ during Escherichia coli cell division. Mol. Microbiol. 42:395–413 [DOI] [PubMed] [Google Scholar]
- 15.Chen JC, Weiss DS, Ghigo JM, Beckwith J. 1999. Septal localization of FtsQ, an essential cell division protein in Escherichia coli. J. Bacteriol. 181:521–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14 [DOI] [PubMed] [Google Scholar]
- 17.Crozat E, Grainge I. 2010. FtsK DNA translocase: the fast motor that knows where it's going. Chembiochem 11:2232–2243 [DOI] [PubMed] [Google Scholar]
- 18.Dale JW, Greenaway PJ. 1985. Preparation of chromosomal DNA from E. coli. Methods Mol. Biol. 2:197–200 [DOI] [PubMed] [Google Scholar]
- 19.de Boer PA. 2010. Advances in understanding E. coli cell fission. Curr. Opin. Microbiol. 13:730–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Lallo G, Fagioli M, Barionovi D, Ghelardini P, Paolozzi L. 2003. Use of a two-hybrid assay to study the assembly of a complex multicomponent protein machinery: bacterial septosome differentiation. Microbiology (Reading) 149:3353–3359 [DOI] [PubMed] [Google Scholar]
- 21.Durand-Heredia JM, Yu HH, De Carlo S, Lesser CF, Janakiraman A. 2011. Identification and characterization of ZapC, a stabilizer of the FtsZ ring in Escherichia coli. J. Bacteriol. 193:1405–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gassel M, Möllenkamp T, Puppe W, Altendorf K. 1999. The KdpF subunit is part of the K(+)-translocating Kdp complex of Escherichia coli and is responsible for stabilization of the complex in vitro. J. Biol. Chem. 274:37901–37907 [DOI] [PubMed] [Google Scholar]
- 23.Gerding MA, Ogata Y, Pecora ND, Niki H, de Boer PA. 2007. The trans-envelope Tol-Pal complex is part of the cell division machinery and required for proper outer-membrane invagination during cell constriction in E. coli. Mol. Microbiol. 63:1008–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goehring NW, Gonzalez MD, Beckwith J. 2006. Premature targeting of cell division proteins to midcell reveals hierarchies of protein interactions involved in divisome assembly. Mol. Microbiol. 61:33–45 [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez MD, Akbay EA, Boyd D, Beckwith J. 2010. Multiple interaction domains in FtsL, a protein component of the widely conserved bacterial FtsLBQ cell division complex. J. Bacteriol. 192:2757–2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griffith KL, Wolf RE. 2002. Measuring beta-galactosidase activity in bacteria: cell growth, permeabilization, and enzyme assays in 96-well arrays. Biochem. Biophys. Res. Commun. 290:397–402 [DOI] [PubMed] [Google Scholar]
- 27.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hale CA, et al. 2011. Identification of Escherichia coli ZapC (YcbW) as a component of the division apparatus that binds and bundles FtsZ polymers. J. Bacteriol. 193:1393–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harry E, Monahan L, Thompson L. 2006. Bacterial cell division: the mechanism and its precison. Int. Rev. Cytol. 253:27–94 [DOI] [PubMed] [Google Scholar]
- 30.Hobbs EC, Astarita JL, Storz G. 2010. Small RNAs and small proteins involved in resistance to cell envelope stress and acid shock in Escherichia coli: analysis of a bar-coded mutant collection. J. Bacteriol. 192:59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hobbs EC, Fontaine F, Yin X, Storz G. 2011. An expanding universe of small proteins. Curr. Opin. Microbiol. 14:167–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karimova G, Dautin N, Ladant D. 2005. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J. Bacteriol. 187:2233–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karimova G, Pidoux J, Ullmann A, Ladant D. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. U. S. A. 95:5752–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karimova G, Robichon C, Ladant D. 2009. Characterization of YmgF, a 72-residue inner membrane protein that associates with the Escherichia coli cell division machinery. J. Bacteriol. 191:333–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karimova G, Ullmann A, Ladant D. 2001. Protein-protein interaction between Bacillus stearothermophilus tyrosyl-tRNA synthetase subdomains revealed by a bacterial two-hybrid system. J. Mol. Microbiol. Biotechnol. 3:73–82 [PubMed] [Google Scholar]
- 36.Lara B, Ayala JA. 2002. Topological characterization of the essential Escherichia coli cell division protein FtsW. FEMS Microbiol. Lett. 216:23–32 [DOI] [PubMed] [Google Scholar]
- 37.Margolin W. 2009. Sculpting the bacterial cell. Curr. Biol. 19:R812–R822 doi:10.1016/j.cub.2009.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 39.Modell JW, Hopkins AC, Laub MT. 2011. A DNA damage checkpoint in Caulobacter crescentus inhibits cell division through a direct interaction with FtsW. Genes Dev. 25:1328–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paradis-Bleau C, et al. 2010. Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell 143:1110–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peters NT, Dinh T, Bernhardt TG. 2011. A fail-safe mechanism in the septal ring assembly pathway generated by the sequential recruitment of cell separation amidases and their activators. J. Bacteriol. 193:4973–4983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phoenix P, Drapeau GR. 1988. Cell division control in Escherichia coli K-12: some properties of the ftsZ84 mutation and suppression of this mutation by the product of a newly identified gene. J. Bacteriol. 170:4338–4342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reddy M. 2007. Role of FtsEX in cell division of Escherichia coli: viability of ftsEX mutants is dependent on functional SufI or high osmotic strength. J. Bacteriol. 189:98–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ricard M, Hirota Y. 1973. Process of cellular division in Escherichia coli: physiological study on thermosensitive mutants defective in cell division. J. Bacteriol. 116:314–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rico AI, García-Ovalle M, Palacios P, Casanova M, Vicente M. 2010. Role of Escherichia coli FtsN protein in the assembly and stability of the cell division ring. Mol. Microbiol. 76:760–771 [DOI] [PubMed] [Google Scholar]
- 46.Robichon C, Karimova G, Beckwith J, Ladant D. 2011. Role of leucine zipper motifs in association of the Escherichia coli cell division proteins FtsL and FtsB. J. Bacteriol. 193:4988–4992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sambrook J, Russell DW. 2006. The condensed protocols from molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 48.Storts DR, Markovitz A. 1991. A novel rho promoter::Tn10 mutation suppresses an ftsQ1(Ts) missense mutation in an essential Escherichia coli cell division gene by a mechanism not involving polarity suppression. J. Bacteriol. 173:655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Typas A, et al. 2010. Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell 143:1097–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uehara T, Dinh T, Bernhardt TG. 2009. LytM-domain factors are required for daughter cell separation and rapid ampicillin-induced lysis in Escherichia coli. J. Bacteriol. 191:5094–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weiss DS, Chen JC, Ghigo JM, Boyd D, Beckwith J. 1999. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J. Bacteriol. 181:508–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wong RS, McMurry LM, Levy SB. 2000. ‘Intergenic’ blr gene in Escherichia coli encodes a 41-residue membrane protein affecting intrinsic susceptibility to certain inhibitors of peptidoglycan synthesis. Mol. Microbiol. 37:364–370 [DOI] [PubMed] [Google Scholar]