

Abstract
While β-adrenergic receptor (β-AR) stimulation leads to positive inotropic effects, it can also induce arrhythmogenic Ca2+ waves. β-AR stimulation increases mitochondrial oxygen consumption and, thereby, the production of reactive oxygen species (ROS). We therefore investigated the role of ROS in the generation of Ca2+ waves during β-AR stimulation in rabbit ventricular myocytes. Isoproterenol (ISO) increased Ca2+ transient amplitude during systole, sarcoplasmic reticulum (SR) Ca2+ load and the occurrence of Ca2+ waves during diastole. These effects, however, developed at different time points during ISO application. While SR Ca2+ release and load reached a maximum level after 3 min, Ca2+ waves occurred at the highest frequency only after 6 min of ISO application. Measurement of intra-SR-free Ca2+ concentration ([Ca2+]SR) showed an initial increase of SR Ca2+ load followed by a gradual decline over time during ISO application. This decline of [Ca2+]SR was not due to decreased SR Ca2+ uptake, but instead was the result of increased SR Ca2+ leak mainly in the form of Ca2+ waves. ISO application led to significant RyR phosphorylation at the protein kinase A (PKA)-specific site, which remained relatively stable throughout β-AR activation. Moreover, β-AR stimulation significantly increased ROS production after 4–6 min of ISO application. The ROS scavenger Tiron and the superoxide dismutase mimetic MnTBPA abolished the ISO-mediated ROS production. The mitochondria-specific antioxidant Mito-Tempo and an inhibitor of the electron transport chain, rotenone, also effectively prevented the ISO-mediated ROS production. Scavenging ROS during ISO application decreased the occurrence of Ca2+ waves and partially prevented augmentation of SR Ca2+ leak, but did not affect the increase of Ca2+ transient amplitude. Treatment of myocytes with ISO for 15 min significantly reduced the free thiol content in RyRs. These data suggest that increased mitochondrial ROS production during β-AR stimulation causes RyR oxidation. Together with RyR phosphorylation, oxidation of RyRs increases diastolic SR Ca2+ leak to a critical level leading to the generation of arrhythmogenic Ca2+ waves.
Key point
β-Adrenergic receptor (β-AR) stimulation is the most important positive inotropic effect on the heart, but it can also induce cardiac arrhythmias.
In rabbit ventricular myocytes, short-term β-AR stimulation induced a positive inotropic effect that was associated with increased ryanodine receptor phosphorylation.
However, prolonged β-AR stimulation increased the occurrence of calcium waves during diastole. This effect was associated with an increase in the reactive oxygen species production and oxidation of thiol groups on ryanodine receptors.
These results suggest that phosphorylation combined with oxidation of ryanodine receptors during β-AR stimulation increases the receptor activity to a critical level leading to the generation of arrhythmogenic calcium waves.
Thus, attenuating reactive oxygen species production during β-AR stimulation may be a promising therapeutic strategy to prevent the occurrence of arrhythmias, while at the same time preserving cardiac positive inotropy.
Introduction
β-Adrenergic receptor (β-AR) stimulation initiates a signalling cascade that produces the physiologically important positive inotropic effect on the heart (Dzimiri, 1999; Bers, 2001; Rockman et al. 2002). This effect is mainly mediated via the adenylate cyclase–cAMP–protein kinase A (PKA) signalling pathway. PKA phosphorylates an array of important proteins involved in excitation–contraction coupling (ECC), such as a sarcoplasmic reticulum (SR) Ca2+-ATPase (SERCA) regulator, phospholamban (PLB), an L-type Ca2+ channel (LTCC) and ryanodine receptors (RyRs) (Bers, 2001). This results in an increase of Ca2+ flux through LTCC, elevation of SR Ca2+ load, and synchronization of SR Ca2+ release during systole (Hussain & Orchard, 1997; Callewaert et al. 1988; Song et al. 2001; Ginsburg & Bers, 2004). These modifications of Ca2+ transport systems play a critical role in positive inotropy by allowing adjustment of the strength of ECC to match the physiological demands of the heart. However, prolonged or excessive β-AR activation can lead to the generation of cardiac arrhythmias (Nam et al. 2005; Katra et al. 2007; Venetucci et al. 2007). The cellular mechanisms underlying the β-AR-mediated progression of Ca2+ cycling from positive inotropic to arrhythmogenic remain obscure.
A growing body of evidence suggests that β-AR-mediated cardiac arrhythmias can be triggered by increased SR Ca2+ efflux during diastole commonly termed SR Ca2+ leak (Ellison et al. 2007; Curran et al. 2007; Morimoto et al. 2009; Ogrodnik & Niggli, 2010). Excessive diastolic SR Ca2+ leak, particularly in the form of Ca2+ waves (Venetucci et al. 2007; Curran et al. 2010), can generate delayed afterdepolarizations (DADs), an effective substrate for cardiac arrhythmias (Schlotthauer & Bers, 2000). It has been suggested that increased SR Ca2+ leak during β-AR stimulation is mediated by RyR phosphorylation by PKA (Yoshida et al. 1992; Marx et al. 2000; Wehrens et al. 2006; Morimoto et al. 2009). Specifically, phosphorylation of the RyR at Ser 2808 (2809 in rabbit) can cause dissociation of the auxiliary protein FKBP 12.6 from RyRs, leading to an increase in the channel's sensitivity to Ca2+ (Marx et al. 2000). This mechanism has been shown to be involved in β-AR-mediated Ca2+ arrhythmias observed in multicellular ventricular preparations (Katra et al. 2007). However, the concept that PKA is solely responsible for augmentation of SR Ca2+ leak during β-AR stimulation has been recently challenged. Several studies have shown that Ca2+–calmodulin-dependent kinase type II (CaMKII) also plays an important role in enhancing SR Ca2+ leak during β-AR stimulation (Curran et al. 2007; Ferrero et al. 2007; Ogrodnik & Niggli, 2010). It has been shown that RyR phosphorylation at a single site (Ser 2814; 2815 in rabbit) is entirely responsible for CaMKII effects on SR Ca2+ leak and ventricular arrhythmias (van Oort et al. 2010). Despite the controversy about the contribution of PKA and CaMKII to the β-AR response, phosphorylation is not the only type of post-translational modification that affects the channel's function during β-AR stimulation. β-AR stimulation also increases cellular metabolism and therefore mitochondrial oxygen consumption. This results in a higher rate of mitochondrial reactive oxygen species (ROS) production (Opie et al. 1979; Remondino et al. 2003). Chronic β-AR activation is commonly associated with myocardial injury induced by oxidative stress and apoptosis (Communal et al. 1998; Zhang et al. 2005; Srivastava et al. 2007). Recent studies showed that even acute β-AR activation can rapidly increase ROS production (Kohlhaas et al. 2010) and these free radicals play a critical role in augmentation of LTCC current during β-AR activation (Andersson et al. 2011). ROS also are known to alter RyR activity by oxidation of thiol groups of cysteine residues in the channel (Kourie, 1998; Zima & Blatter, 2006). Oxidation of RyR thiols appears able to enhance the channel's activity, augment SR Ca2+ leak (Terentyev et al. 2008) and increase Ca2+ spark frequency (Yan et al. 2008). Furthermore, it has been shown that the arrhythmogenic effect of cardiac glycosides involves the generation of mitochondrial ROS and oxidation of RyRs (Ho et al. 2011). Although previous studies (Kohlhaas et al. 2010; Andersson et al. 2011) showed increased ROS production after ISO application, the potential role of ROS in augmentation of SR Ca2+ leak and generation of Ca2+ waves during β-AR stimulation has received little attention.
In this work, we tested the hypothesis that enhanced ROS production during β-AR stimulation plays an important role in the progression from the positive inotropic to arrhythmogenic effect. We found that short-term β-AR stimulation induced a positive inotropic effect that was associated with increased RyR phosphorylation at the PKA- and CaMKII-specific sites in rabbit ventricular myocytes. However, prolonged β-AR stimulation significantly increased mitochondrial ROS production and the occurrence of Ca2+ waves. Scavenging ROS during β-AR stimulation normalized SR Ca2+ leak and significantly decreased the occurrence of Ca2+ waves, without affecting the positive inotropic effect. Together, these data suggest that increased mitochondrial ROS production during β-AR stimulation causes redox modification of the RyR resulting in enhanced channel activity. Together with RyR phosphorylation, the redox modification of RyRs increases diastolic SR Ca2+ leak to a critical level that leads to the generation of arrhythmogenic Ca2+ waves. This finding is important because attenuating ROS production during β-AR stimulation may be a promising new therapeutic strategy to prevent the occurrence of arrhythmias, while at the same time preserving cardiac positive inotropy. Part of this work has been published in abstract form (Bovo et al. 2011b).
Methods
Myocyte isolation
Ventricular myocytes were isolated from New Zealand White rabbits (39 animals, 2–2.5 kg; Myrtle's Rabbitry, TN, USA) according to the procedure described previously (Domeier et al. 2009). Briefly, rabbits were anaesthetized with sodium pentobarbital (50 mg kg−1 i.v.). Following thoracotomy hearts were quickly excised, mounted on a Langendorff apparatus, and retrogradely perfused with Liberase Blendzyme (Roche Applied Science, IN, USA)-containing solution at 37°C for 16 min. The cell isolation procedure was approved by the Institutional Animal Care and Use Committee, and complies with US and UK regulations on animal experimentation (Drummond, 2009). Chemicals and reagents were purchased from Sigma-Aldrich (St Louis, MO, USA) unless otherwise stated. All experiments were performed at room temperature (22–24°C).
Confocal microscopy
Changes in cytosolic free Ca2+ concentration ([Ca2+]i), intra-SR free Ca2+ concentration ([Ca2+]SR) and ROS were measured with laser scanning confocal microscopy (Radiance 2000 MP, Bio-Rad, UK and LSM 410, Zeiss, Germany) equipped with a ×40 oil-immersion objective lens (N.A., 1.3).
Measurements of [Ca2+]i
To record [Ca2+]i we used the high-affinity Ca2+ indicator Fluo-4 (Molecular Probes/Invitrogen, Carlsbad, CA, USA). To load the cytosol with Ca2+ indicator, cells were incubated at room temperature with 10 μm Fluo-4 AM for 15 min in Tyrode solution (in mm: NaCl 140; KCl 4; CaCl2 2; MgCl2 1; glucose 10; Hepes 10; pH 7.4), followed by a 20 min wash. Fluo-4 was excited with the 488 nm line of an argon laser and fluorescence was measured at >515 nm. Action potentials were induced by electrical field stimulation using a pair of platinum electrodes, which were connected to a Grass stimulator (Astro-Med. Inc., USA) set at a voltage ∼50% above the threshold. Fluo-4 images were acquired in line-scan mode (3 ms per scan; pixel size 0.12 μm). Ca2+ wave propensity was analysed as numbers of waves per cardiac cycle (as a percentage).
Measurements of [Ca2+]SR and SR Ca2+ leak
To record [Ca2+]SR we used the low-affinity Ca2+ indicator Fluo-5N (Molecular Probes/Invitrogen). To load the SR with Ca2+ indicator, myocytes were incubated with 5 μm Fluo-5N AM for 2.5 h at 37°C as described before (Zima et al. 2008). SR Ca2+ leak was measured as a function of [Ca2+]SR according to the protocol described previously (Zima et al. 2010). Briefly, Fluo-5N was excited with minimum energy of the 488 nm line of an argon laser to minimize dye photobleaching. To improve the signal-to-noise ratio of the low-intensity Fluo-5N signal, fluorescence was collected with an open pinhole at >515 nm and averaged over the entire cellular width of 2-D image (pixel size 0.2 μm). Changes in [Ca2+]SR were calculated by the formula (Cannell et al. 1994): [Ca2+]SR=Kd×R/(Kd/[Ca2+]SRdiast–R+ 1), where R is the normalized Fluo-5N fluorescence (R=[F–Fmin]/[F0–Fmin]); F0 and Fmin are the fluorescence level at rest and after depletion of the SR with caffeine, respectively; Kd (Fluo-5N Ca2+ dissociation constant) was 390 μm based on in situ calibrations (Zima et al. 2010), and [Ca2+]SRdiast (diastolic [Ca2+]SR at 0.75 Hz) was 900 μm (Domeier et al. 2009). SR Ca2+ leak was measured as the changes of total [Ca2+]SR ([Ca2+]SRT) over time (d[Ca2+]SRT/dt) after complete SERCA inhibition with thapsigargin (TG). [Ca2+]SRT was calculated as: [Ca2+]SRT=Bmax/(1 +Kd/[Ca2+]SR) +[Ca2+]SR; where Bmax and Kd were 2700 μm and 630 μm, respectively (Shannon et al. 2000). The rate of SR Ca2+ leak (d[Ca2+]SRT/dt) was plotted as a function of [Ca2+]SR for each time point (15 s) during [Ca2+]SR decline.
Measurements of intracellular ROS level
ROS production was measured with a ROS-sensitive fluorescent dye, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluore scein diacetate (H2DCFDA; Molecular Probes/Invitrogen). Myocytes were loaded with 20 μm H2DCFDA for 40 min at room temperature. H2DCFDA was excited by the 488 nm line of an argon laser and emitted fluorescence was collected at wavelengths >600 nm. 2-D images were acquired at 15 s intervals. Fluorescence intensity (F) was integrated over the entire volume of the cell. Changes of ROS level were presented as background-subtracted normalized fluorescence (F/F0) where F0 is the initial fluorescence recorded under steady-state conditions at the beginning of an experiment. To minimize the photoproduction of ROS by laser illumination, all experiments were performed at the lowest laser power and fluorescence was collected with an open pinhole. The relative changes in ROS production was estimated a linear fit of the H2DCFDA signal under different experimental conditions. In the end of each experiment xanthine (0.1 mm) and xanthine oxidase (0.2 U ml−1) were applied to estimate the maximal rate of ROS production.
Western blot analysis of RyR and PLB phosphorylation
Equal amounts of a myocyte suspension were treated for different periods of time with ISO under the same experimental conditions used for our [Ca2+] measurements. After treatment, cells were quickly settled down by centrifugation and lysed in Laemmli buffer (Sigma, USA). The same amount of total lysate from each sample was subjected to 4–15% SDS-PAGE and transferred to nitrocellulose membranes. RyR phosphorylation level at PKA (Ser 2809) and CaMKII sites (Ser 2815) was quantified using phospho-specific antibodies. Western blot assay was performed with primary antibody RyR-PS2809 (Badrilla, UK) and RyR-PS2815 (kindly provided by Dr Wehrens; Baylor College of Medicine, USA). The signal was normalized to total RyR level measured with a primary antibody C34 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA, USA). PLB phosphorylation at the PKA site (Ser 16) was measured with a phospho-specific antibody, PLB-PS16 (Badrilla, UK). Secondary antibodies were all HRP conjugated, therefore RyR and PLB bands were visualized using the Luminata Forte Western HRP substrate (Millipore, USA) and signals were quantified using the UVP EpiChemi3 imaging system and ImageJ software.
Measurements of free thiol content of RyRs
The content of free thiols in RyRs was measured with the monobromobimane (mBB) fluorescence method as described previously (Kosower & Kosower, 1987; Terentyev et al. 2008). Ventricular myocytes were electrically stimulated and treated with ISO for 15 min. The maximal free thiol content (maximal reduction) was determined after treatment of cells with a strong reducing agent, dithiothreitol (DTT; 5 mm). The minimal free thiol content (maximal oxidation) was measured after incubation of cells with a strong oxidant, 2.2′-dithiodipyridine (DTDP; 0.5 mm). Afterwards, cells were permeabilized with saponin, incubated with mBB (400 μm) for 1 h, and then washed 3 times to eliminate any extra unbound mBB. Cells were than lysed in Laemmli buffer and an equal amount of lysate was loaded on two separate 4–15% SDS-PAGE gels. One gel was used to measure mBB fluorescence with the UVP EpiChemi3 imaging system. The second gel was stained with Coomassie Blue and was used to normalize the mBB signal to the total RyR level.
Statistics
Data are presented as mean ± SEM of n measurements. Statistical comparisons between groups were performed with Student's t test. Differences were considered statistically significant at P < 0.05.
Results
β-AR stimulation exerts complex effects on SR Ca2+ release during ECC
We studied how stimulation of β-ARs with isoproterenol (ISO) affects Ca2+ handling during ECC in rabbit ventricular myocytes. Figure 1A shows that ISO (0.1 μm) increased systolic Ca2+ transient amplitude, accelerated the decay of Ca2+ transients and decreased diastolic [Ca2+]i (second image). The recordings were made at 0.75 Hz pacing frequency. β-AR stimulation also led to the occurrence of spontaneous Ca2+ waves during diastole (third image). The positive inotropic and arrhythmogenic effects of ISO developed at different times. Systolic Ca2+ transient amplitude reached a maximum within 3 min (Fig. 1B; open circles). On average, Ca2+ transient amplitude increased by 48 ± 5% (n= 18; P < 0.05). Spontaneous Ca2+ waves were not very frequent events at the beginning of ISO application. However, Ca2+ wave propensity significantly increased after 6 min of ISO application (Fig. 1B filled squares). As Ca2+ wave propensity increased, the amplitude of systolic Ca2+ transients began to gradually decrease. ISO effects were completely reversed after 5 min of ISO washout (Fig. 1A far right image). We also found that the arrhythmogenic effect of ISO depends on stimulation frequency. At 0.3 Hz spontaneous Ca2+ waves were relatively rare in the presence of ISO, but their occurrence significantly increased at frequencies >0.5 Hz (online Supplemental Fig. S1). These results indicate that the arrhythmogenic effect of ISO is time- and frequency-dependent.
Figure 1. Effects of β-AR stimulation on SR Ca2+ release.
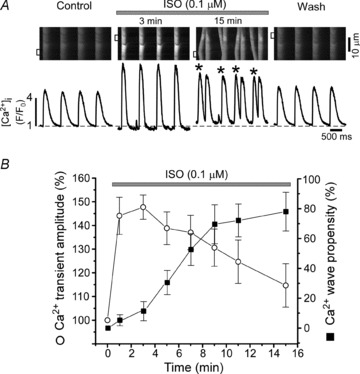
A, confocal line-scan images of Fluo-4 fluorescence and corresponding F/F0 profiles recorded from an electrically paced (at 0.75 Hz) ventricular myocyte. The recordings were made in control, during ISO (0.1 μm) application (after 3 and 15 min) and ISO washout. Ca2+ transients (F/F0 profiles) were obtained by averaging fluorescence from the 3=μm-wide regions marked by the white boxes. The asterisks indicate spontaneous Ca2+ waves. B, time-course of ISO effects on the Ca2+ transient amplitude (open circles) and on the propensity of Ca2+ waves (filled squares). Ca2+ wave propensity was presented as numbers of waves per cardiac cycle.
We examined to what extent ISO effects were due to β-AR1 or β-AR2 stimulation. In the presence of the β-AR2 inhibitor ICI 118,551 (0.3 μm), the effects of ISO on SR Ca2+ release were virtually unaffected (data not shown) suggesting that β-AR2 is not involved. The selective agonist of β-AR2, zinterol (1 μm), evoked only a small inotropic (∼11%; n= 6), but no arrhythmogenic effect (online Supplemental Fig. S1). Thus, in rabbit ventricular myocytes the arrhythmogenic effect of ISO is primarily mediated by β-AR1 stimulation.
β-AR stimulation increases diastolic [Ca]SR and Ca2+ leak
In the following experiments we studied whether the progression from positive inotropic to arrhythmogenic effect during ISO application was associated with alteration in SR Ca2+ load. SR Ca2+ load is proportional to diastolic [Ca]SR measured with Fluo-5N. Figure 2A and B show representative examples of diastolic [Ca2+]SR recording in control conditions, in the presence of ISO (0.1 μm), and after subsequent application of the SERCA inhibitor, thapsigargin (TG; 10 μm). Cells were continuously stimulated at 0.75 Hz, until TG application. β-AR stimulation led to a significant increase of SR Ca2+ load. Diastolic [Ca2+]SR reached a maximum level after 3 min of ISO application, increasing from 0.9 to 2.2 mm. The initial increase of diastolic [Ca2+]SR was followed by a gradual decline over time during β-AR stimulation (Fig. 2C). This decline of [Ca]SR was not due to alteration of SERCA, NCX or LTCC activity (online Supplemental Fig. S2), but instead was the result of increased SR Ca2+ leak. SR Ca2+ leak was measured as the changes of total [Ca2+]SR after complete SERCA inhibition in control conditions, after 3 min (Fig. 2A) and 15 min (Fig. 2B) of ISO application. SR Ca2+ leak was measured and plotted as a function of free [Ca2+]SR. SR Ca2+ leak progressively increased during ISO application (Fig. 2D). The initial augmentation of SR Ca2+ leak was, in part, due to the appearance of Ca2+ sparks (online Supplemental Fig. S3). The late augmentation of SR Ca2+ leak was temporally associated with the occurrence of Ca2+ waves (Fig. 1A). This suggests that excessive diastolic Ca2+ leak in the form of Ca2+ waves depletes SR Ca2+ content and, therefore, decreases Ca2+ transient amplitude during systole (Fig. 1B). These results also indicate that Ca2+ waves occur as a result of increased RyR activity, and not solely because of SR Ca2+ overload, otherwise Ca2+ waves would be more frequent at the beginning of ISO application, when SR Ca2+ load is maximal.
Figure 2. Effects of β-AR stimulation on diastolic [Ca2+]SR and Ca2+ leak.

The recordings of diastolic [Ca2+]SR in control conditions, during ISO (0.1 μm) application, and after subsequent application of thapsigargin (TG; 10 μm). SR Ca2+ leak was measured as the changes of total [Ca2+]SR over time in the presence of TG (see Methods). In these two examples, SR Ca2+ leak was measured after 3 min (A) and 15 min (B) of ISO application. The dashed lines show SR Ca2+ leak in control conditions. The asterisks indicate spontaneous Ca2+ waves. At the end of the experiments caffeine (10 mm) was applied to verify complete depletion of the SR. C, time-course of ISO effect on diastolic [Ca2+]SR. D, relationships between SR Ca2+ leak rate and [Ca2+]SR measured in control conditions (open circles), 3 min (black circles; #P < 0.05 vs. control), and 15 min after ISO application (grey squares; *P < 0.05 vs. ISO 3 min).
Changes of RyR phosphorylation during β-AR stimulation
Stimulation of β-ARs leads to RyR phosphorylation by PKA and CaMKII (Marx et al. 2000; Wehrens et al. 2004; Huke & Bers, 2008). By using phospho-specific antibodies, we studied changes of RyR phosphorylation by PKA and CaMKII during ISO application. Figure 3 (top) shows images of representative Western blots using phospho-specific antibodies against PKA (Ser 2809) and CaMKII (Ser 2815) specific sites, respectively. Electrical pacing (0.75 Hz) significantly increased the level of phosphorylation at the CaMKII site (Fig. 3B); however, it did not affect the PKA site (Fig. 3A). During electrical pacing, ISO application (3 min) led to robust phosphorylation of RyR at Ser 2809 (Fig. 3A), but not at Ser 2815 (Fig. 3B). Longer treatment (15 min) with ISO increased RyR phosphorylation at Ser 2815 by only 25% over the control level (Fig. 3B). For both sites, the RyR phosphorylation level at PKA and CaMKII sites did not significantly change (P > 0.05) between 3 and 15 min of ISO treatments. These results indicate that the RyR is phosphorylated by PKA during β-AR stimulation, whereas CaMKII phosphorylates the channel primarily as a result of increased [Ca2+]i during electrical pacing. The relatively stable level of RyR phosphorylation during ISO application suggests that while RyR phosphorylation might play a role in the initial inotropic effect of β-AR stimulation, the latter arrhythmogenic effect cannot be solely explained by this post-translational modification.
Figure 3. Effects of β-AR stimulation on RyR phosphorylation by PKA and CaMKII.
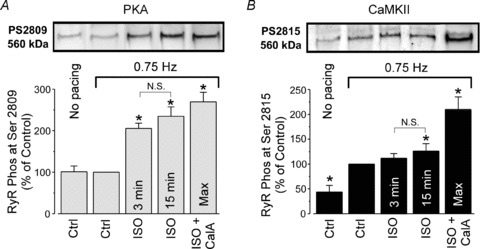
Representative Western blots (top) and statistical analysis (bottom) showing the effects of electrical pacing (0.75 Hz) and ISO application (3 and 15 min) on RyR phosphorylation at the PKA (A) and at the CaMKII specific sites (B). The changes in RyR phosphorylation were compared with RyR phosphorylation in control conditions during electrical pacing (0.75 Hz). Maximal RyR phosphorylation at PKA and CaMKII sites were obtained after treatment of cells with ISO (0.1 μm) and the phosphatase inhibitor Calyculin A (CalA; 1 μm) during electrical pacing. *P < 0.05 vs. control during electrical pacing (0.75 Hz). N.S., not statistically significant.
β-AR stimulation increases intracellular ROS production
By increasing cellular metabolism, β-AR stimulation can lead to a higher rate of mitochondrial ROS production. Intracellular ROS production rate was measured with a ROS-sensitive fluorescence dye (H2DCFDA) under the same experimental conditions used for our [Ca2+] measurements. We found that during electrical pacing the ROS-related signal monotonically increased, presumably due to basal endogenous ROS production (Fig. 4A). The application of ISO (0.1 μm) significantly increased the rate of ROS production (indicated by dashed lines; Fig. 4A). ROS production was highly dependent on electrical stimulation, as ISO did not increase ROS levels in quiescent myocytes (Fig. 4B). Pretreatment of cells with superoxide dismutase (SOD) mimetic MnTBPA (20 μm) almost completely prevented the increase in ROS elicited during ISO application (Fig. 4A). Similar results were obtained when cells were treated with the ROS scavenger Tiron (10 mm; Fig. 4B). We also studied whether an increase of ROS production during ISO application was mediated by mitochondria. We found that treatment of cells with a mitochondria-targeted ROS scavenger Mito-Tempo (25 μm; Fig. 4A and B) or an inhibitor of complex I of the mitochondrial electron transport chain (ETC), rotenone (0.3 μm), significantly decreased the ROS production elicited during ISO application (Fig. 4B). These data suggest that β-AR stimulation leads to an increase of intracellular ROS. This mainly occurs due to facilitation of electron flux through the mitochondrial ETC. Another important observation is that the ROS level significantly increased only after 4–6 min of ISO application. This delay is important because it correlates with the time needed for β-AR activation to produce spontaneous Ca2+ waves (Fig. 1B).
Figure 4. Effects of β-AR stimulation on intracellular ROS production.
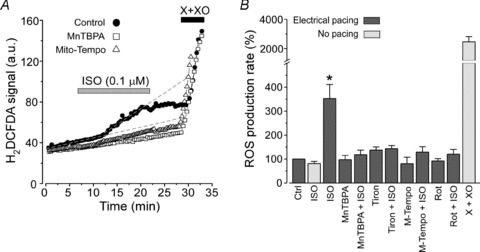
A, changes of H2DCFDA signal during ISO (0.1 μm) application in control (filled circles), in the presence of ROS scavengers MnTBPA (20 μm; open squares) and Mito-Tempo (25 μm; open triangles). Cardiomyocytes were continuously paced at 0.75 Hz. At the end of each experiment xanthine (0.1 mm) and xanthine oxidase (0.2 U ml−1) (X+XO) were applied to estimate the maximal rate of ROS production. B, relative changes of ROS production during ISO application in control conditions and in the presence of ROS scavengers or in the presence of the ETC inhibitor rotenone (Rot). Tiron and rotenone concentrations were 10 mm and 0.3 μm, respectively. *P < 0.05 vs. control during electrical pacing.
ROS scavengers prevent the generation of Ca2+ waves during β=AR stimulation
To elucidate whether increased ROS production was involved in the generation of Ca2+ waves during β-AR stimulation, we studied the effects of ISO on SR Ca2+ release in the presence of ROS scavengers. Figure 5A shows representative examples of Ca2+ transients recorded in control conditions, 6 min after treatment with Tiron (10 mm), and after subsequent ISO application. Summary results showed that Tiron significantly suppressed the occurrence of Ca2+ waves during β-AR activation (Fig. 5B; filled squares). The propensity of Ca2+ waves induced by ISO decreased from 78 ± 13% (n= 18) in control to 20 ± 11% (n= 14) after treatment of cells with Tiron. However, the ROS scavenger did not appear to limit the ISO-mediated positive inotropic action (Fig. 5B; open circles): in the presence of Tiron, Ca2+ transient amplitude increased by 41 ± 9% (n= 14) after 3 min of ISO application. This effect was similar to that induced by ISO alone (48 ± 5%; n= 18). Tiron did not affect the decay of the caffeine- and the AP-induced Ca2+ transients in control and in the presence of ISO (online Supplemental Fig. S2). These results indicate that ROS produced during ISO application affected neither SERCA nor NCX activity. Furthermore, scavenging ROS with Tiron partially prevented the decline of the positive inotropic effect of β-AR stimulation (Fig. 5B). A similar anti-arrhythmic effect was observed when cells were treated with MnTBPA (20 μm; Fig. 5C) or Mito-Tempo (25 μm; Fig. 5D). These ROS scavengers also did not limit ISO's positive inotropic effect. These data indicate that attenuating mitochondrial ROS production effectively prevents the generation of Ca2+ waves during β-AR stimulation.
Figure 5. Effects of β-AR stimulation on SR Ca2+ release in the presence of ROS scavengers.

A, Ca2+ transients recorded in control conditions, in the presence of Tiron (10 mm), and after subsequent application of ISO (0.1 μm). B, time-course of ISO effects on the Ca2+ transient amplitude (open circles) and on the propensity of Ca2+ waves (filled squares) in the presence of Tiron. C, effects of ISO on SR Ca2+ release in the presence of MnTBPA (20 μm). D, effects of ISO on SR Ca2+ release in the presence of Mito-Tempo (25 μm).
ROS scavengers normalize SR Ca2+ leak during β-AR stimulation
In the next experiments, we studied whether ROS scavengers can prevent the augmentation of SR Ca2+ leak during β-AR stimulation. As before, we used the low-affinity Ca2+ dye Fluo-5N to track Ca2+ within the SR (Fig. 2). Application of Tiron alone did not significantly affect diastolic [Ca2+]SR (Fig. 6A). In the presence of the scavenger, ISO (0.1 μm) application increased [Ca2+]SR to the same level (2.0 ± 0.2 mm; n= 8) as with ISO alone (2.2 ± 0.3 mm; n= 9). It is important to note that after reaching the maximum level, diastolic [Ca2+]SR declined significantly slower in the presence of Tiron than in the presence of ISO alone (Fig. 6A; dashed line). Similar results were obtained when myocytes were treated with MnTBPA or Mito-Tempo before β-AR stimulation (data not shown). Thus, attenuating ROS levels allowed the maintenance of increased SR Ca2+ load during β-AR stimulation. We also measured SR Ca2+ leak after 15 min of ISO application in the presence of Tiron. Summary results are presented in Fig. 6B. The ROS scavenger significantly decreased SR Ca2+ leak augmented by β-AR stimulation. Furthermore, the ROS scavenger normalized the Ca2+ leak rate to a level which is comparable to the leak rate measured after 3 min of ISO application (Fig. 2D; filled symbols). During this period of ISO's action, Ca2+ waves were very rare events (Fig. 1B). These data suggest that ROS scavengers prevent the generation of Ca2+ waves during β-AR stimulation by decreasing SR Ca2+ leak.
Figure 6. Effects of β-AR activation on diastolic [Ca2+]SR and SR Ca2+ leak in the presence of ROS scavengers.
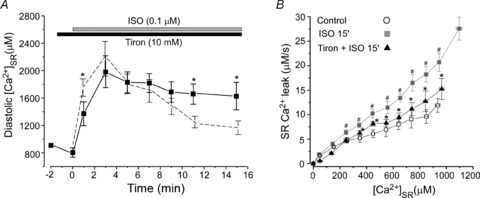
A, time-course of ISO effect on diastolic [Ca2+]SR in the presence of Tiron (10 mm). The dashed line shows the effect of ISO on diastolic [Ca2+]SR in control conditions (Fig. 2C). *P < 0.05 vs. ISO in control conditions. B, relationships between SR Ca2+ leak rate and [Ca2+]SR measured in control conditions (open circles), 15 min after ISO application (grey squares; #P < 0.05 vs. control) and 15 min after ISO application in the presence of Tiron (black triangles; *P < 0.05 vs. ISO 15 min).
β-AR stimulation causes oxidation of thiol groups on RyRs
The RyR protein contains at least 80 cysteine residues per monomer (Xu et al. 1998) and some of these residues are sensitive to redox modification (Liu et al. 1994). Oxidation of thiol groups of cysteine residues by ROS increases the channel's activity (Zima & Blatter, 2006). By using an mBB assay, we found that treatment of cells for 15 min with ISO (0.1 μm) significantly decreased the content of free thiols on RyRs (Fig. 7A). The mBB signal was expressed as a percentage of the maximal free thiol content (obtained after the DTT treatment). On average, ISO decreased the content of free thiols on RyRs from 91 ± 7 to 63 ± 11% (n= 5; P < 0.05) or by 30%. The observed ISO effect was abolished by treatment of cells with ROS scavengers (data not shown). These data suggest that, during β-AR stimulation, increased ROS production causes oxidation of RyR thiol groups. This RyR redox modification is probably involved in increasing SR Ca2+ leak and generating arrhythmogenic Ca2+ waves.
Figure 7. Effects of β-AR stimulation on content of free thiols of RyRs.
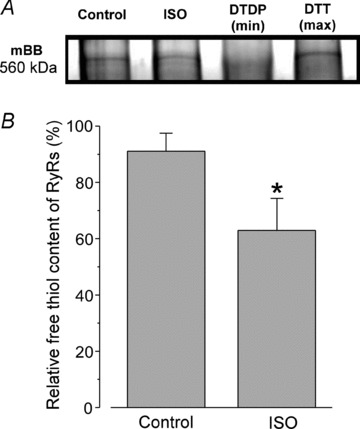
A, mBB fluorescence intensity signals of RyRs measured under control conditions and after 15 min of ISO (0.1 mm) treatment. For both groups, myocytes were electrically stimulated at 0.75 Hz. The maximal and the minimal free thiol content were determined after treatment of cells with DTT (5 mm) and DTDP (0.5 mm), respectively. B, relative free thiol content of RyRs from control and ISO-treated samples were obtained by normalizing mBB fluorescence to RyR amount determined using Coomassie Blue staining of the gel run in parallel. *P < 0.05 vs. control.
Discussion
The novel finding of this study is a new mechanism that is involved in the clinically relevant progression of Ca2+ cycling from positive inotropic to arrhythmogenic during β=AR stimulation. We found that short-term β-AR stimulation induced a positive inotropic effect which was associated with increased RyR phosphorylation at PKA- and CaMKII-specific sites and enhanced SR Ca2+ leak. Thus, the increase in RyR phosphorylation is necessary to develop the important inotropic effect, but alone is not sufficient to increase RyR activity to a high enough level to induce Ca2+ waves. However, prolonged β-AR stimulation increased mitochondrial ROS production which, in turn, causes oxidation of thiol residues on the RyR. This post-translational modification of RyR further increased SR Ca2+ leak to a critical level causing the generation of Ca2+ waves. Thus, RyR oxidation combined with RyR phosphorylation produces an enhanced effect on RyR-mediated Ca2+ leak that leads to the development of Ca2+-dependent arrhythmias.
Mechanism of ROS production during β-AR stimulation
The important finding of this study is that acute β-AR stimulation significantly increases intracellular ROS levels in rabbit ventricular myocytes (Fig. 4). In cardiomyocytes, ROS can be generated by several enzymatic systems including the mitochondrial ETC, NADPH oxidase, xanthine oxidase and nitric oxide synthase (Zima & Blatter, 2006). We found that the mitochondria-specific antioxidant Mito-Tempo and inhibitor of the ETC rotenone effectively prevented the ISO-mediated ROS production. These results strongly support the conclusion that mitochondria are the primary source of these oxygen free radicals. Our finding is consistent with several recent reports (Kohlhaas et al. 2010; Andersson et al. 2011) showing increased mitochondrial ROS production in ventricular myocytes during acute β-AR stimulation. The mechanisms underlying this effect, however, are not fully understood. Under normal conditions, up to 2% of consumed oxygen by mitochondria forms superoxide anions as a result of the electron leakage from the ETC (Turrens, 2003). Thus, enhanced mitochondrial oxygen consumption during β-AR stimulation (Opie et al. 1979) would lead to higher ROS production. An important factor that might play a key role in the β-AR-mediated ROS production is mitochondrial Ca2+. As SR Ca2+ release sites and mitochondria are in close physical proximity (Csordas et al. 2001), the released Ca2+ can be effectively sequestered into the mitochondria via the electrogenic Ca2+ uniporter. It has been shown that enhanced SR Ca2+ turnover during β-AR stimulation increases [Ca2+] within mitochondria (Kohlhaas et al. 2010). The increased mitochondrial [Ca2+] would enhance ROS production by facilitating specific Krebs cycle reactions at pyruvate, isocitrate and α-ketoglutarate dehydrogenases and, therefore, electron flux through the ETC. Furthermore, uncoupling of oxidative phosphorylation during cytosolic Ca2+ overload can lead to a higher rate of mitochondrial ROS production (Aon et al. 2010). Alternatively, β-AR stimulation can increase mitochondrial ROS production via a Ca2+-independent mechanism (Andersson et al. 2011). In permeabilized cardiomyocytes, the PKA catalytic subunit increased ROS production leading to alteration of mitochondrial function (Nagasaka et al. 2007). We found that ROS production significantly increased only after 4–6 min of ISO application. This effect occurred afterβ-AR stimulation produced a maximal effect on systolic SR Ca2+ release. On the other hand, we did not find significant changes in ROS level after activation of PKA with ISO in quiescent myocytes (Fig. 4B). Thus, these results suggest that PKA activation alone is not sufficient to increase ROS production during β-AR stimulation. On the other hand, the increase of [Ca2+]i alone (with a LTCC agonist BayK or increased external [Ca2+]) had only minor effect on ROS production (data not shown). Thus, it is likely that the combination of increased cytosolic Ca2+ cycling and PKA activation is required for a significant increase in the mitochondrial ROS production during acute β-AR stimulation.
The role of ROS in the progression of Ca2+ cycling from inotropic to arrhythmogenic during β-AR stimulation
β-AR stimulation elicits positive inotropy by enhancing intracellular Ca2+ cycling (Bers, 2001). This effect is mediated by PKA- and CaMKII-dependent phosphorylation of several important proteins involved in ECC, including the RyR (Marx et al. 2000; Wehrens et al. 2004; Ferrero et al. 2007; Huke & Bers, 2008). In accordance with previous studies, we found that β-AR stimulation led to significant RyR phosphorylation at the PKA-specific site (Fig. 3). On the other hand, RyR phosphorylation at the CaMKII site was significantly increased primarily by electrical stimulation under control conditions. We did not find any significant alteration in RyR phosphorylation at the CaMKII site immediately after ISO application; however, a small increase (∼25%) in RyR phosphorylation was detected after prolonged β-AR stimulation. This effect can be a result of Ca2+-independent activation of CaMKII by ROS (Erickson et al. 2008). Although the functional importance of RyR phosphorylation remains highly controversial, it has been suggested that RyR phosphorylation either by PKA or CaMKII can increase the channel's sensitivity to Ca2+ (Marx et al. 2000; van Oort et al. 2010). This post-translational modification functionally translates into a faster rate of SR Ca2+ release during systole (Ginsburg & Bers, 2004), but also into higher SR Ca2+ leak during diastole (Curran et al. 2010). As SR Ca2+ leak highly depends on SR Ca2+ load (Shannon et al. 2002; Zima et al. 2010), it is difficult to determine if changes in SR Ca2+ leak during β-AR stimulation are due to changes in RyR gating or due to an increase in SR Ca2+ load. We recently developed a new approach to directly and simultaneously track [Ca2+]SR and SR Ca2+ leak (Zima et al. 2010; Bovo et al. 2011a). Using this approach, we defined the relationship between SR Ca2+ leak and diastolic [Ca2+]SR (which is proportional to SR Ca2+ load) in control conditions and at different time points during β-AR stimulation. Thus, we were able to accurately compare the changes in SR Ca2+ leak at the same SR Ca2+ loads during β-AR stimulation. We found that during the initial phase of β-AR stimulation (≤3 min) SR Ca2+ leak significantly increased (Fig. 2A and D), in part due to the occurrence of Ca2+ sparks (online Supplemental Fig. S3). At the same time, Ca2+ waves were not very frequent (Fig. 1B). At this stage of β-AR stimulation, RyR phosphorylation at PKA and CaMKII sites reached a steady-state level (Fig. 3), but ROS production did not change significantly (Fig. 4A). Thus, the increase in RyR phosphorylation (to 80% and to 60% for PKA and CaMKII sites, respectively) does not increase RyR activity to a sufficiently high level to induce propagating Ca2+ waves. At the same time, increased SERCA activity (due to PLB phosphorylation; online Supplemental Fig. S2) effectively counterbalanced the enhanced SR Ca2+ leak allowing maintenance of the relatively high SR Ca2+ load (Fig. 2C). However, the positive inotropic effect plays an important role in setting the conditions for the development of Ca2+-dependent arrhythmias.
The positive inotropic effect of β-AR stimulation increases cellular energy demand, mitochondrial metabolism and ROS production (Fig. 4). It has been shown that ROS can increase RyR activity as a result of oxidation of thiol residues on the channel (Terentyev et al. 2008). We found that prolonged β-AR stimulation (15 min) significantly reduced the free thiol content on RyRs (Fig. 7). This redox modification further increased diastolic SR Ca2+ leak (Fig. 2B and D). This would lead to higher energy consumption, because more ATP will be used by SERCA to maintain a pump–leak balance. These conditions create a ‘vicious cycle’ (SR Ca2+ leak increases ROS production and ROS further increase SR Ca2+ leak) ultimately leading to an increase in diastolic SR Ca2+ leak to the level that promotes Ca2+ waves. These Ca2+ waves are not only a powerful substrate for cardiac arrhythmias (Schlotthauer & Bers, 2000), but also cause significant depletion of SR Ca2+ load during diastole (Fig. 2C), thus diminishing systolic Ca2+ release (Fig. 1). By scavenging ROS during β-AR stimulation we were able to significantly inhibit the generation of Ca2+ waves without limiting the positive inotropic effect (Fig. 6). In the presence of ROS scavengers, the ISO-mediated augmentation of Ca2+ transient amplitude was only 14% smaller than in control conditions. This can be attributed to augmentation of LTCC current by ROS during β-AR stimulation (Andersson et al. 2011). Although ROS can affect function of SERCA and NCX (Zima & Blatter, 2006), we did not observe any significant effect of ROS scavengers on the decay of the caffeine- and the AP-induced Ca2+ transients before and after ISO application (online Supplemental Fig. S2). We concluded that the level of ROS produced during ISO application is not high enough to affect NCX and SERCA activity. Interestingly, scavenging ROS normalized SR Ca2+ leak during the late stage of β-AR stimulation to the same level as during the initial phase of β-AR stimulation (Fig. 6) when only RyR phosphorylation occurred. This finding further supports our conclusion that simultaneous phosphorylation and oxidation causes an enhanced effect on RyR-mediated Ca2+ leak leading to the generation of Ca2+ waves. A similar mechanism of alterations in RyR activity and generation of Ca2+-dependent arrhythmias was described during progression of heart failure (Belevych et al. 2011), a clinical syndrome that is commonly induced by chronically elevated adrenergic tone.
Conclusion
The results of this study reveal a novel mechanism underlying the development of arrhythmogenic Ca2+ waves during β-AR stimulation. Increased mitochondrial ROS production during β-AR stimulation is a key step in the transition from positive inotropy to arrhythmias. High ROS levels change the redox status of thiol residues on the RyR, leading to augmentation of SR Ca2+ leak. Together with RyR phosphorylation, this redox modification of the channel increases SR Ca2+ leak to a critical level, resulting in the generation of Ca2+ waves. Thus, attenuating ROS production may be a promising therapeutic strategy to prevent the occurrence of arrhythmias and preserve cardiac positive inotropy during pathological conditions associated with chronic β-AR stimulation.
Acknowledgments
This work was supported by the McCormick Foundation and The Schweppe Foundation (to AVZ). We thank Stefan Mazurek for excellent technical assistance. We would like to thank Dr Wehrens for providing the phospho-specific antibody against Ser2815. The authors would also like to thank Drs Seth L. Robia and Joshua Maxwell for critical reading of the manuscript.
Glossary
- β-AR
β-adrenergic receptor
- [Ca2+]i
cytosolic free calcium concentration
- CaMKII
Ca2+–calmodulin-dependent kinase type II
- CICR
Ca2+-induced Ca2+ release
- ETC
electron transport chain
- ECC
excitation–contraction coupling
- ISO
isoproterenol
- mBB
monobromobimane
- MnTBPA
Mn-(III)-tetrakis (4-benzoic acid) porphyrin
- LTCC
L-type Ca2+ channel
- NCX
Na+–Ca2+ exchange
- PLB
phospholamban
- ROS
reactive oxygen species
- RyR
ryanodine receptor
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+-ATPase
- SR
sarcoplasmic reticulum
- [Ca2+]SR
sarcoplasmic reticulum free calcium concentration
- TG
thapsigargin
Author contributions
All authors contributed to the conception and design of the study, interpretation of data and writing of the manuscript. E.B. and A.V.Z. performed the experimental work and analysis of results. All authors have approved the version to be published.
Supplementary material
Supplemental Fig. S1
Supplemental Fig. S2
Supplemental Fig. S3
References
- Andersson DC, Fauconnier J, Yamada T, Lacampagne A, Zhang SJ, Katz A, Westerblad H. Mitochondrial production of reactive oxygen species contributes to the β-adrenergic stimulation of mouse cardiomycytes. J Physiol. 2011;589:1791–1801. doi: 10.1113/jphysiol.2010.202838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, O’Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta. 2010;1797:865–877. doi: 10.1016/j.bbabio.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Terentyev D, Terentyeva R, Nishijima Y, Sridhar A, Hamlin RL, Carnes CA, Gyorke S. The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc Res. 2011;90:493–502. doi: 10.1093/cvr/cvr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Dordrecht: Kluwer Academic Publishers; 2001. [Google Scholar]
- Bovo E, Mazurek SR, Blatter LA, Zima AV. Regulation of sarcoplasmic reticulum Ca2+ leak by cytosolic Ca2+ in rabbit ventricular myocytes. J Physiol. 2011a;589:6039–6050. doi: 10.1113/jphysiol.2011.214171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovo E, Mazurek SR, Lipsius SL, Zima AV. β-Adrenergic receptor stimulation of ROS production generates spontaneous Ca waves in rabbit ventricular myocytes. Biophys J. 2011b;100:559a–560a. Ref Type: Abstract. [Google Scholar]
- Callewaert G, Cleemann L, Morad M. Epinephrine enhances Ca2+ current-regulated Ca2+ release and Ca2+ reuptake in rat ventricular myocytes. Proc Natl Acad Sci U S A. 1988;85:2009–2013. doi: 10.1073/pnas.85.6.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell MB, Cheng H, Lederer WJ. Spatial non-uniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Biophys J. 1994;67:1942–1956. doi: 10.1016/S0006-3495(94)80677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the β-adrenergic pathway. Circulation. 1998;98:1329–1334. doi: 10.1161/01.cir.98.13.1329. [DOI] [PubMed] [Google Scholar]
- Csordas G, Thomas AP, Hajnoczky G. Calcium signal transmission between ryanodine receptors and mitochondria in cardiac muscle. Trends Cardiovasc Med. 2001;11:269–275. doi: 10.1016/s1050-1738(01)00123-2. [DOI] [PubMed] [Google Scholar]
- Curran J, Brown KH, Santiago DJ, Pogwizd S, Bers DM, Shannon TR. Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca2+-calmodulin-dependent protein kinase II. J Mol Cell Cardiol. 2010;49:25–32. doi: 10.1016/j.yjmcc.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. β-Adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- Domeier TL, Blatter LA, Zima AV. Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J Physiol. 2009;587:5197–5209. doi: 10.1113/jphysiol.2009.177576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzimiri N. Regulation of β-adrenoceptor signaling in cardiac function and disease. Pharmacol Rev. 1999;51:465–501. [PubMed] [Google Scholar]
- Ellison GM, Torella D, Karakikes I, Purushothaman S, Curcio A, Gasparri C, et al. Acute β-adrenergic overload produces myocyte damage through calcium leakage from the ryanodine receptor 2 but spares cardiac stem cells. J Biol Chem. 2007;282:11397–11409. doi: 10.1074/jbc.M607391200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero P, Said M, Sanchez G, Vittone L, Valverde C, Donoso P, et al. Ca2+/calmodulin kinase II increases ryanodine binding and Ca2+-induced sarcoplasmic reticulum Ca2+ release kinetics during β-adrenergic stimulation. J Mol Cell Cardiol. 2007;43:281–291. doi: 10.1016/j.yjmcc.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg KS, Bers DM. Modulation of excitation–contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J Physiol. 2004;556:463–480. doi: 10.1113/jphysiol.2003.055384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho HT, Stevens SC, Terentyeva R, Carnes CA, Terentyev D, Gyorke S. Arrhythmogenic adverse effects of cardiac glycosides are mediated by redox modification of ryanodine receptors. J Physiol. 2011;589:4697–4708. doi: 10.1113/jphysiol.2011.210005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huke S, Bers DM. Ryanodine receptor phosphorylation at Serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem Biophys Res Commun. 2008;376:80–85. doi: 10.1016/j.bbrc.2008.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain M, Orchard CH. Sarcoplasmic reticulum Ca2+ content, L-type Ca2+ current and the Ca2+ transient in rat myocytes during β-adrenergic stimulation. J Physiol. 1997;505:385–402. doi: 10.1111/j.1469-7793.1997.385bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katra RP, Oya T, Hoeker GS, Laurita KR. Ryanodine receptor dysfunction and triggered activity in the heart. Am J Physiol Heart Circ Physiol. 2007;292:H2144–H2151. doi: 10.1152/ajpheart.00924.2006. [DOI] [PubMed] [Google Scholar]
- Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, O’Rourke B, Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–1613. doi: 10.1161/CIRCULATIONAHA.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosower NS, Kosower EM. Thiol labeling with bromobimanes. Methods Enzymol. 1987;143:76–84. doi: 10.1016/0076-6879(87)43015-2. [DOI] [PubMed] [Google Scholar]
- Kourie JI. Interaction of reactive oxygen species with ion transport mechanisms. Am J Physiol Cell Physiol. 1998;275:C1–C24. doi: 10.1152/ajpcell.1998.275.1.C1. [DOI] [PubMed] [Google Scholar]
- Liu G, Abramson JJ, Zable AC, Pessah IN. Direct evidence for the existence and functional role of hyperreactive sulfhydryls on the ryanodine receptor-triadin complex selectively labeled by the coumarin maleimide 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin. Mol Pharmacol. 1994;45:189–200. [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Morimoto S, Uchi J, Kawai M, Hoshina T, Kusakari Y, Komukai K, et al. Protein kinase A-dependent phosphorylation of ryanodine receptors increases Ca2+ leak in mouse heart. Biochem Biophys Res Commun. 2009;390:87–92. doi: 10.1016/j.bbrc.2009.09.071. [DOI] [PubMed] [Google Scholar]
- Nagasaka S, Katoh H, Niu CF, Matsui S, Urushida T, Satoh H, Watanabe Y, Hayashi H. Protein kinase A catalytic subunit alters cardiac mitochondrial redox state and membrane potential via the formation of reactive oxygen species. Circ J. 2007;71:429–436. doi: 10.1253/circj.71.429. [DOI] [PubMed] [Google Scholar]
- Nam GB, Burashnikov A, Antzelevitch C. Cellular mechanisms underlying the development of catecholaminergic ventricular tachycardia. Circulation. 2005;111:2727–2733. doi: 10.1161/CIRCULATIONAHA.104.479295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogrodnik J, Niggli E. Increased Ca2+ leak and spatiotemporal coherence of Ca2+ release in cardiomyocytes during β-adrenergic stimulation. J Physiol. 2010;588:225–242. doi: 10.1113/jphysiol.2009.181800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opie LH, Thandroyen FT, Muller C, Bricknell OL. Adrenaline-induced ‘oxygen-wastage’ and enzyme release from working rat heart. Effects of calcium antagonism, β-blockade, nicotinic acid and coronary artery ligation. J Mol Cell Cardiol. 1979;11:1073–1094. doi: 10.1016/0022-2828(79)90395-x. [DOI] [PubMed] [Google Scholar]
- Remondino A, Kwon SH, Communal C, Pimentel DR, Sawyer DB, Singh K, Colucci WS. β-Adrenergic receptor-stimulated apoptosis in cardiac myocytes is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of the mitochondrial pathway. Circ Res. 2003;92:136–138. doi: 10.1161/01.res.0000054624.03539.b4. [DOI] [PubMed] [Google Scholar]
- Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ Res. 2000;87:774–780. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Ginsburg KS, Bers DM. Reverse mode of the sarcoplasmic reticulum calcium pump and load-dependent cytosolic calcium decline in voltage-clamped cardiac ventricular myocytes. Biophys J. 2000;78:322–333. doi: 10.1016/S0006-3495(00)76595-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- Song LS, Wang SQ, Xiao RP, Spurgeon H, Lakatta EG, Cheng H. β-Adrenergic stimulation synchronizes intracellular Ca2+ release during excitation-contraction coupling in cardiac myocytes. Circ Res. 2001;88:794–801. doi: 10.1161/hh0801.090461. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Chandrasekar B, Gu Y, Luo J, Hamid T, Hill BG, Prabhu SD. Downregulation of CuZn-superoxide dismutase contributes to β-adrenergic receptor-mediated oxidative stress in the heart. Cardiovasc Res. 2007;74:445–455. doi: 10.1016/j.cardiores.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, et al. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venetucci LA, Trafford AW, Eisner DA. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ Res. 2007;100:105–111. doi: 10.1161/01.RES.0000252828.17939.00. [DOI] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- Yan Y, Liu J, Wei C, Li K, Xie W, Wang Y, Cheng H. Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc Res. 2008;77:432–441. doi: 10.1093/cvr/cvm047. [DOI] [PubMed] [Google Scholar]
- Yoshida A, Takahashi M, Imagawa T, Shigekawa M, Takisawa H, Nakamura T. Phosphorylation of ryanodine receptors in rat myocytes during β-adrenergic stimulation. J Biochem. 1992;111:186–190. doi: 10.1093/oxfordjournals.jbchem.a123735. [DOI] [PubMed] [Google Scholar]
- Zhang GX, Kimura S, Nishiyama A, Shokoji T, Rahman M, Yao L, et al. Cardiac oxidative stress in acute and chronic isoproterenol-infused rats. Cardiovasc Res. 2005;65:230–238. doi: 10.1016/j.cardiores.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Zima AV, Bovo E, Bers DM, Blatter LA. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2010;588:4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Picht E, Bers DM, Blatter LA. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ Res. 2008;103:e105–e115. doi: 10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.