

Abstract
Ageing causes arterial endothelial dysfunction that increases the risk of cardiovascular diseases (CVD), but the underlying mechanisms are incompletely understood. The aim of the present study was to determine the role of autophagy, the cellular process of recycling damaged biomolecules, in endothelial dysfunction with ageing. In older humans, expression of autophagy markers in arterial endothelial cells was impaired by ∼50% (P < 0.05) and was associated with an ∼30% (P < 0.05) reduction in arterial endothelium-dependent dilatation (EDD). Similarly, in C57BL/6 control mice ageing was associated with an ∼40% decrease (P < 0.05) in arterial markers of autophagy and an ∼25% reduction (P < 0.05) in EDD. In both humans and mice, impaired EDD was mediated by reduced nitric oxide (NO) bioavailability and was associated with increased oxidative stress and inflammation (P < 0.05). In old mice, treatment with the autophagy-enhancing agent trehalose restored expression of autophagy markers, rescued NO-mediated EDD by reducing oxidative stress, and normalized inflammatory cytokine expression. In cultured endothelial cells, inhibition of autophagy increased oxidative stress and reduced NO production, whereas trehalose enhanced NO production via an autophagy-dependent mechanism. These results provide the first evidence that autophagy is impaired with ageing in vascular tissues. Our findings also suggest that autophagy preserves arterial endothelial function by reducing oxidative stress and inflammation and increasing NO bioavailability. Autophagy-enhancing strategies may therefore have therapeutic efficacy for ameliorating age-associated arterial dysfunction and preventing CVD.
Key points
Advancing age is the major risk factor for the development of cardiovascular diseases.
Arterial endothelial dysfunction, characterized by impaired endothelium-dependent dilatation (EDD), is a key antecedent to age-associated clinical cardiovascular disease.
We tested the hypothesis that changes in autophagy, the process by which cells recycle damaged biomolecules, may be an underlying cause of the age-related reduction in EDD.
We show that autophagy is impaired in arteries of older humans and mice with reduced EDD, and that enhancing autophagy restores EDD by reducing superoxide-dependent oxidative stress and inflammation, and increasing nitric oxide bioavailability.
Our results identify impaired autophagy as a potential cause of age-related arterial dysfunction and suggest that boosting autophagy may be a novel strategy for the treatment of arterial endothelial dysfunction and prevention of cardiovascular diseases with ageing.
Introduction
Advancing age is the major risk factor for cardiovascular diseases (CVD) and this risk is strongly related to dysfunction of arteries (Lakatta & Levy, 2003). One key change to arteries that increases the risk of CVD with ageing is the development of vascular endothelial dysfunction (Widlansky et al. 2003), the central feature of which is impaired endothelium-dependent dilatation (EDD). Impaired EDD results primarily from reduced bioavailability of the dilating molecule nitric oxide (NO) (Luscher & Barton, 1997; Taddei et al. 2001). The age-associated reduction in NO is mediated by oxidative stress and chronic low-grade inflammation, both of which contribute to elevated production of reactive oxygen species (e.g. superoxide) and the accumulation of damaged macromolecules (Brandes et al. 2005; Seals et al. 2011). However, the mechanisms by which these processes develop with ageing and strategies that can be employed to prevent them are incompletely understood.
One unexplored hypothesis is that impairments in the regulation and/or cellular machinery of autophagy, a process that has been related to enhanced longevity (Yen & Klionsky, 2008), underlie the development of vascular endothelial dysfunction with ageing. Autophagy is the major process by which cells break down and recycle damaged proteins, macromolecules and organelles. This occurs either by delivery to a lysosomal receptor (chaperone-mediated autophagy) or via the formation of autophagosomes, specialized double-membrane vesicles that envelop target organelles/macromolecules and later fuse with a lysosome (macroautophagy) (Mizushima, 2007). Ultimately, the lysosome breaks down the autophagic targets, recycling them into substrates (amino acids, etc.) for use by the cell.
Impaired vascular autophagy could play a key role in the development of oxidative stress, inflammation and endothelial dysfunction with ageing by reducing the ability to eliminate dysfunctional proteins/organelles and allowing the buildup of damaged biomolecules that interfere with normal cellular function. If so, agents that improve autophagy represent potentially useful treatments for vascular ageing. While the translational potential of many pharmacological autophagy inducers is limited by non-specific and, in some cases, deleterious physiological effects (Sudarsanam & Johnson, 2010), certain dietary strategies/components may be a viable means of enhancing autophagy to prevent disease (Hannigan & Gorski, 2009). In this context, the nutraceutical trehalose, a natural disaccharide that enhances autophagy independent of mammalian target of rapamycin (mTOR) and other canonical autophagy-regulating pathways (Sarkar et al. 2007), may have significant translational therapeutic potential.
Although autophagy may decline with ageing in selective tissues (Hubbard et al. 2011; Rubinsztein et al. 2011), nothing is known about changes in autophagy with age in arteries, its association with vascular endothelial dysfunction, the mechanisms involved or the potential for pharmacological/dietary agents that stimulate autophagy to reverse arterial ageing. Insight into these issues is particularly important given that autophagy can be either protective or detrimental in different settings of CVD (Gottlieb & Mentzer, 2011; Nemchenko et al. 2011; Schrijvers et al. 2011).
In the present study, we tested the hypothesis that impaired autophagy contributes to arterial endothelial dysfunction with ageing. To do so, we first examined EDD, markers of autophagy and possible mechanisms in arteries of young and older human subjects. We then assessed the functional and mechanistic connections between age-related changes in autophagy and EDD in young and old mice, and mice treated with trehalose to enhance autophagy. Finally, we utilized a cell culture model to provide more direct, cause and effect evidence of the mechanisms by which autophagy influences vascular endothelial function.
Methods
Studies in humans
Subjects
Ten healthy young (20–31 years, n= 5, 2 male) and older (61–71 years, n= 5, 3 male) adults were studied. Subjects were non-obese, non-smokers, non-diabetic and free of clinical diseases as assessed by medical history, physical examination, blood chemistry and resting and exercise ECG. The study conformed to the Declaration of Helsinki. All procedures were approved by the Institutional Review Board of the University of Colorado at Boulder, and written informed consent was obtained from all subjects.
NO-mediated EDD
EDD and endothelium-independent dilatation were determined as the forearm blood flow (FBF) responses to incremental intrabrachial artery infusion of acetylcholine (ACh; 1.0, 2.0, 4.0 and 8.0 μg (100 ml forearm volume)−1 min−1) and the NO donor sodium nitroprusside (SNP; 0.5, 1.0, 2.0 and 4.0 μg (100 ml forearm volume)−1 min−1) using standard strain-gauge venous occlusion plethysmography (Hokanson, Bellevue, WA, USA) as previously described (Walker et al. 2010; Donato et al. 2011). FBF values are presented as percentage change from baseline (saline) condition. To assess NO-mediated EDD, the endothelial NO synthase (eNOS) inhibitor NG-monomethyl l-arginine (l-NMMA; Clinalfa AG, Laeufelingen, Switzerland) was infused for 5 min (5 mg min−1) before and continuously during ACh infusion.
Vascular endothelial cell protein expression
Endothelial cells were obtained from the brachial artery and analysed for protein expression by quantitative immunofluorescence as previously described (Donato et al. 2008, 2011). Cells were collected on sterile J-wires advanced into the brachial artery and withdrawn through an 18-gauge catheter. Wires were transferred to a dissociation buffer, and cells were recovered by washing and centrifugation, fixed in 3.7% formaldehyde, and plated on poly-l-lysine slides. For immunofluorescence analysis, slides were analysed in parallel with cultured human umbilical vein endothelial cell (HUVEC, passage 6–9) controls. To characterize autophagy, subject and HUVEC slides were incubated with primary antibodies for beclin 1 (1:500 dilution, Cell Signaling) and the adaptor protein p62 (1:500; MBL International, Woburn, MA, USA), established markers of autophagy that have been reported to change with age (Shibata et al. 2006; Rubinsztein et al. 2011), followed by Alexa Fluor 555-conjugated secondary antibody (Invitrogen, Carlsbad, CA, USA). Slides were then incubated with antibody for von Willebrand factor (1:1000; Dako North America Inc., Carpinteria, CA, USA) followed by Alexa Fluor 488-conjugated secondary antibody (Invitrogen). Vectashield DAPI (Vector Laboratories, Inc., Burlingame, CA, USA) was applied and samples were incubated overnight at 4°C before viewing with a fluorescence microscope (Eclipse 600, Nikon). Thirty endothelial cells were identified on each slide by positive staining for von Willebrand factor and nuclear integrity was confirmed with DAPI. Images were captured and analysed with Metamorph software (Universal Imaging, Downingtown, PA, USA) to quantify Alexa Fluor 555 staining, reported as a ratio of endothelial cell to HUVEC control staining intensity to control for differences among staining sessions (Donato et al. 2008, 2011).
Circulating markers
Plasma C-reactive protein was measured with a high-sensitivity Chemistry Immuno Analyzer (AU400e, Olympus). Interleukin 6 (IL-6; R&D Systems, Minneapolis, MN, USA) and oxidized low-density lipoprotein (oxLDL; Alpco Diagnostics, Salem, NH, USA) were measured by standard ELISA. All assays were performed by the University of Colorado Clinical Research core laboratory.
Studies in mice
Animals
Young (4–6 months, n= 5–7 per group) and old (27–28 months, ∼50% survival age, n= 7 per group) male C57BL/6 mice, an established model of ageing and vascular dysfunction (Sprott & Ramirez, 1997; Blackwell et al. 2004; Brown et al. 2007), were obtained from the National Institute on Aging rodent colony. Control animals received regular drinking water, whereas treated animals received 2% trehalose (Sigma-Aldrich, St Louis, MO, USA) supplemented water, a dose previously reported to be effective (Tanaka et al. 2004; Rodriguez-Navarro et al. 2010), for 4 weeks. Mice were housed in an animal care facility at the University of Colorado at Boulder on a 12:12 h light–dark cycle. All procedures conformed to the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85-23, revised 2011) and were approved by the University of Colorado at Boulder Animal Care and Use Committee.
Arterial protein expression
Western blot analyses were performed on cleaned mouse aortas to provide sufficient tissue for analysis. Thoracic aortas were excised and cleaned of surrounding tissue. Intact aortas were frozen in liquid nitrogen before storage and later homogenized in ice-cold radio-immunoprecipitation assay lysis buffer with protease and phosphatase inhibitors. Ten micrograms of protein was loaded onto 4–12% polyacrylamide gels, separated by electrophoresis and transferred to nitrocellulose membranes for standard immunoblot analysis as previously described (Rippe et al. 2010; Sindler et al. 2011). Blots were probed for the same proteins studied in human cells, as well as additional markers of autophagy and vascular function that have been reported to decline with age (Rippe et al. 2010; Rubinsztein et al. 2011). Primary antibodies included: beclin 1 (1:1000), p62 (1:2000), WD-repeat protein interacting with phosphoinositides (WIPI-1, 1:1000; Sigma), lipid-modified microtubule-associated protein1 light chain 3 (LC3-II, 1:2000, Cell Signaling Technology, Inc., Danvers, MA, USA), lysosome-associated membrane protein 2a (LAMP-2a, 1:1000; Abcam Inc., Cambridge, MA, USA), and eNOS (1:500; BD Biosciences, Franklin Lakes, NJ, USA). Proteins were detected using horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) and ECL chemiluminescent substrate (Pierce, Rockford, IL, USA). Protein expression is presented normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Cell Signaling, 1:1000), and data are expressed as a ratio of the mean of the young control group. Concentrations of the cytokines interleukin 1β (IL-1β), IL-6 and tumour necrosis factor α (TNF-α) were determined in aortic lysates containing 10 μg total protein by multiplex ELISA (Searchlight Mouse Inflammatory Cytokine Kit, Aushon Biosystems, Billerica, MA, USA) according to manufacturer's instructions (Rippe et al. 2010; Sindler et al. 2011).
NO-mediated EDD
EDD and endothelium-independent dilatation were determined ex vivo in isolated carotid arteries as previously described (Rippe et al. 2010; Sindler et al. 2011). Mice were anaesthetized using isoflurane and killed by exsanguination via cardiac puncture. Carotid arteries were excised and cannulated onto glass micropipettes in myograph chambers (DMT Inc., Aarhus, Denmark) using nylon (11-0) suture. Arteries were pressurized to 50 mmHg at 37°C in physiological saline solution and allowed to equilibrate for 1 h. EDD: after preconstriction with phenylephrine (2 μm), increases in luminal diameter in response to ACh (1 × 10−9–1 × 10−4 m) with and without co-administration of the eNOS inhibitor NG-nitro-l-arginine methyl ester (l-NAME; 0.1 mm, 30 min incubation to block NO production) or the superoxide dismutase mimetic TEMPOL (1 mm, 60 min incubation to scavenge superoxide) were measured. Endothelium-independent dilatation was determined similarly by measuring vasodilatation in response to SNP (1 × 10−10–1 × 10−4 m). All dose–response data are presented on a percentage basis to account for differences in maximal carotid artery diameter between young and old animals. NO-dependent dilatation was determined from maximal EDD in the absence or presence of l-NAME according to the following formula: NO-dependent dilatation (%) = max dilatationACh− max dilatationACh+L-NAME.
Aortic superoxide production
Measures of superoxide production were made by electron paramagnetic resonance (EPR) spectroscopy using the spin probe 1-hydroxy-3-methoxycarbonly-2,2,5,5-tetramethylpyrrolidine (CMH; Alexis Biochemicals, Farmingdale, NY, USA) as previously described (Rippe et al. 2010; Sindler et al. 2011). Freshly cleaned and dissected 2 mm aortic segments were incubated for 60 min at 37°C in Krebs–Hepes buffer with 0.5 mm CMH, and the EPR signal amplitude was analysed immediately on an MS300 X-band EPR spectrometer (Magnettech GmbH, Berlin, Germany) with the following settings: centrefield, 3350 G; sweep, 80 G; microwave modulation, 3000 mG; microwave attenuation, 7 dB. Stock solutions of CMH were prepared in ice-cold de-oxygenated buffer to minimize auto-oxidation of the spin probe. Data are expressed relative to the mean of the young control group.
Studies in cell culture and isolated arteries
Cell culture
HUVECs were used for cell culture experiments because they are a relevant and established model of vascular endothelial biology, and are sufficiently robust to endure treatments such as nucleic acid transfection (Bouis et al. 2001). Pooled (multiple donors) primary HUVEC cultures (Lonza, Walkersville, MD, USA) were grown in an endothelial cell growth media system (EGM-2, Lonza) at 37°C, 5% humidity. Cells were subcultured at 70% confluency, and all experiments were performed on passage 3–4 cells. Treatments were applied by adding freshly prepared medium containing reagents (10 mm trehalose, 10 mm 3-methyladenine (3-MA)) for 24 h. All experiments were repeated four times. Protein expression was determined in whole-cell lysates by standard western blotting analysis as described above. Primary antibodies: autophagy protein 12 (Atg12, 1:1000, Cell Signaling), eNOS (1:1000), p62 (1:1000).
Nucleic acid transfection
Cells were grown to 60% confluency and transfected with small interfering RNA (siRNA) specific to Atg12 (GenBank accession number CN400030, Invitrogen) to directly inhibit autophagy, or non-targeting siRNA (Invitrogen) using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's instructions. Mixtures (50:50) of Lipofectamine (10 μl ml−1) and siRNA (10 nm) were prepared in OPTI-MEM (Gibco) reduced serum medium, incubated for 20 min at 20°C, then added to fresh EGM-2 medium (1:5 dilution) and applied to HUVEC cultures. Cells were monitored for toxicity and allowed to expand to 90% confluency (∼48 h) before measurements of Atg12 protein expression and/or superoxide and NO production.
Cellular superoxide production
HUVEC superoxide production was measured by techniques similar to those used for mouse aorta. Cells were collected by gentle scraping, resuspended at a concentration of 106 cells ml−1 in Krebs–Hepes buffer, and incubated for 1 h with CMH. Cell suspension (50 μl) was loaded into a capillary tube and analysed by EPR (Cai et al. 2007). Data are expressed relative to the mean of the control condition.
Cellular NO production
Endothelial cell (HUVEC) NO production was measured by fluorescence microscopy with the NO-specific dye 4-amino-5-methylamino-2′,7′- difluorofluorescein diacetate (DAF-FM-DA, Invitrogen) according to manufacturer's instructions (Rathel et al. 2003). Cells grown on 4-well glass culture slides were pre-incubated in Hanks’ balanced salt solution (HBSS) containing 10 μm DAF-FM-DA, 1 mg ml−1 glucose and 1 mm l-arginine with or without l-NAME (0.1 mm) for 30 min at 37°C. Cells were then washed and incubated in fresh HBSS for 30 min before the addition of ACh (0.1 mm, 20 min). Images of each quadrant in each well were captured on a fluorescence microscope (Eclipse 600, Nikon) and mean fluorescence intensity per cell was analysed. Data are expressed relative to the mean of the control condition.
Isolated arteries
Autophagic flux was determined by monitoring p62 degradation in aortas that were excised, cleaned and incubated in physiological saline solution at 37°C with or without trehalose (10 mm) (Bjorkoy et al. 2009; Mizushima et al. 2010). Segments of aorta were snap frozen each hour and later lysed and analysed for p62 expression by Western blotting. Autophagic protein degradation (flux) is expressed as change in p62 levels relative to β-actin.
Statistical analyses
Results are presented as means ± SEM. Statistical analysis was performed with SPSS 19.0 software. For dose responses, group differences were determined by repeated measures ANOVA. For maximal dilatation, protein expression, superoxide and NO production, and human and animal characteristics, comparisons between groups were made using ANOVA. Significance was determined using P < 0.05.
Results
Studies in humans
EDD and markers of autophagy
Maximal FBF in response to intrabrachial infusion of ACh (FBFACh), a measure of EDD, was lower in the older (63 ± 2 years) compared with the young (22 ± 2 years) adults, and these group differences were abolished by NO inhibition (Fig. 1A), indicating impaired NO-mediated EDD with ageing. No group differences were observed in the FBF responses to SNP (endothelium-independent dilatation, Fig. 1B), suggesting an endothelium-specific impairment in NO-dependent function. Protein expression of beclin 1, a key member of the lipid-kinase complex involved in initiation and regulation of macroautophagy (Cao & Klionsky, 2007), was lower, while levels of p62, a marker of undegraded autophagy substrates (Mizushima et al. 2010), were greater in endothelial cells isolated from the brachial artery of older compared with young adults (Fig. 1C). In the overall group, FBFACh was positively related to expression of beclin 1 in brachial artery endothelial cells (Fig. 1D). The older adults also had greater (P < 0.05) plasma concentrations of oxLDL, a circulating marker of oxidative stress (58 ± 3 vs. 50 ± 2 U l−1), and the inflammatory markers IL-6 (1.2 ± 0.2 vs. 0.6 ± 0.1 pg ml−1) and high-sensitivity C-reactive protein (1.1 ± 0.2 vs. 0.4 ± 0.2 mg l−1) compared with the young controls. Both groups had clinically normal blood pressure and blood chemistries, lipids and glucose.
Figure 1. Impaired EDD in older humans is associated with reduced markers of autophagy.
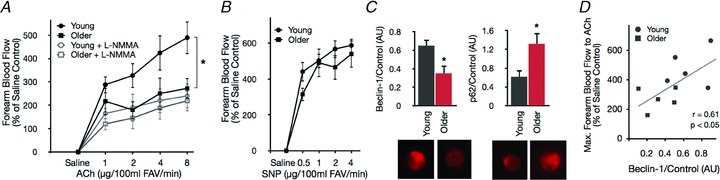
A and B, forearm blood flow responses to intra-brachial infusion of the endothelium-dependent dilator acetylcholine (ACh) in the absence and presence of the eNOS inhibitor NG-monomethyl-l-arginine (l-NMMA), and to the endothelium-independent dilator sodium nitroprusside (SNP) in young and older healthy adults. FAV, forearm volume. C, expression of the key autophagy proteins beclin 1 and p62 in endothelial cells isolated from the brachial artery. D, correlation between maximal forearm blood flow to ACh and beclin 1 protein expression. Protein expression values normalized to HUVEC control cells. All values are means ± SEM (n= 5 per group). *P < 0.05 vs. young.
Studies in mice
Markers of autophagy
Although some of the core autophagic machinery (e.g. autophagy proteins Atg5, Atg7 and Atg12) appeared intact with age in vascular tissue from mice, we observed significant age-related changes in several important markers of autophagy. Similar to our observations in humans, beclin 1 was lower and p62 was greater in old compared with young mice (Fig. 2A and B). This was accompanied by lower expression of WIPI-1 and LC3-II, key downstream mediators/indexes of macroautophagy (Fig. 2C and D) (Mizushima et al. 2010; Nair et al. 2010), and lower levels of LAMP-2a, the critical lysosomal receptor involved in chaperone-mediated autophagy (Fig. 2E) (Cuervo & Dice, 2000).
Figure 2. Autophagy is impaired in the vasculature of old mice and restored by trehalose supplementation.
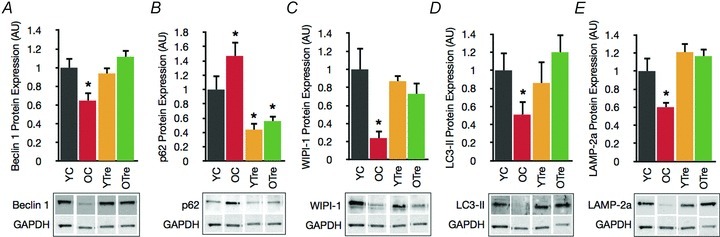
A, key autophagy mediator beclin 1 in aorta of young and old control (YC and OC) and young and old trehalose supplemented (YTre and OTre) mice. B, p62, a marker of undegraded autophagy substrates. C and D, WIPI-1 and LC3-II, markers/indexes of macroautophagy. E, LAMP-2a, critical mediator of chaperone-mediated autophagy. Data expressed relative to GAPDH and normalized to YC mean value. Representative Western blot images below. Values are means ± SEM (n= 5–7 per group). *P < 0.05 vs. YC.
Trehalose supplementation
In aorta of old mice supplemented with trehalose, the autophagy markers beclin 1, WIPI-1, LC3-II and LAMP-2a were restored to levels observed in young mice, and p62 levels were reduced (Fig. 2).
NO-mediated EDD
As observed in our human subjects, EDD to ACh was lower in old compared with young control mice as a result of a smaller NO dilatory influence, indicated by a smaller reduction in EDD in the presence vs. absence of l-NAME (Fig. 3A and B). Trehalose supplementation restored EDD to ACh in old mice by restoring NO-mediated dilatation, but had no effect in the young animals (Fig. 3A and B). There were no differences in NO-mediated EDD in the young control and young and old trehalose treated animals. Endothelium-independent dilatation to SNP, a measure of vascular smooth muscle sensitivity to NO, did not differ among the groups (Fig. 3C), indicating that the impaired EDD in old control mice and the restored EDD in old trehalose treated animals were the result of differences in endothelial production and bioavailability of NO. TEMPOL, a superoxide scavenger, restored EDD to ACh in old control animals, while not affecting responses in young control or young or old trehalose treated animals (Fig. 3D). Consistent with the reduced NO bioavailability and TEMPOL-mediated improvements observed in old control mice, protein expression of eNOS (the enzyme responsible for NO production) was lower (Fig. 3E) and aortic superoxide production was markedly greater (Fig. 3F) in aorta of old compared with young control animals. Importantly, trehalose supplementation restored eNOS and reduced superoxide production in old animals to levels observed in young mice (Fig. 3E and F).
Figure 3. Impaired EDD in old mice is mediated by excessive superoxide, reduced NO bioavailability and increased inflammation, and is restored by trehalose supplementation.
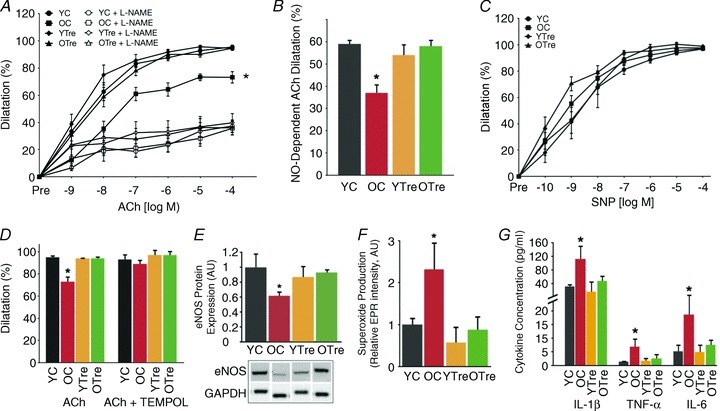
A, dose responses to the endothelium-dependent dilator acetylcholine (ACh) in the absence and presence of the eNOS inhibitor l-NAME in carotid arteries of young and old control (YC and OC) and young and old trehalose-supplemented (YTre and OTre) mice. B, NO-dependent dilatation (Max DilatationACh– Max DilatationACh + L-NAME). C, dose responses to the endothelium-independent dilator sodium nitroprusside (SNP). D, maximal dilatation of carotid arteries to ACh in the presence/absence of TEMPOL a superoxide dismutase mimetic. E, aortic protein expression of eNOS expressed relative to GAPDH and normalized to YC mean value. Representative Western blot image below. F, mean EPR signal for superoxide from aortic rings. G, aortic expression of the inflammatory cytokines IL-1β, TNFα, and IL-6. Values are means ± SEM (n= 5–7 per group). *P < 0.05 vs. YC.
Vascular inflammation
We observed marked increases in expression of the inflammatory cytokines IL-1β, IL-6 and TNF-α in aorta of old compared with young mice (Fig. 3G). Trehalose supplementation reduced aortic inflammatory cytokines only in old mice, normalizing expression to levels observed in young controls.
Studies in cell culture and isolated arteries
Autophagy and the effects of trehalose in vascular endothelial cells
In cultured HUVECs, treatment with the autophagy blocker 3-MA inhibited autophagy, as indicated by increased p62 levels, and was accompanied by an increase in superoxide production (Fig. 4A–C). The addition of trehalose to the medium reduced p62, lowered superoxide production and increased eNOS and NO bioavailability (Fig. 4A–D). These effects of trehalose were abolished upon co-incubation with 3-MA (Fig. 4A–D). siRNA transfection efficiently and specifically reduced Atg12 protein expression (Fig. 5A), increased superoxide production, reduced NO bioavailability and suppressed the effects of trehalose in HUVEC (Fig. 5B and C).
Figure 4. Autophagy influences human endothelial cell function by modulating oxidative stress and NO bioavailability.
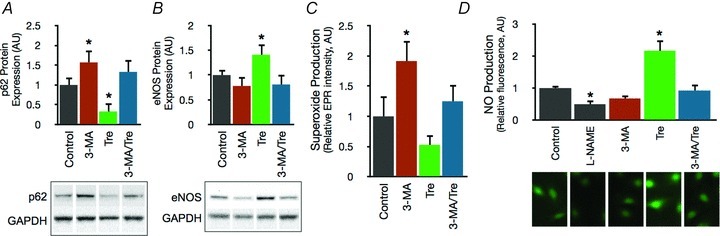
A and B, protein expression of p62 and eNOS in control HUVECs and HUVECs treated for 24 h with the autophagy inhibitor 3-MA (10 mm) and/or trehalose (10 mm). Data expressed relative to GAPDH and normalized to control mean value. Representative Western blot images below. C, mean EPR signal for superoxide in control HUVECs and cells treated with 3-MA and/or trehalose. D, NO production assessed by fluorescence microscopy with the NO-specific dye DAF-FM-DA in control HUVECs and HUVECs treated with the eNOS inhibitor l-NAME, 3-MA and/or trehalose. Representative fluorescence images below. Values are means ± SEM. *P < 0.05 vs. control.
Figure 5. Autophagy is directly linked with NO homeostasis and the effects of trehalose.
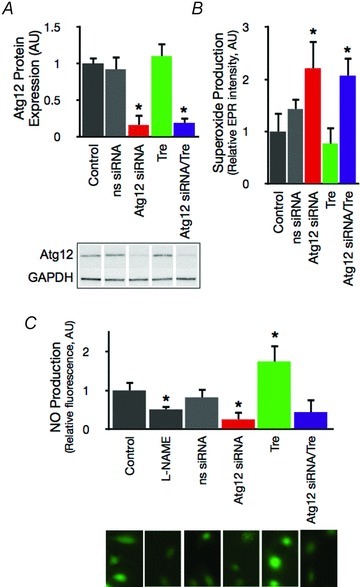
A, Atg12 protein expression in control HUVECs and HUVECs transfected with nonsense (ns) or Atg12-specific siRNA and/or treated with trehalose. Representative Western blot image below. B, mean EPR signal for superoxide in control HUVECs and cells treated with siRNA and/or trehalose. C, NO production assessed with DAF-FM-DA in control HUVECs and cells treated with siRNA and/or trehalose. Representative fluorescence images below. Values are means ± SEM. *P < 0.05 vs. control.
Autophagic flux
Autophagic flux was greater in aorta of young compared with old control mice, as indicated by a greater reduction (via autophagic degradation) in p62 protein levels (Fig. 6). Trehalose enhanced degradation of p62 in aorta from old mice (Fig. 6).
Figure 6. Autophagic flux is reduced in vascular tissue of old mice and enhanced by trehalose.

Top, representative Western blot images of p62 protein in aorta from young and old control mice (YC and OC). Isolated aortas were incubated at 37°C with or without trehalose (Tre, 10 mm), and segments of aorta were snap frozen at the indicated time points. Bottom, autophagic protein degradation expressed as change in p62 levels relative to β-actin. Values are means ± SEM (n= 4 per group). *P < 0.05 vs. YC.
Discussion
Autophagy currently is a compelling focus within the biology of ageing because of its potential relevance to cellular and organismic ageing and age-related dysfunction and disease (Yen & Klionsky, 2008; Madeo et al. 2010; Hubbard et al. 2011; Rubinsztein et al. 2011). However, the role of autophagy in arterial ageing, a major antecedent to clinical CVD (Lakatta & Levy, 2003), is completely unknown. That autophagy can have protective or adverse effects in pathophysiological cardiovascular settings such as atherosclerosis and heart disease (Gottlieb & Mentzer, 2011; Nemchenko et al. 2011; Schrijvers et al. 2011), perhaps depending on the stage of pathology, lends further uncertainty as to its effects in vascular ageing.
The present findings suggest that autophagy is reduced in arteries of older mice and humans and contributes to impaired vascular endothelial function, a clinically important expression of arterial ageing. Importantly, our parallel findings in mice and humans indicate that autophagy protects vascular endothelial function with ageing by reducing oxidative stress and inflammation and increasing NO bioavailability. These results provide a basis for translational research aimed at enhancing autophagy to reverse arterial ageing and reduce the risk of age-associated CVD in humans.
Autophagy and arterial ageing
Impaired cellular housekeeping as a result of reduced autophagy is implicated in neurological diseases, cancers and myopathies, and age-associated decreases in autophagy have been described in a variety of tissues (Mizushima et al. 2008; Yen & Klionsky, 2008; Hubbard et al. 2011). Tissue-specific patterns vary, but in general the expression of critical autophagic mediators such as beclin 1 and LAMP-2a declines with age (Cuervo & Dice, 2000; Shibata et al. 2006; Rubinsztein et al. 2011). Moreover, the degradation rate of both macro- and chaperone-mediated autophagy targets is reduced in aged cells and organisms (Cuervo & Dice, 2000; Del Roso et al. 2003; Hubbard et al. 2011). Our results demonstrate for the first time that age-associated reductions in these established markers of autophagy occur in vascular tissues and endothelial cells of mice and humans. Moreover, we showed that autophagy itself (i.e. autophagic flux) was reduced in arteries of old mice. Given the critical role of autophagy in cellular homeostasis, these findings suggest that reduced autophagy may be an important mechanism underlying age-related changes in the vasculature.
Oxidative stress, autophagy and arterial ageing
A key mechanism underlying vascular endothelial dysfunction and CVD is oxidative stress, characterized by excessive superoxide production and oxidative protein/organelle damage (Lakatta & Levy, 2003; Seals et al. 2011). Because autophagy is a major mechanism by which cells are protected from oxidative stress and dispose of organelles, proteins and other macromolecules subjected to oxidant damage (Mizushima, 2007; Kroemer et al. 2010), impaired autophagy may contribute to increased vascular oxidative stress with ageing. In support of this idea, we observed that reduced vascular autophagy was associated with oxidative stress-mediated impairment of NO-dependent EDD in old mice. This was indicated by increased arterial superoxide production measured directly by EPR spectroscopy and selective rescue of function in arteries from old mice by administration of TEMPOL, a superoxide dismutase mimetic. eNOS protein also was reduced in aorta of old animals and may have contributed to impaired NO-mediated dilatation. Consistent with these observations in mice, we found that reduced expression of autophagy markers in arterial endothelial cells was associated with impaired NO-dependent EDD (FBFACh) in older humans. We also showed that circulating oxLDL, a marker of systemic oxidative stress, was elevated in the older adults, in agreement with previous reports from our laboratory (Eskurza et al. 2004, 2006) and others (Taddei et al. 2001) linking oxidative stress to impaired endothelial function with ageing in humans.
Collectively, these findings are in keeping with an established role of autophagy in cellular homeostasis and defence against oxidative stress (Kroemer et al. 2010), as well as observations that cultured human endothelial cells respond to oxidant and biochemical stressors by increasing autophagy (Zhang et al. 2010; Wang et al. 2011). Moreover, our cell culture experiments extend these findings by demonstrating that extended inhibition of autophagy reduces eNOS and ACh-mediated NO production and increases superoxide production in vascular endothelial cells. These results support the idea that the associations between autophagy and NO-mediated vascular endothelial function we observed in mice and humans are mechanistically linked. Autophagy may, therefore, directly influence endothelial cell function by modulating cellular redox status and maintaining NO bioavailability.
Inflammation, autophagy and arterial ageing
Inflammation is implicated in the aetiology of CVD (Pearson et al. 2003) and contributes to vascular ageing and dysfunction in mice and humans (Csiszar et al. 2003; Donato et al. 2008; Lesniewski et al. 2011). Autophagy is a critical modulator of the cellular inflammatory response (Kroemer et al. 2010) and impaired autophagy could, therefore, play an important role in mediating age-associated vascular inflammation. Recent reports indicate that cultured human endothelial cells respond to inflammatory stimuli by increasing autophagy (Patschan et al. 2008; Xie et al. 2011), but the exact role of this process in mediating vascular inflammation in vivo is unknown. Previous results from our laboratory and others show that endothelial dysfunction with age is associated with increased vascular and systemic inflammation, indicated by elevated levels of pro-inflammatory cytokines such as TNF-α and IL-6 (Csiszar et al. 2003; Donato et al. 2008; Lesniewski et al. 2011). Here, we extend these findings by showing that reduced vascular autophagy is associated with increased inflammation and impaired EDD in older mice and humans. As such, our results provide the first evidence for a link between age-related impairments in vascular autophagy and inflammation.
Trehalose, autophagy and arterial ageing
As confirmed in the present study, vascular endothelial dysfunction with ageing is mediated by reduced NO bioavailability as a result of increases in superoxide-associated oxidative stress and inflammation (Taddei et al. 2001; Rippe et al. 2010; Seals et al. 2011; Sindler et al. 2011). Accordingly, strategies aimed at reducing oxidative stress and inflammation may have therapeutic efficacy for treating vascular endothelial dysfunction with advancing age (Rippe et al. 2010; Sindler et al. 2011). In this context, the natural disaccharide trehalose may have potential as a nutraceutical for intervention in the ageing process. In cell culture and animal models, trehalose enhances autophagy (Sarkar et al. 2007; Rodriguez-Navarro et al. 2010), improves function in models of age-related neurological diseases (Tanaka et al. 2004; Rodriguez-Navarro et al. 2010), protects against diet-induced insulin resistance and extends longevity (Tanaka et al. 2004; Arai et al. 2010; Honda et al. 2010).
In the present study, we found that trehalose supplementation restored the expression of autophagy markers and rescued vascular endothelial function by increasing NO bioavailability and eNOS, reducing superoxide production and normalizing inflammatory cytokines in arteries of old mice. Importantly, our studies in isolated arteries suggest that trehalose enhances autophagic flux, and our cell culture experiments demonstrate that trehalose stimulates NO production and reduces superoxide production in vascular endothelial cells via an autophagy-dependent mechanism.
Taken together, these findings suggest that enhancing autophagy with trehalose may be a promising intervention for treating arterial ageing by increasing NO bioavailability and reducing oxidative stress and inflammation. Moreover, because trehalose is a widely available nutraceutical with low toxicity and its effects were observed after a relatively short treatment period (4 weeks), it may have significant translational potential for treating arterial ageing in humans.
Conclusions
The results of this study show for the first time that autophagy is reduced with ageing in vascular tissues, and that enhancing autophagy with age ameliorates vascular endothelial dysfunction. Our findings also demonstrate that autophagy plays an important role in preserving vascular endothelial function by reducing superoxide-associated oxidative stress, increasing NO bioavailability and exerting an anti-inflammatory influence on arteries. Importantly, our data provide the first in vitro and in vivo evidence for antioxidant, anti-inflammatory and NO-enhancing effects of the nutraceutical trehalose on vascular tissue and specifically in the setting of arterial ageing.
Acknowledgments
This work was supported by the National Institutes of Health: AG013038, AG039210. We would like to thank Melanie Connell for her technical assistance. We have no conflicts of interest or disclosures.
Glossary
- ACh
acetylcholine
- Atg12
autophagy-related protein 12
- CMH
1-hydroxy-3-methoxycarbonly-2,2,5,5-tetramethylpyrrolidine
- CVD
cardiovascular disease
- DAF-FM-DA
4-amino-5-methylamino-2′,7′- difluorofluorescein diacetate
- EDD
endothelium-dependent dilatation
- eNOS
endothelial nitric oxide synthase
- EPR
electron paramagnetic resonance
- FBF
forearm blood flow
- HUVEC
human umbilical vein endothelial cell
- IL-1β
interleukin 1β
- IL-6
interleukin 6
- 3-MA
3-methyladenine
- l-NAME
NG-nitro-l-arginine methyl ester
- l-NMMA
NG-monomethyl l-arginine
- LAMP-2a
lysosome-associated membrane protein 2a
- LC3-II
lipid-modified microtuble-associated protein light chain 3
- NO
nitric oxide
- oxLDL
oxidized low-density lipoprotein
- siRNA
small interfering RNA
- SNP
sodium nitroprusside
- TNF-α
tumor necrosis factor α
- vWVF
von Willebrand Factor
- WIPI-1
WD-repeat protein interacting with phosphoinositides 1
Author contributions
T.J.L., G.D.H., A.T., A.L.S., G.L.P. and D.R.S. contributed to the conception and design, analysis and interpretation of data. T.J.L. and D.R.S. contributed to the drafting and revision of the article. All authors provided final approval of the version to be published. All experiments were carried out at the University of Colorado at Boulder.
References
- Arai C, Arai N, Mizote A, Kohno K, Iwaki K, Hanaya T, Arai S, Ushio S, Fukuda S. Trehalose prevents adipocyte hypertrophy and mitigates insulin resistance. Nutr Res. 2010;30:840–848. doi: 10.1016/j.nutres.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- Blackwell KA, Sorenson JP, Richardson DM, Smith LA, Suda O, Nath K, Katusic ZS. Mechanisms of aging-induced impairment of endothelium-dependent relaxation: role of tetrahydrobiopterin. Am J Physiol Heart Circ Physiol. 2004;287:H2448–2453. doi: 10.1152/ajpheart.00248.2004. [DOI] [PubMed] [Google Scholar]
- Bouis D, Hospers GA, Meijer C, Molema G, Mulder NH. Endothelium in vitro: a review of human vascular endothelial cell lines for blood vessel-related research. Angiogenesis. 2001;4:91–102. doi: 10.1023/a:1012259529167. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Fleming I, Busse R. Endothelial aging. Cardiovasc Res. 2005;66:286–294. doi: 10.1016/j.cardiores.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Brown KA, Didion SP, Andresen JJ, Faraci FM. Effect of aging, MnSOD deficiency, and genetic background on endothelial function: evidence for MnSOD haploinsufficiency. Arterioscler Thromb Vasc Biol. 2007;27:1941–1946. doi: 10.1161/ATVBAHA.107.146852. [DOI] [PubMed] [Google Scholar]
- Cai H, Dikalov S, Griendling KK, Harrison DG. Detection of reactive oxygen species and nitric oxide in vascular cells and tissues: comparison of sensitivity and specificity. Methods Mol Med. 2007;139:293–311. doi: 10.1007/978-1-59745-571-8_20. [DOI] [PubMed] [Google Scholar]
- Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res. 2007;17:839–849. doi: 10.1038/cr.2007.78. [DOI] [PubMed] [Google Scholar]
- Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Aging-induced proinflammatory shift in cytokine expression profile in coronary arteries. FASEB J. 2003;17:1183–1185. doi: 10.1096/fj.02-1049fje. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000;275:31505–31513. doi: 10.1074/jbc.M002102200. [DOI] [PubMed] [Google Scholar]
- Del Roso A, Vittorini S, Cavallini G, Donati A, Gori Z, Masini M, Pollera M, Bergamini E. Ageing-related changes in the in vivo function of rat liver macroautophagy and proteolysis. Exp Gerontol. 2003;38:519–527. doi: 10.1016/s0531-5565(03)00002-0. [DOI] [PubMed] [Google Scholar]
- Donato AJ, Black AD, Jablonski KL, Gano LB, Seals DR. Aging is associated with greater nuclear NFκB, reduced IκBα, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging Cell. 2008;7:805–812. doi: 10.1111/j.1474-9726.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato AJ, Magerko KA, Lawson BR, Durrant JR, Lesniewski LA, Seals DR. SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J Physiol. 2011;589:4545–4554. doi: 10.1113/jphysiol.2011.211219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskurza I, Kahn ZD, Seals DR. Xanthine oxidase does not contribute to impaired peripheral conduit artery endothelium-dependent dilatation with ageing. J Physiol. 2006;571:661–668. doi: 10.1113/jphysiol.2005.102566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskurza I, Monahan KD, Robinson JA, Seals DR. Effect of acute and chronic ascorbic acid on flow-mediated dilatation with sedentary and physically active human ageing. J Physiol. 2004;556:315–324. doi: 10.1113/jphysiol.2003.057042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb RA, Mentzer RM., Jr Cardioprotection through autophagy: ready for clinical trial? Autophagy. 2011;7:434–435. doi: 10.4161/auto.7.4.14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannigan AM, Gorski SM. Macroautophagy: the key ingredient to a healthy diet? Autophagy. 2009;5:140–151. doi: 10.4161/auto.5.2.7529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda Y, Tanaka M, Honda S. Trehalose extends longevity in the nematode Caenorhabditis elegans. Aging Cell. 2010;9:558–569. doi: 10.1111/j.1474-9726.2010.00582.x. [DOI] [PubMed] [Google Scholar]
- Hubbard VM, Valdor R, Macian F, Cuervo AM. Selective autophagy in the maintenance of cellular homeostasis in aging organisms. Biogerontology. 2011;13:21–35. doi: 10.1007/s10522-011-9331-x. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139–146. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- Lesniewski LA, Durrant JR, Connell ML, Folian BJ, Donato AJ, Seals DR. Salicylate treatment improves age-associated vascular endothelial dysfunction: potential role of nuclear factor κB and forkhead Box O phosphorylation. J Gerontol A Biol Sci Med Sci. 2011;66:409–418. doi: 10.1093/gerona/glq233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher TF, Barton M. Biology of the endothelium. Clin Cardiol. 1997;20:II-3–10. [PubMed] [Google Scholar]
- Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol. 2010;12:842–846. doi: 10.1038/ncb0910-842. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair U, Cao Y, Xie Z, Klionsky DJ. Roles of the lipid-binding motifs of Atg18 and Atg21 in the cytoplasm to vacuole targeting pathway and autophagy. J Biol Chem. 2010;285:11476–11488. doi: 10.1074/jbc.M109.080374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemchenko A, Chiong M, Turer A, Lavandero S, Hill JA. Autophagy as a therapeutic target in cardiovascular disease. J Mol Cell Cardiol. 2011;51:584–593. doi: 10.1016/j.yjmcc.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patschan S, Chen J, Polotskaia A, Mendelev N, Cheng J, Patschan D, Goligorsky MS. Lipid mediators of autophagy in stress-induced premature senescence of endothelial cells. Am J Physiol Heart Circ Physiol. 2008;294:H1119–1129. doi: 10.1152/ajpheart.00713.2007. [DOI] [PubMed] [Google Scholar]
- Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO, 3rd, Criqui M, Fadl YY, Fortmann SP, Hong Y, Myers GL, Rifai N, Smith SC, Jr, Taubert K, Tracy RP, Vinicor F. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499–511. doi: 10.1161/01.cir.0000052939.59093.45. [DOI] [PubMed] [Google Scholar]
- Rathel TR, Leikert JJ, Vollmar AM, Dirsch VM. Application of 4,5-diaminofluorescein to reliably measure nitric oxide released from endothelial cells in vitro. Biol Proced Online. 2003;5:136–142. doi: 10.1251/bpo55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippe C, Lesniewski L, Connell M, LaRocca T, Donato A, Seals D. Short-term calorie restriction reverses vascular endothelial dysfunction in old mice by increasing nitric oxide and reducing oxidative stress. Aging Cell. 2010;9:304–312. doi: 10.1111/j.1474-9726.2010.00557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Navarro JA, Rodriguez L, Casarejos MJ, Solano RM, Gomez A, Perucho J, Cuervo AM, Garcia de Yebenes J, Mena MA. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis. 2010;39:423–438. doi: 10.1016/j.nbd.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–5652. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- Schrijvers DM, De Meyer GR, Martinet W. Autophagy in atherosclerosis: a potential drug target for plaque stabilization. Arterioscler Thromb Vasc Biol. 2011;31:2787–2791. doi: 10.1161/ATVBAHA.111.224899. [DOI] [PubMed] [Google Scholar]
- Seals DR, Jablonski KL, Donato AJ. Aging and vascular function in humans. Clin Sci (Lond) 2011;120:357–375. doi: 10.1042/CS20100476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, MacDonald M, Yankner B, Yuan J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem. 2006;281:14474–14485. doi: 10.1074/jbc.M600364200. [DOI] [PubMed] [Google Scholar]
- Sindler AL, Fleenor BS, Calvert JW, Marshall KD, Zigler ML, Lefer DJ, Seals DR. Nitrite supplementation reverses vascular endothelial dysfunction and large elastic artery stiffness with aging. Aging Cell. 2011;10:429–437. doi: 10.1111/j.1474-9726.2011.00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprott RL, Ramirez I. Current inbred and hybrid rat and mouse models for gereontological research. ILAR J. 1997;38:104–109. doi: 10.1093/ilar.38.3.104. [DOI] [PubMed] [Google Scholar]
- Sudarsanam S, Johnson DE. Functional consequences of mTOR inhibition. Curr Opin Drug Discov Devel. 2010;13:31–40. [PubMed] [Google Scholar]
- Taddei S, Virdis A, Ghiadoni L, Salvetti G, Bernini G, Magagna A, Salvetti A. Age-related reduction of NO availability and oxidative stress in humans. Hypertension. 2001;38:274–279. doi: 10.1161/01.hyp.38.2.274. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, Kurosawa M, Nekooki M, Nukina N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med. 2004;10:148–154. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- Walker AE, Seibert SM, Donato AJ, Pierce GL, Seals DR. Vascular endothelial function is related to white blood cell count and myeloperoxidase among healthy middle-aged and older adults. Hypertension. 2010;55:363–369. doi: 10.1161/HYPERTENSIONAHA.109.145870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Liang B, Shirwany NA, Zou MH. 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS One. 2011;6:e17234. doi: 10.1371/journal.pone.0017234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlansky ME, Gokce N, Keaney JF, Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003;42:1149–1160. doi: 10.1016/s0735-1097(03)00994-x. [DOI] [PubMed] [Google Scholar]
- Xie Y, You SJ, Zhang YL, Han Q, Cao YJ, Xu XS, Yang YP, Li J, Liu CF. Protective role of autophagy in AGE-induced early injury of human vascular endothelial cells. Mol Med Report. 2011;4:459–464. doi: 10.3892/mmr.2011.460. [DOI] [PubMed] [Google Scholar]
- Yen WL, Klionsky DJ. How to live long and prosper: autophagy, mitochondria, and aging. Physiology (Bethesda) 2008;23:248–262. doi: 10.1152/physiol.00013.2008. [DOI] [PubMed] [Google Scholar]
- Zhang YL, Cao YJ, Zhang X, Liu HH, Tong T, Xiao GD, Yang YP, Liu CF. The autophagy-lysosome pathway: a novel mechanism involved in the processing of oxidized LDL in human vascular endothelial cells. Biochem Biophys Res Commun. 2010;394:377–382. doi: 10.1016/j.bbrc.2010.03.026. [DOI] [PubMed] [Google Scholar]