

Abstract
Intrauterine growth restriction is a risk factor for cardiovascular disease in adulthood. We have previously shown that intrauterine growth restriction caused by uteroplacental insufficiency programmes uterine vascular dysfunction and increased arterial stiffness in adult female rat offspring. The aim of this study was to investigate vascular adaptations in growth restricted female offspring when they in turn become pregnant. Uteroplacental insufficiency was induced in WKY rats by bilateral uterine vessel ligation (Restricted) or sham surgery (Control) on day 18 of pregnancy. F0 pregnant females delivered naturally at term. F1 Control and Restricted offspring were mated at 4 months of age and studied on day 20 of pregnancy. Age-matched non-pregnant F1 Control and Restricted females were also studied. Wire and pressure myography were used to test endothelial and smooth muscle function, and passive mechanical wall properties, respectively, in uterine, mesenteric, renal and femoral arteries of all four groups. Collagen and elastin fibres were quantified using polarized light microscopy and qRT-PCR. F1 Restricted females were born 10–15% lighter than Controls (P < 0.05). Non-pregnant Restricted females had increased uterine and renal artery stiffness compared with Controls (P < 0.05), but this difference was abolished at day 20 of pregnancy. Vascular smooth muscle and endothelial function were preserved in all arteries of non-pregnant and pregnant Restricted rats. Collagen and elastin content were unaltered in uterine arteries of Restricted females. Growth restricted females develop compensatory vascular changes during late pregnancy, such that region-specific vascular deficits observed in the non-pregnant state did not persist in late pregnancy.
Key points
Uteroplacental insufficiency programmes uterine vascular dysfunction in female offspring born growth restricted. The vascular adaptations in these female offspring when they in turn become pregnant are poorly understood.
Females born small and later become pregnant have compensatory vascular adaptations, such that the increased uterine and renal arterial stiffness observed in the non-pregnant state was resolved in late pregnancy.
Vascular smooth muscle and endothelial function was normal in pregnant growth restricted female offspring. There was a reduced sensitivity to angiotensin II, but an increased sensitivity to phenylephrine in uterine arteries during pregnancy, and enhanced endothelium-mediated relaxation in uterine and mesenteric arteries. Importantly, arteries of growth restricted females adapted to these changes.
Pregnancy was associated with increased outside and internal diameters in uterine and mesenteric arteries, but not renal and femoral arteries, and being born growth restricted did not alter this process.
These findings may assist our understanding of the maternal vascular adaptations to pregnancy in growth restricted female offspring.
Introduction
Intrauterine growth restriction occurs in about 7–10% of pregnancies and is a major cause of perinatal morbidity and mortality. Uteroplacental insufficiency is the leading cause of intrauterine growth restriction in the Western world and is characterised by compromised uteroplacental blood flow and reduced oxygen and nutrient delivery to the developing fetus. Epidemiological and experimental studies have shown a strong association between low birth weight, an indicator of intrauterine growth restriction, and risk of higher blood pressure and cardiovascular disease in adulthood (Barker et al. 2006). Uteroplacental insufficiency causes fetal growth restriction in both male and female offspring. However, there is a sexually dimorphic adult cardiovascular phenotype, with males but not females developing hypertension and glomerular hypertrophy (Grigore et al. 2008; Moritz et al. 2010).
During pregnancy, the maternal cardiovascular system undergoes remarkable adaptive changes. At a systemic level, increased blood flow to the uteroplacental circulation is achieved by elevating maternal blood volume and increasing cardiac output (Poston et al. 1995; Thornburg et al. 2000). To accommodate the increased blood supply, vascular tone is shifted in favour of vasodilatation. Accordingly, vascular responsiveness to vasopressors is attenuated in some vascular beds and vasodilator responses are enhanced via endothelium-dependent and –independent mechanisms (Magness et al. 2001; Gillham et al. 2003). The increase in endothelium-dependent vasodilatation is mediated by nitric oxide (NO), prostacyclin (PGI2) and endothelium-derived hyperpolarising factor (EDHF). In addition, one of the most dramatic changes that occurs during pregnancy is remodelling of the uterine vasculature to ensure adequate uteroplacental perfusion to the developing fetus (Osol & Mandala, 2009). During normal pregnancy, the uterine artery vascular wall undergoes hypertrophic and hyperplasic changes. Accordingly, the main uterine artery doubles in size (outside and internal diameters) in pregnant humans and increases 2- to 3-fold in rodents. Importantly, inappropriate adaptation of the uterine vasculature in pregnancy is associated with compromised uteroplacental blood flow, intrauterine growth restriction and pregnancy complications, including pre-eclampsia (Reslan & Khalil, 2010).
Our laboratory uses a rat model of uteroplacental insufficiency induced by bilateral uterine vessel ligation in late gestation, which results in offspring that are born 10–15% smaller (Wlodek et al. 2005, 2007, 2008). We have previously shown that 18-month-old virgin growth restricted female offspring have impaired uterine endothelial function manifested by reduced EDHF-mediated relaxation (Mazzuca et al. 2010). These rats have reduced uterine artery diameter and increased wall stiffness, and this is associated with increased proportion of thick, less compliant collagen and elastin fibre content (Mazzuca et al. 2010).
There is little information regarding the vascular adaptations to pregnancy in females who themselves had been born small. Thus, we have extended our studies to investigate whether or not adult females born growth restricted have impaired vascular adaptations in their pregnancy in the absence of any further uteroplacental insufficiency or other challenge. We predicted that the physiological challenge of pregnancy would exacerbate existing vascular phenotypes in growth restricted females. Given the regional heterogeneity in vascular function, this study aimed to provide a more comprehensive analysis of the vascular adaptations to pregnancy following growth restriction in female offspring by studying uterine, mesenteric, renal and femoral arteries.
Methods
Ethics
All procedures were approved by The University of Melbourne Pharmacology, Physiology, Biochemistry & Molecular Biology and Bio21 Institute Animal Ethics Committee. All of the authors have read the article ‘Reporting ethical matters in The Journal of Physiology: standards and advice’ (Drummond, 2009), and our experiments comply with the policies and regulations.
Animals
Wistar–Kyoto rats were housed in an environmentally controlled room with a 12 h light–dark cycle and access to food and tap water ad libitum. Virgin female rats aged 12–19 weeks were mated with normal males (Wlodek et al. 2007; O’Dowd et al. 2008). On day 18 of gestation, F0 pregnant rats were randomly allocated to a sham (offspring termed Control) or uteroplacental insufficiency (offspring termed Restricted) group (n= 10/group). The Restricted group underwent bilateral uterine vessel (artery and vein) ligation surgery (Wlodek et al. 2005, 2007; O’Dowd et al. 2008; Moritz et al. 2009; Mazzuca et al. 2010). The F0 pregnant females delivered naturally at term on day 22 of gestation. We have previously shown that uteroplacental insufficiency reduces total (male and female) litter size (Gallo et al. 2012). Body weights from Control and Restricted female offspring (F1) were recorded at postnatal days 1, 6, 14 and 35. At 12 weeks of age, F1 Control and Restricted female offspring were allocated to either non-pregnant or pregnant groups (1/litter/group; n= 10/group). Those allocated to the pregnant group were weighed and mated (12–19 weeks) with different normal males. Mating a Restricted female with a normal male resulted in a ∼100% pregnancy success rate on the first attempt at mating. It is important to note that the pregnant offspring did not undergo uteroplacental insufficiency or sham surgery or any other experimental manipulation. At day 20 of gestation, pregnant female rats and age matched non-pregnant rats were weighed and anaesthetized with intraperitoneal injection of ketamine (100 mg kg−1 body weight) and Ilium Xylazil-20 (30 mg (kg body weight)−1). The main uterine and small mesenteric, renal and femoral arteries were isolated and used for vascular function studies (Mazzuca et al. 2010). Remaining uterine and renal artery were immediately snap frozen and stored at −80°C for later molecular analysis. The remainder of the uterine artery tissue was immersion fixed with 10% neutral buffered formalin for histological analysis (Mazzuca et al. 2010).
In vitro vascular reactivity
Rings of uterine (outside diameter (OD) ∼350 μm), mesenteric (OD ∼300 μm), renal (OD ∼400 μm), femoral (OD ∼400 μm) arteries ∼1–2 mm in length were mounted on a four channel wire myograph (Model 610 M, Danish Myo Technology, Aarhus, Denmark) for measurement of isometric tension (Mazzuca et al. 2010). Vascular segments from all rats came from the same location along the vascular tree. We used the main uterine artery, 2nd order mesenteric arteries, renal interlobar arteries and the saphenous branch of the femoral artery. Functional reactivity experiments were performed on the same vascular segment.
Briefly, arteries were bathed in physiological saline solution (PSS; mm: 120 NaCl, 5 KCl, 2.5 CaCl2, 25 NaHCO3, 11 glucose, 1 KH2PO4, 1.2 MgSO4) bubbled with 95% O2 and 5% CO2 at ∼36°C. Prior to the commencement of each experiment the viability of the smooth muscle in each segment was tested using a brief application of high K+ PSS (isotonic replacement of Na+ with 100 mm K+). Endothelial function was tested using 10−5 m acetylcholine (ACh) applied for 2 min in arteries preconstricted with phenylephrine (PE).
To test smooth muscle contraction, arteries were exposed to cumulative concentrations of PE (10−9–10−4 m) and angiotensin II (Ang II, 10−10–10−7 m). Contractions were either expressed as a percentage of the contraction evoked by high K+ PSS or by the maximum contraction evoked by that constrictor for that individual artery. Endothelial function was studied in arteries submaximally preconstricted (∼70% of maximal) with PE (3 × 10−7− 3 × 10−6 m). The level of preconstriction was not different in arteries from non-pregnant or pregnant Control and Restricted groups. The endothelium was stimulated with increasing concentrations of ACh (10−9–10−5 m). To elucidate the role of EDHF, ACh curves were repeated in the presence of Nω-nitro-l-arginine methyl ester (l-NAME, 2 × 10−4 m) and indomethacin (Indo, 10−6 m) to block nitric oxide synthase (NOS) and cyclooxygenase (COX), respectively. Smooth muscle relaxation was tested using the NO donor sodium nitroprusside (SNP, 10−9–10−5 m) and ATP-sensitive potassium (KATP) channel opener levcromakalim (LEV, 10−9–10−6 m).
Passive mechanical wall properties
In separate segments of arteries from these rats, uterine, mesenteric, renal and femoral arteries (3–4 mm long) were assessed for their passive mechanical wall properties using a pressure myograph (Living Systems Instrumentation, Burlington, VT, USA), as previously described (Mazzuca et al. 2010). Briefly, arteries were continuously superfused at ∼15 ml min−1 with 0 mm Ca2+-free, 1 mm EGTA PSS, gassed with 95% O2, 5% CO2 at ∼36°C. Each artery was pressurised from 0 to 200 mmHg, with 10 mmHg increases in intraluminal pressure. Arterial dimensions (length, OD and wall thickness (WT)) were measured at each 10 mmHg increment. Wall stress and strain were derived as follows: wall stress (kPa) = (intraluminal pressure × internal diameter)/(2 × wall thickness); wall strain = (internal diameter – internal diameter extrapolated to 10 mmHg pressure)/internal diameter extrapolated to 10 mmHg pressure (Wigg et al. 2001). The media-to-lumen ratio was determined by calculating wall thickness (WT)/internal diameter (ID).
Histological examination of collagen and elastin in uterine arteries
Transverse fixed segments of uterine artery were cut at 5 μm thickness from two locations along the artery from both non-pregnant and pregnant groups (Mazzuca et al. 2010). Collagen and elastin fibres were stained with picrosirius red or Gomori fuschin aldehyde, respectively. Artery sections were examined under a polarised microscope (Olympus10 BX51 Pol) using conventional bright-field illumination and circular crossed-polarised light setup. Total collagen and elastin content were quantified using bright-field (Mazzuca et al. 2010). The proportion of thick (visualised as red, orange and yellow fibres under circular crossed-polarised light) and thin (green fibres) collagen fibres were quantified with circular crossed-polarised light (Mazzuca et al. 2010).
Gene expression analysis in uterine and renal arteries
RNA from frozen uterine and renal arteries was extracted and converted to cDNA and gene expression quantified using fluorescence-based qRT-PCR primers and TaqMan probes on the Rotor Gene 3000 system (Corbett Research, Mortlake, VIC, Australia) as previously described (Moritz et al. 2009; Mazzuca et al. 2010). Only the uterine and renal arteries were selected for molecular analysis as the largest vascular effects between non-pregnant and pregnant groups were detected in these arteries. The primer-probe design strategy was to situate primers and probes within the protein-coding region, exon spanning where possible to avoid genomic DNA contamination. The genes of interest included collagen I, III, IV (α1), elastin, fibronectin, endothelial NOS, small-conductance calcium-activated potassium channel (Kca2.3) and intermediate-conductance calcium-activated potassium channel (Kca3.1). Optimal concentrations for all primers and probes were 300 and 100 nm, respectively. Relative quantification of gene expression was performed by the comparative CT (ΔΔCT) method with ribosomal 18S as the endogenous control. 18S expression in the uterine and renal artery was not different between Control and Restricted from either the pregnant or non-pregnant groups as verified by the cycle threshold values (Ct) (Control vs. Restricted: Uterine, 18S, P= 0.60; Renal, P= 0.40; by two-way ANOVA). Although 18S was used as our housekeeper, we also verified the validity of our results with other common housekeepers such as cyclophilin A and β-actin. The Ct value of these housekeepers also did not change between Control and Restricted groups. Primer and probe sequences are given in the online Supplemental Material (Table S1).
Drugs and chemicals
The following agents were used: angiotensin II (Auspep, Melbourne, VIC, Australia); acetylcholine, indomethacin, levcromakalim, Nω-nitro-l-arginine methyl ester, phenylephrine (Sigma-Aldrich, Castle Hill, NSW, Australia); ilium xylazil-20 (Troy Laboratories, Pty Ltd, Smithfield, NSW, Australia); ketamine (Parnell Laboratories, Pty Ltd, Alexandria, NSW, Australia); primers and probes (Biosearch Technologies, Novato, CA, USA); sodium nitroprusside (Ajax Chemicals, Auburn, SA, Australia); Sirius red F3B and DPX (BDH Laboratory Supplies, Poole, UK); Superscript III RT (Invitrogen, VIC, Australia); Tri Reagent (Ambion, Inc., Austin, TX, USA).
Statistical analysis
Values are expressed as means ±sem with n representing the number of offspring from separate mothers per group. Concentration–response curves were constructed using Prism (v.5.01; GraphPad Software, San Diego, CA, USA) and sigmoidal curves fitted to the data using the least squares method. Contractions to Ang II and PE were expressed as a percentage of contraction to high-K+ PSS or as a percentage of their own maximum contraction evoked by that constrictor. Relaxations evoked by ACh, SNP and LEV were expressed as a percentage of pre-contraction evoked by PE. Where appropriate, data were analysed using a two-way ANOVA to determine the main effects of uteroplacental insufficiency (Control and Restricted) and pregnancy (non-pregnant and pregnant), or Student's unpaired t test, using SPSS (v18, SPSS Inc., Chicago, IL, USA). Two-way ANOVA with repeated measures was performed for vascular dose–response curves. If significant interactions were observed, then individual group means were compared, with the level of significance adjusted by Bonferroni's method to account for multiple comparisons, or Student's unpaired t test were performed for post hoc comparisons. For dose–response curves, if a main effect or significant interaction was observed, the EC50 was determined and the pD2 (–log EC50) and maximum response (Emax) were then compared between groups. In renal arteries, higher concentrations of ACh tended to cause contraction, and the EC50 and Emax values for ACh-induced relaxation were derived from sigmoidal curves fitted to the relaxation component of the dose–response curve (Mazzuca et al. 2010). Stress–strain relationships, OD and ID, WT, media-to-lumen ratio, histological examination of collagen and elastin were analysed using a two-way ANOVA or Student's unpaired t test, where appropriate. Results were considered significant when P < 0.05.
Results
Body weights
Uteroplacental insufficiency in F0 females reduced F1 female body weight at postnatal day 1 (by ∼15%, P < 0.05, Table 1) compared with sham surgery Controls. Restricted female offspring remained lighter for the first 6 days of postnatal life (by 12–17%, P < 0.05, Table 1) but there was no difference in body weight by day 14 when compared with Controls (Table 1). Body weight was not different between Control and Restricted at the time of mating (4 mo) or at postmortem (Table 1). As expected, body weight was greater in pregnant compared to age-matched non-pregnant rats (+29%, P < 0.05) with no differences between Control and Restricted groups (Table 1).
Table 1.
F1 female body weights
| Group allocation | ||||
|---|---|---|---|---|
| Non-pregnant | Pregnant | |||
| Control | Restricted | Control | Restricted | |
| F1 body weight (g) | ||||
| Postnatal day 1 | 4.03 ± 0.07 | 3.55 ± 0.09* | 4.00 ± 0.08 | 3.42 ± 0.07* |
| Postnatal day 6 | 8.53 ± 0.27 | 7.28 ± 0.49* | 7.67 ± 0.42 | 6.15 ± 0.35*# |
| Postnatal day 14 | 20.57 ± 0.50 | 19.57 ± 1.40 | 20.20 ± 1.03 | 17.82 ± 1.07 |
| Postnatal day 35 | 73.67 ± 1.45 | 67.33 ± 3.15 | 66.80 ± 2.20 | 63.48 ± 1.87 |
| Mating | — | — | 204 ± 4 | 203 ± 7 |
| Postmortem | 212 ± 5 | 214 ± 4 | 288 ± 5 | 278 ± 9# |
| Pregnancy weight gain | — | — | 84 ± 3 | 75 ± 3 |
Values are expressed as means ± SEM; n= 9–11/group. *P < 0.05 vs. Control (main effect); #P < 0.05 vs. non-pregnant (main effect). — indicates no measure.
Smooth muscle contraction
Sensitivity or maximal contraction evoked by Ang II or PE in uterine, mesenteric, renal and femoral arteries was not different between Control and Restricted females from either non-pregnant or pregnant groups (Fig. 1, Tables 2 and 3). Sensitivity to Ang II was reduced in uterine arteries of the pregnant groups compared with non-pregnant groups (P < 0.05; Fig. 1A, Table 3). Ang II did not evoke significant contraction in mesenteric, renal or femoral arteries (<0.2% of K+-PSS contraction) from either non-pregnant or pregnant animals (Tables 2 and 3). In late pregnancy there was an increase in sensitivity to PE (by ∼6-fold) in uterine arteries compared with non-pregnant groups (P < 0.05; Fig. 1B, Tables 2 and 3), with no changes in sensitivity to PE in mesenteric, renal and femoral arteries (Fig. 1C–E, Tables 2 and 3).
Figure 1. Smooth muscle contraction in arteries.
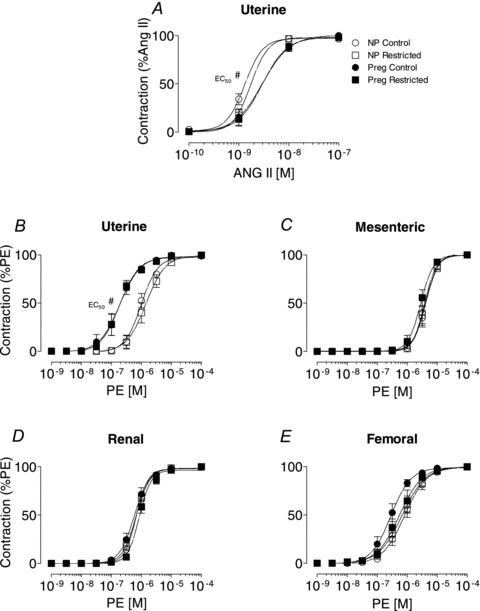
A, concentration–contraction curves to Ang II in uterine arteries (A) from non-pregnant and pregnant (E20) rats. B–E, concentration–contraction curves to PE in uterine (B), mesenteric (C), renal (D) and femoral (E) arteries from non-pregnant and pregnant (E20) animals. Values are expressed as means ± SEM; uterine (n = 8–10/group); mesenteric (n = 6–10/group); renal (n = 8–10/group); femoral (n = 6–10/group). #P < 0.05 vs. non-pregnant (main effect).
Table 2.
Smooth muscle and endothelial function in arteries
| Non-pregnant Control | Non-pregnant Restricted | |||||||
|---|---|---|---|---|---|---|---|---|
| Arteries | Uterine | Mesenteric | Renal | Femoral | Uterine | Mesenteric | Renal | Femoral |
| Smooth muscle function | ||||||||
| 100 mmol l−1 K+ (mN mm−1) | 10.5 ± 0.2 | 7.0 ± 0.4 | 11.2 ± 0.6 | 13.4 ± 0.7 | 11.3 ± 0.3 | 6.5 ± 0.3 | 10.6 ± 0.3 | 14.1 ± 0.6 |
| Maximum PE (mN mm−1) | 11.3 ± 0.2 | 6.5 ± 0.3 | 11.5 ± 0.6 | 13.4 ± 0.5 | 11.8 ± 0.4 | 6.2 ± 0.3 | 10.9 ± 0.3 | 14.1 ± 0.8 |
| Maximum Ang II (mN mm−1) | 5.5 ± 0.6 | 0.02 ± 0.01 | 0.04 ± 0.02 | 0.02 ± 0.01 | 5.5 ± 0.4 | 0.03 ± 0.01 | 0.04 ± 0.01 | 0.04 ± 0.03 |
| Ang II, Emax (% K+) | 55 ± 6 | — | — | — | 49 ± 4 | — | — | — |
| PE, Emax (% K+) | 108 ± 2 | 93 ± 3 | 103 ± 1 | 101 ± 2 | 105 ± 2 | 95 ± 2 | 103 ± 2 | 99 ± 2 |
| SNP, Emax (%) | 96 ± 1 | 97 ± 1 | 88 ± 2 | 86 ± 2 | 93 ± 1 | 95 ± 1 | 89 ± 2 | 81 ± 4 |
| LEV, Emax (%) | 95 ± 1 | 91 ± 1 | 80 ± 4 | 85 ± 7 | 94 ± 1 | 97 ± 1 | 85 ± 3 | 89 ± 3 |
| Endothelium-dependent relaxation | ||||||||
| ACh, Emax (%) | 96 ± 1 | 99 ± 1 | 96 ± 1 | 90 ± 2 | 95 ± 1 | 98 ± 1 | 94 ± 1 | 90 ± 2 |
| ACh+l-NAME+Indo, Emax (%) | 34 ± 9 | 95 ± 1 | 90 ± 1 | — | 53 ± 10 | 94 ± 2 | 87 ± 2 | — |
| Pregnant Control | Pregnant Restricted | |||||||
|---|---|---|---|---|---|---|---|---|
| Arteries | Uterine | Mesenteric | Renal | Femoral | Uterine | Mesenteric | Renal | Femoral |
| Smooth muscle function | ||||||||
| 100 mmol l−1 K+ (mN mm−1) | 8.5 ± 0.5 # | 6.7 ± 0.5 | 9.3 ± 0.6 | 11.0 ± 0.9 # | 8.9 ± 0.5 # | 6.0 ± 0.5 | 10.9 ± 0.8 | 13.1 ± 0.9 # |
| Maximimum PE (mN mm−1) | 10.3 ± 0.6 # | 6.4 ± 0.5 | 9.5 ± 0.6 | 11.1 ± 1.2 # | 10.2 ± 0.5 # | 6.4 ± 0.5 | 10.3 ± 0.7 | 12.5 ± 0.8 # |
| Maximum Ang II (mN mm−1) | 8.2 ± 1.0 # | 0.06 ± 0.05 | 0.1 ± 0.04 | 0.04 ± 0.04 | 8.2 ± 0.9 # | 0.09 ± 0.06 | 0.1 ± 0.05 | 0.09 ± 0.08 |
| Ang II, Emax (% K+) | 88 ± 4 # | — | — | — | 83 ± 3 # | — | — | — |
| PE, Emax (% K+) | 122 ± 2 # | 94 ± 3 | 104 ± 4 | 100 ± 2 | 114 ± 1 # | 105 ± 3 | 95 ± 2 | 94 ± 4 |
| SNP, Emax (%) | 99 ± 1 | 97 ± 2 | 92 ± 2 | 81 ± 8 | 95 ± 1 | 98 ± 2 | 86 ± 2 | 79 ± 5 |
| LEV, Emax (%) | 91 ± 1 | 97 ± 1 | 84 ± 4 | 93 ± 1 | 87 ± 2 | 94 ± 1 | 75 ± 5 | 77 ± 7 |
| Endothelium-dependent relaxation | ||||||||
| ACh, Emax (%) | 99 ± 0.4 | 100 ± 0.2 | 96 ± 1 | 94 ± 3 | 98 ± 1 | 100 ± 0.4 | 93 ± 1 | 94 ± 5 |
| ACh+l-NAME+Indo, Emax (%) | 86 ± 2 # | 98 ± 1 | 89 ± 2 | — | 83 ± 2 # | 92 ± 2 | 86 ± 2 | — |
Responses in arteries from non-pregnant and pregnant (E20) animals. Values are expressed as mean ± SEM; uterine (n = 8–10/group); mesenteric (n = 6–10/group); renal (n = 8–10/group); femoral (n = 6–10/group). Emax, maximal response. #P < 0.05 vs. non-pregnant (main effect). — indicates no response.
Table 3.
Sensitivities (pD2) to vasoconstrictors and vasodilators in arteries
| Non-pregnant Control | Non-pregnant Restricted | |||||||
|---|---|---|---|---|---|---|---|---|
| Arteries | Uterine | Mesenteric | Renal | Femoral | Uterine | Mesenteric | Renal | Femoral |
| pD2 | ||||||||
| AngII (% K+) | 8.92 ± 0.23 | — | — | — | 8.66 ± 0.14 | — | — | — |
| PE (% K+) | 6.01 ± 0.04 | 5.38 ± 0.03 | 6.17 ± 0.01 | 6.08 ± 0.05 | 5.87 ± 0.07 | 5.39 ± 0.03 | 6.17 ± 0.03 | 6.16 ± 0.07 |
| Ang II (% max Ang II) | 8.83 ± 0.15 | — | — | — | 8.77 ± 0.09 | — | — | — |
| PE (% max PE) | 6.01 ± 0.04 | 5.38 ± 0.04 | 6.17 ± 0.01 | 6.09 ± 0.05 | 5.85 ± 0.06 | 5.39 ± 0.03 | 6.17 ± 0.03 | 6.17 ± 0.07 |
| SNP | 7.77 ± 0.04 | 7.45 ± 0.07 | 7.90 ± 0.06 | 7.51 ± 0.11 | 7.68 ± 0.04 | 7.30 ± 0.07 | 7.82 ± 0.06 | 7.33 ± 0.10 |
| ACh | 7.71 ± 0.04 | 8.13 ± 0.02 | 8.04 ± 0.03 | 7.53 ± 0.05 | 7.71 ± 0.03 | 8.08 ± 0.04 | 7.88 ± 0.03 | 7.61 ± 0.05 |
| ACh+l-AME+Indo | 6.99 ± 0.05 | 7.28 ± 0.02 | 7.42 ± 0.03 | — | 6.92 ± 0.07 | 7.18 ± 0.03 | 7.40 ± 0.03 | — |
| Pregnant Control | Pregnant Restricted | |||||||
|---|---|---|---|---|---|---|---|---|
| Arteries | Uterine | Mesenteric | Renal | Femoral | Uterine | Mesenteric | Renal | Femoral |
| pD2 | ||||||||
| AngII (% K+) | 8.55 ± 0.12 | — | — | — | 8.54 ± 0.11 | — | — | — |
| PE (% K+) | 6.71 ± 0.06 # | 5.42 ± 0.05 | 6.24 ± 0.01 | 6.53 ± 0.05 | 6.68 ± 0.05 # | 5.55 ± 0.03 | 6.09 ± 0.02 | 6.26 ± 0.08 |
| Ang II (% max Ang II) | 8.53 ± 0.09 # | — | — | — | 8.53 ± 0.10 # | — | — | — |
| PE (% max PE) | 6.71 ± 0.06 # | 5.42 ± 0.05 | 6.23 ± 0.03 | 6.54 ± 0.05 | 6.72 ± 0.05 # | 5.55 ± 0.03 | 6.07± 0.02 | 6.27 ± 0.07 |
| SNP | 7.71 ± 0.04 | 7.57 ± 0.09 | 7.99 ± 0.06 | 7.60 ± 0.20 | 7.88 ± 0.07 | 7.54 ± 0.15 | 7.95 ± 0.08 | 7.67 ± 0.19 |
| ACh | 8.12 ± 0.02 # | 8.23 ± 0.03 # | 8.05 ± 0.03 | 7.36 ± 0.08 | 8.07 ± 0.05 # | 8.22 ± 0.02 # | 7.94 ± 0.04 | 7.43 ± 0.10 |
| ACh+l-NAME+Indo | 7.58 ± 0.02 # | 7.36 ± 0.03 # | 7.55 ± 0.04 | — | 7.55 ± 0.06 # | 7.48 ± 0.04 # | 7.45 ± 0.03 | — |
Responses in arteries from non-pregnant and pregnant (E20) animals. Values are expressed as means ± SEM; uterine (n = 8–10/group); mesenteric (n = 6–10/group); renal (n = 8–10/group); femoral (n = 6–10/group). Max, maximal contraction. #P < 0.05 vs. non-pregnant (main effect). — indicates no response.
Maximal absolute contraction evoked by K+-PSS, PE or Ang II (mN mm−1) was not different in arteries from Control and Restricted groups (Table 2). Pregnancy was associated with a ∼50% increase in maximal absolute Ang II-mediated contraction in the uterine artery compared with non-pregnant groups (P < 0.05; Table 2) with no differences in mesenteric, renal and femoral arteries (Table 2). Absolute contractions to K+-PSS or PE were smaller in uterine and femoral arteries of the pregnant groups when compared with those from non-pregnant (P < 0.05, Table 2), but were unchanged for the mesenteric and renal arteries (Table 2).
Endothelium-dependent and –independent relaxation
In the absence of blockers, sensitivity and maximal total endothelium-dependent relaxation evoked by ACh was not different in uterine, mesenteric, renal or femoral arteries between Control and Restricted groups (Fig. 2A–D, Tables 2 and 3). However, in the renal artery, higher concentrations of ACh (10−6–10−5 m) evoked contraction that was greater in Restricted compared with Controls for both non-pregnant and pregnant groups (10−5 m, P < 0.05, Fig. 2C). There was a pregnancy-induced increase in sensitivity to ACh in the uterine (by ∼2.5-fold, P < 0.0001; Fig. 2A, Table 3) and mesenteric arteries (by ∼1.3-fold, P < 0.05; Fig. 2B, Table 3) but not in renal or femoral arteries compared with arteries from non-pregnant rats (Fig. 2C and D, Tables 2 and 3).
Figure 2. Total endothelium-dependent and EDHF-mediated relaxation in arteries.
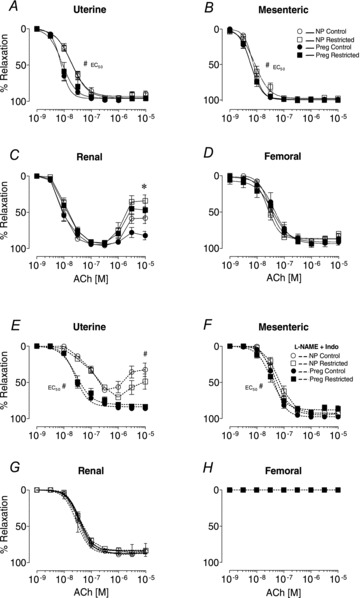
Concentration–relaxation curves to ACh in the absence or presence l-NAME and Indo in uterine (A and E), mesenteric (B and F), renal (C and G) and femoral (D and H) arteries from non-pregnant and pregnant (E20) animals. Values are expressed as means ± SEM; uterine (n = 8–10/group); mesenteric (n = 6–10/group); renal (n = 8–10/group); femoral (n = 6–10/group). *P < 0.05 vs. Control (main effect); #P < 0.05 vs. non-pregnant (main effect).
The relaxation attributed to EDHF was revealed following the blockade of NOS and COX with l-NAME and Indo, respectively. In the presence of l-NAME and Indo, concentration–relaxation curves to ACh were shifted to the right in the uterine, mesenteric and renal arteries of all four groups (Fig. 2E–G). In femoral arteries, ACh failed to evoke relaxation in the presence of l-NAME and Indo, reflecting the absence of EDHF-mediated relaxation (Fig. 2H; Tables 2 and 3) (Wigg et al. 2001; Mazzuca et al. 2010). In late pregnant rats, sensitivity to ACh and maximal EDHF-mediated relaxation in the uterine arteries was greater (pD2, ∼4-fold; Emax, 41%) compared with arteries from the non-pregnant groups (P < 0.05; Fig. 2E, Tables 2 and 3). Furthermore, the constriction evoked by higher concentrations of ACh (10−6–10−5 m) in uterine arteries from non-pregnant animals was absent in arteries from the pregnant groups (Fig. 2E). In mesenteric arteries, there was a modest leftward shift (by ∼1.5-fold) for the EDHF-mediated relaxation in pregnant compared with non-pregnant rats (P < 0.05; Fig. 2F, Table 3) with no differences in maximal relaxation (Table 2). Pregnancy was without effect on the EDHF-mediated relaxation in renal arteries of either group (Fig. 2C, Tables 2 and 3).
Sensitivities and/or maximal relaxations evoked by SNP or KATP channel opener LEV were not different in any of the arteries of all experimental groups (Tables 2 and 3).
Passive mechanical wall properties
Stress–strain relationships for uterine and renal arteries of the non-pregnant Restricted group were shifted to the left compared with those for non-pregnant Controls (P < 0.05; Fig. 3A and C), indicating increased arterial wall stiffness. There were no differences in the stress–strain relationships for the mesenteric and femoral arteries in non-pregnant Restricted compared with non-pregnant Controls (Fig. 3B and D). Both pregnant Control and Restricted females had similar stress–strain relationships for all arteries (Fig. 3A–D). In late pregnant rats, the stress–strain relationships for all arteries were shifted to the left compared with those from non-pregnant (P < 0.05; Fig. 3A–D) indicating increased arterial stiffness.
Figure 3. Passive mechanical wall properties in arteries.
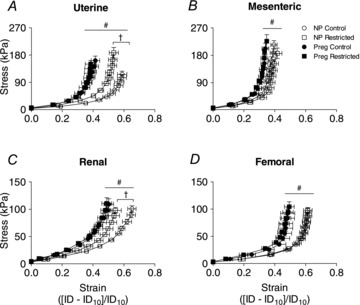
Stress–strain relationships for uterine (A), mesenteric (B), renal (C) and femoral (D) arteries from non-pregnant and pregnant (E20) animals. Values are expressed as means ± SEM; uterine (n = 8–10/group); mesenteric (n = 6–10/group); renal (n = 8–10/group); femoral (n = 6–10/group). #P < 0.05 vs. non-pregnant (main effect). #P < 0.05 vs. non-pregnant Control (following significant interaction).
In late pregnancy, absolute OD and ID of the uterine arteries were significantly larger (by 42% and 44%, respectively) compared with those from non-pregnant rats (P < 0.05; Fig. 4A, Table 4). For the mesenteric arteries, OD and IDs were modestly larger in pregnant rats compared with non-pregnant rats (by 12%, P < 0.05; Fig. 4B, Table 4). Absolute WT was reduced in the uterine artery of non-pregnant Restricted females (P < 0.05; Table 4). During pregnancy, uterine artery WT in Restricted females increased to become similar to that in their Control pregnant counterparts. The media-to-lumen ratio (at 100 mmHg) was smaller in uterine arteries of non-pregnant Restricted compared with non-pregnant Controls (P < 0.05; Table 4). In pregnant Controls, there was a uterine artery specific decrease in the media-to-lumen ratio compared with non-pregnant Controls (P < 0.05; Table 4).
Figure 4. Outside diameters in arteries.
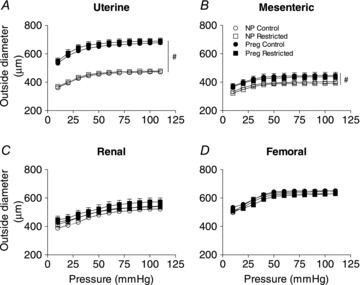
Absolute ODs for uterine (A), mesenteric (B), renal (C) and femoral (D) arteries from non-pregnant and pregnant (E20) animals.Values are expressed as means ± SEM; uterine (n = 8–10/group); mesenteric (n = 6–10/group); renal (n = 8–10/group); femoral (n = 6–10/group). #P < 0.05 vs. non-pregnant (main effect).
Table 4.
Passive mechanical wall properties in arteries at 100 mmHg
| Non-pregnant control | Non-pregnant restricted | |||||||
|---|---|---|---|---|---|---|---|---|
| Arteries | Uterine | Mesenteric | Renal | Femoral | Uterine | Mesenteric | Renal | Femoral |
| OD (μm) | 473 ± 9 | 403 ± 15 | 522 ± 12 | 627 ± 5 | 479 ± 6 | 394 ± 6 | 540 ± 18 | 644 ± 7 |
| ID (μm) | 417 ± 11 | 377 ± 15 | 455 ± 12 | 539 ± 7 | 442 ± 7 | 364 ± 6 | 469 ± 17 | 559 ± 8 |
| WT (μm) | 28 ± 2 | 14 ± 1 | 34 ± 1 | 44 ± 3 | 19 ± 1† | 16 ± 1 | 36 ± 2 | 43 ± 2 |
| M-L ratio | 0.068 ± 0.007 | 0.035 ± 0.002 | 0.075 ± 0.005 | 0.081 ± 0.006 | 0.042 ± 0.004 † | 0.041 ± 0.003 | 0.077 ± 0.005 | 0.077 ± 0.004 |
| Pregnant control | Pregnant restricted | |||||||
|---|---|---|---|---|---|---|---|---|
| Arteries | Uterine | Mesenteric | Renal | Femoral | Uterine | Mesenteric | Renal | Femoral |
| OD (μm) | 678 ± 10# | 447 ± 23# | 543 ± 21 | 652 ± 6 | 689 ± 22# | 441 ± 20# | 573 ± 28 | 631 ± 12 |
| ID (μm) | 617 ± 11# | 416 ± 23# | 474 ± 17 | 560 ± 8 | 626 ± 21# | 414 ± 21# | 504 ± 27 | 551 ± 11 |
| WT (μm) | 31 ± 3 | 17 ± 1 | 35 ± 3 | 47 ± 3 | 32 ± 2 § | 14 ± 1 | 36 ± 3 | 40 ± 3 |
| M-L ratio | 0.050 ± 0.005 ‡ | 0.039 ± 0.005 | 0.073 ± 0.006 | 0.083 ± 0.006 | 0.050 ± 0.004 | 0.034 ± 0.003 | 0.070 ± 0.006 | 0.073 ± 0.006 |
Outside diameter (OD), internal diameter (ID), wall thickness (WT) and media-to-lumen (M-L) ratio in arteries from non-pregnant and pregnant (E20) animals. Values are expressed as means ± SEM; uterine (n = 8–10/group); mesenteric (n = 6–10/group); renal (n = 8–10/group); femoral (n = 6–10/group). #P < 0.05 vs. non-pregnant (main effect). †P < 0.05 vs. non-pregnant Control (following significant interaction). §P < 0.05 vs. non-pregnant Restricted (following significant interaction). ‡P < 0.05 vs. non-pregnant Control (following significant interaction). — indicates no response.
Quantitative histological analysis of collagen and elastin in uterine arteries
There were no differences in total collagen and elastin content or in the proportion of thick and thin birefringent collagen fibres in the media or adventitia in uterine arteries of Restricted compared with Controls (Fig. 5A–D). Interestingly, pregnancy caused a reduction in total collagen (by 5%) and elastin content (by 8%) in the adventitia of uterine arteries, compared with non-pregnant groups (P < 0.05; Fig. 5A and B). Pregnant rats had fewer thick collagen fibres (by 11%), and more thin collagen fibres (by 10%) in the adventitia compared with their non-pregnant counterparts (P < 0.05; Fig. 5D). There were no changes in the proportions of thick and thin collagen fibres in the media between non-pregnant and pregnant groups (Fig. 5C).
Figure 5. Collagen and elastin content in the uterine artery.
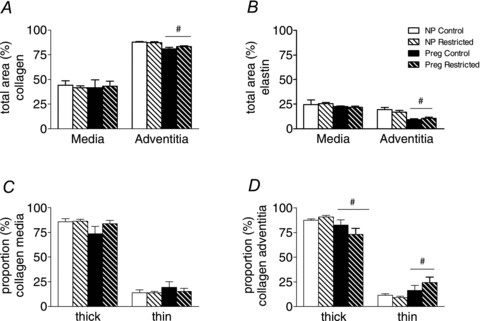
Total collagen (A) and elastin (B) content, and thick and thin collagen content in media (C) and adventitia (D) in uterine arteries from non-pregnant and pregnant (E20) animals. Values are expressed as means ± SEM; uterine (n = 6–8/group). #P < 0.05 vs. non-pregnant (main effect).
Uterine and renal artery gene expression
There were no differences in the relative mRNA expression of collagens type I (α1), III (α1), IV (α1), elastin, fibronectin, eNOS, KCa2.3 and KCa3.1 in uterine or renal arteries between Control and Restricted offspring (data not shown).
Discussion
In this study, uteroplacental insufficiency programmed region-specific vascular dysfunction in non-pregnant growth restricted females. Arterial wall stiffness was increased in the uterine and renal arteries, but was unaffected in mesenteric and femoral arteries of non-pregnant Restricted females. In late pregnancy, growth restricted females had compensatory vascular adaptations, such that uterine and renal arteries were no longer stiffer than Controls. Furthermore, vascular smooth muscle and endothelial function was essentially preserved in all arteries of non-pregnant and late pregnant Restricted females. Overall, this study highlights that females born growth restricted experience normal vascular adaptations to pregnancy such that the differences in arterial wall stiffness and wall thickness in the non-pregnant group did not persist in late pregnancy.
Passive mechanical wall properties
Uterine and renal arteries of Restricted virgin females have increased passive wall stiffness. One of the most striking findings of this study was that differences in arterial wall stiffness between Control and Restricted groups disappeared with pregnancy. In fact, the Restricted pregnant females exhibited normal vascular adaptations such that the stress–strain and pressure–diameter relationships were indistinguishable compared with late pregnant Controls. The fact that Restricted females do not have increased arterial stiffness in the major resistance beds including the mesenteric arteries may in part explain why these females do not develop high blood pressure during pregnancy (Gallo et al. 2012).
Another important point is that pregnancy caused a leftward shift in the stress–strain relationships for uterine, mesenteric, renal and femoral arteries when compared with non-pregnant, suggesting an increase in arterial stiffness. Increased stiffness of uterine and renal arteries in pregnancy is consistent with previous studies in pregnant rats and sheep (Griendling et al. 1985; Gandley et al. 1997; St Louis et al. 1997; Gandley et al. 2010). However, in some studies of mesenteric and renal arteries there was a rightward shift in the stress–strain relationship (increase compliance) with pregnancy in rats (Mackey et al. 1992; Gandley et al. 1997). It is likely that methodological differences may account for the conflicting results. In our study, passive mechanical wall properties were determined in the presence of solution containing zero Ca2+ and 1 mm EGTA (in both the lumen and bathing solutions) to remove any active influence of the smooth muscle. In the previous studies, mechanical wall properties were determined in the presence of papaverine, a smooth muscle relaxant, but in a Ca2+ containing solution, and thus may not reflect truly passive properties. Our study demonstrates that there is a marked increase in uterine artery diameter accompanied by enhanced arterial wall stiffness in late pregnancy.
Consistent with previous findings (Osol & Mandala, 2009), pregnancy was associated with increases in both OD and ID in uterine arteries by ∼43% and in mesenteric arteries by ∼12%. The hypertrophic growth occurred without a change in WT. Importantly, blood vessels of growth restricted females adapted normally to these structural changes during late pregnancy. The increase in ID and OD in uterine and mesenteric arteries observed during pregnancy was associated with reduced distensibility (increased arterial stiffness), which may reflect the fact that the arteries were already maximally distended and had reached their physiological limits of relaxation. The increased uterine and mesenteric arterial stiffness, as indicated by a leftward shift in the stress–strain relationship during pregnancy may not be detrimental in a physiological sense, because both diameter and endothelial vasodilator function were enhanced to accommodate the expected increase in uteroplacental blood flow to the fetal placental unit.
The underlying mechanisms responsible for changes in the structural composition of the artery wall during pregnancy likely involve alterations in extracellular matrix proteins, including collagen and/or elastin (Osol & Mandala, 2009). Although this study failed to show differences in the relative mRNA expression of collagen type I(α1), III(α1), IV(α1), elastin, and fibronectin in uterine and renal arteries between Control and Restricted groups, protein levels may be different. Unfortunately, due to the limited amount of vascular tissue available at postmortem, protein expression could not be measured. Studies have shown that collagen was reduced in main uterine arteries of pregnant sheep and pigs, whilst elastin remained unchanged, reducing the collagen to elastin ratio (Griendling et al. 1985). In our study, total adventitial collagen and elastin content in the uterine arteries was reduced in pregnant compared with non-pregnant animals and this correlated with increased OD and ID and increased stiffness during pregnancy. Using circular crossed-polarised light microscopy we demonstrated, for the first time, that pregnancy was associated with a ‘switch’ in the proportion of thick and thin collagen fibres, such that there was a reduction in thick (stiffer collagen) and an increase in thin (resilient collagen) collagen fibres in pregnant uterine arteries. Importantly, being born growth restricted did not alter collagen and elastin composition at 4 months of age.
Smooth muscle contraction
Growth restricted females had normal Ang II and PE-mediated contractions in both the non-pregnant or pregnant state and this, may in part, explain the lack of elevated blood pressure at 4 months of age (Gallo et al. 2012). Absolute maximal contractile responses to K+-PSS, PE or Ang II (mN mm−1) were also not different in any of the arteries of Restricted females. These data confirm our previous observations in our growth restricted 18-month-old female offspring (Mazzuca et al. 2010). Importantly, these data suggest that contractility via the adrenergic or Ang II receptor stimulation pathways or Ca2+ influx through voltage operated Ca2+ channels are intact in these arteries of Restricted female offspring. Although there were no differences between Control and Restricted groups, there was an effect with pregnancy. This study showed that in late pregnancy, uterine arteries of both Control and Restricted groups had reduced sensitivity to Ang II, increased sensitivity to PE, and increased efficacy of both constrictors when compared with non-pregnant groups, and these findings are in accordance with previous studies (Annibale et al. 1989; D’Angelo & Osol, 1993; St Louis et al. 1997, 2001; Zwart et al. 1998; Xiao & Zhang, 2005). Alternatively, pregnancy had no effect on the sensitivity to Ang II and PE in mesenteric, renal and femoral arteries in both groups, highlighting the region-specific vascular effects that occur during pregnancy.
Endothelium-dependent and –independent relaxation
In the absence of NOS and COX inhibitors, total endothelium-dependent relaxation to ACh was unaltered in all arteries of non-pregnant or pregnant Restricted females when compared with Controls. These observations confirm our previous finding in Restricted females (Mazzuca et al. 2010). These data suggest that NO synthesis/bioavailability and/or release from endothelial cells were unaltered in Restricted females. This was further supported with our mRNA results showing no differences in eNOS expression in uterine and renal arteries of Restricted females when compared with Controls. In pregnant growth restricted offspring exposed to a low protein diet during F0 gestation, endothelium-dependent relaxation was impaired in mesenteric arteries (Torrens et al. 2003) but preserved in uterine arteries (Musha et al. 2011). Discrepancies in the results may be related to the nature and timing of the prenatal insult, growth profile trajectories, the location of the vascular bed used and the animal model studied. In our study, high concentrations of ACh caused contraction in renal arteries that were larger in arteries of Restricted females. This could be due to increased production of vasoconstrictor prostanoids, which stimulate thromboxane-prostanoid and/or prostaglandin E2 receptors in the underlying smooth muscle or an interaction resulting from enhanced superoxide anion generation (Vanhoutte & Tang, 2008). Whether the ACh-induced vasoconstriction in the renal artery is endothelium dependent and/or due to a constrictor prostanoid would need to be explored in future studies by denuding the endothelium and/or adding a specific prostanoid receptor antagonists.
Pregnant Restricted females also had normal upregulation of total endothelium-dependent relaxation responses in mesenteric and uterine arteries. Sensitivity to ACh in the uterine arteries was ∼2.5-fold greater in both pregnant groups, when compared with non-pregnant, and these findings agree with previous reports in uterine arteries from late pregnant rat and human (Ni et al. 1997; Nelson et al. 1998, 2000; Dalle Lucca et al. 2000b). This may be due to the greater synthesis and bioavailability of NO released from endothelial cells. This conclusion is supported by considerable work showing that eNOS mRNA expression and protein activity are greater in uterine arteries of pregnant vs. non-pregnant animals (Bird et al. 2003). The increased sensitivity to ACh observed in the mesenteric arteries (by ∼1.3-fold) but not in renal and femoral arteries of pregnant Control and Restricted offspring (∼1.3-fold) also agree with earlier reports (Davidge & McLaughlin, 1992; Kim et al. 1994; Yamasaki et al. 1996; Gerber et al. 1998; Cooke & Davidge, 2003). In the systemic circulation, NO metabolites nitrite and nitrate are known to be upregulated during pregnancy (Conrad et al. 1993) and this may in part explain the increase in sensitivity to endothelial stimulation in the mesenteric artery. Taken together, these data suggest that pregnancy enhances total endothelium-dependent relaxation in a region-specific manner and that growth restriction did not alter this process.
EDHF-mediated relaxation plays an important role in small resistance vessels, which are important for vascular resistance and organ blood flow (Edwards et al. 2010). EDHF generation is underpinned by the activation of endothelial KCa2.3 and KCa3.1, leading to hyperpolarisation of the endothelium and relaxation of vascular smooth muscle. In this study, sensitivity and maximal EDHF-mediated relaxation were also unaltered in all arteries of non-pregnant or pregnant Restricted females compared with their Control counterparts. This is supported by the lack of difference in mRNA expression of KCa2.3 and KCa3.1 in uterine and renal arteries. Pregnancy potentiated EDHF-mediated relaxation in uterine and mesenteric arteries but not in renal arteries in both Control and Restricted. In fact, in late pregnancy we observed ∼3.4-fold rightward shift in sensitivity to ACh in the presence l-NAME and Indo when compared to no blockers in the uterine artery, suggesting that most of the ACh-induced relaxation is largely EDHF mediated. However, we cannot exclude the possibility that PGI2 synthesis may also have been upregulated in the pregnant uterine artery (Poston et al. 1995). Indeed, EDHF-mediated relaxation was increased in uterine and mesenteric resistance arteries of the pregnant rat (Gerber et al. 1998; Dalle Lucca et al. 2000a, b) and subcutaneous, myometrial and omental arteries of pregnant humans (Pascoal & Umans, 1996; Kenny et al. 2002; Gillham et al. 2003; Luksha et al. 2004; Lang et al. 2007).These data suggest that pregnancy enhances EDHF mediated relaxation in a region-specific manner and that growth restriction did not alter this process.
Lastly, relaxations to SNP or KATP channel opener LEV were not different between non-pregnant or pregnant Control and Restricted groups for all arteries tested. These results suggest that the soluble guanylyl cyclase signalling pathways and KATP-dependent membrane hyperpolarisation and relaxation pathways in the smooth muscle are intact in Restricted females and unaffected with pregnancy.
Summary
Late gestation uteroplacental insufficiency does not, by and large, impair maternal vascular adaptations when growth restricted females become pregnant. The major resistance arteries of these females essentially had normal smooth muscle and endothelial function, and the enhanced uterine and renal artery stiffness in non-pregnant growth restricted females was completely resolved during late pregnancy. Although the vascular adaptations during late pregnancy were preserved in pregnant growth restricted offspring, we have recently reported that these females have fetuses (F2) that are 6% lighter at day 20 of gestation when compared to fetuses of F1 control mothers (Gallo et al. 2012). It is likely that other factors including programmed maternal endocrine factors and physiological stressors or epigenetic modifications of the F1 gametes may also be involved in the programming of fetal weight across subsequent generations.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia (Grants nos 400004 and 546087), Heart Foundation of Australia (G 08M 3698) and the March of Dimes Birth Defects Foundation, USA (Grant no. 6-FY08-269). Marc Mazzuca was supported by a Kidney Health Australia Biomedical Scholarship and The University of Melbourne Fee Remission Scholarship. We would like to thank Kerryn Westcott, Andrew Jefferies, Bruce Abaloz and Ian Boundy for their valuable contributions.
Glossary
- Ang II
angiotensin II
- EDHF
endothelium-derived hyperpolarising factor
- eNOS
endothelial nitric oxide synthase
- Emax
maximal response
- Indo
indomethacin
- ID
internal diameter
- LEV
levcromakalim
- NO
nitric oxide
- pD2
−log EC50
- PE
phenylephrine
- PSS
physiological saline solution
- PGI2
prostacyclin
- KCa2.3
small-conductance calcium-activated potassium channel
- KCa3.1
intermediate-conductance calcium-activated potassium channel
- SNP
sodium nitroprusside
- WT
wall thickness
Author contributions
All authors contributed to the conception and design, or analysis and interpretation of data, drafting the article or revising it critically for important intellectual content, and all authors gave final approval of the version to be published. This study was conducted at The University of Melbourne and Monash University.
Supplementary material
Supplemental Table S1
References
- Annibale DJ, Rosenfeld CR, Kamm KE. Alterations in vascular smooth muscle contractility during ovine pregnancy. Am J Physiol Heart Circ Physiol. 1989;256:H1282–H1288. doi: 10.1152/ajpheart.1989.256.5.H1282. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Bagby SP, Hanson MA. Mechanisms of disease: in utero programming in the pathogenesis of hypertension. Nat Clin Pract Nephrol. 2006;2:700–707. doi: 10.1038/ncpneph0344. [DOI] [PubMed] [Google Scholar]
- Bird IM, Zhang L, Magness RR. Possible mechanisms underlying pregnancy-induced changes in uterine artery endothelial function. Am J Physiol Regul Integr Comp Physiol. 2003;284:R245–R258. doi: 10.1152/ajpregu.00108.2002. [DOI] [PubMed] [Google Scholar]
- Conrad KP, Joffe GM, Kruszyna H, Kruszyna R, Rochelle LG, Smith RP, Chavez JE, Mosher MD. Identification of increased nitric oxide biosynthesis during pregnancy in rats. FASEB J. 1993;7:566–571. [PubMed] [Google Scholar]
- Cooke CL, Davidge ST. Pregnancy-induced alterations of vascular function in mouse mesenteric and uterine arteries. Biol Reprod. 2003;68:1072–1077. doi: 10.1095/biolreprod.102.009886. [DOI] [PubMed] [Google Scholar]
- D’Angelo G, Osol G. Regional variation in resistance artery diameter responses to alpha-adrenergic stimulation during pregnancy. Am J Physiol Heart Circ Physiol. 1993;264:H78–H85. doi: 10.1152/ajpheart.1993.264.1.H78. [DOI] [PubMed] [Google Scholar]
- Dalle Lucca JJ, Adeagbo AS, Alsip NL. Influence of oestrous cycle and pregnancy on the reactivity of the rat mesenteric vascular bed. Hum Reprod. 2000a;15:961–968. doi: 10.1093/humrep/15.4.961. [DOI] [PubMed] [Google Scholar]
- Dalle Lucca JJ, Adeagbo AS, Alsip NL. Oestrous cycle and pregnancy alter the reactivity of the rat uterine vasculature. Hum Reprod. 2000b;15:2496–2503. doi: 10.1093/humrep/15.12.2496. [DOI] [PubMed] [Google Scholar]
- Davidge ST, McLaughlin MK. Endogenous modulation of the blunted adrenergic response in resistance-sized mesenteric arteries from the pregnant rat. Am J Obstet Gynecol. 1992;167:1691–1698. doi: 10.1016/0002-9378(92)91763-z. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Feletou M, Weston AH. Endothelium-derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Arch. 2010;459:863–879. doi: 10.1007/s00424-010-0817-1. [DOI] [PubMed] [Google Scholar]
- Gallo LA, Tran M, Moritz KM, Mazzuca MQ, Parry LJ, Westcott KT, Jefferies AJ, Cullen-McEwen LA, Wlodek ME. Cardio-renal and metabolic adaptations during pregnancy in female rats born small: implications for maternal health and second generation fetal growth. J Physiol. 2012;590:617–630. doi: 10.1113/jphysiol.2011.219147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandley RE, Griggs KC, Conrad KP, McLaughlin MK. Intrinsic tone and passive mechanics of isolated renal arteries from virgin and late-pregnant rats. Am J Physiol Regul Integr Comp Physiol. 1997;273:R22–R27. doi: 10.1152/ajpregu.1997.273.1.R22. [DOI] [PubMed] [Google Scholar]
- Gandley RE, Jeyabalan A, Desai K, McGonigal S, Rohland JD, Deloia JA. Cigarette exposure induces changes in maternal vascular function in a pregnant mouse model. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1249–R1256. doi: 10.1152/ajpregu.00274.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber RT, Anwar MA, Poston L. Enchanced acetylcholine induced relaxation in small mesenteric arteries from pregnant rats: an important role for endothelium-derived hyperpolarizing factor (EDHF) Br J Pharmacol. 1998;125:455–460. doi: 10.1038/sj.bjp.0702099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillham JC, Kenny LC, Baker PN. An overview of endothelium-derived hyperpolarising factor (EDHF) in normal and compromised pregnancies. Eur J Obstet Gynecol Reprod Biol. 2003;109:2–7. doi: 10.1016/s0301-2115(03)00044-7. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Fuller EO, Cox RH. Pregnancy-induced changes in sheep uterine and carotid arteries. Am J Physiol Heart Circ Physiol. 1985;248:H658–H665. doi: 10.1152/ajpheart.1985.248.5.H658. [DOI] [PubMed] [Google Scholar]
- Grigore D, Ojeda NB, Alexander BT. Sex differences in the fetal programming of hypertension. Gend Med. 2008;5:S121–S132. doi: 10.1016/j.genm.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny LC, Baker PN, Kendall DA, Randall MD, Dunn WR. Differential mechanisms of endothelium-dependent vasodilator responses in human myometrial small arteries in normal pregnancy and pre-eclampsia. Clin Sci (Lond) 2002;103:67–73. doi: 10.1042/cs1030067. [DOI] [PubMed] [Google Scholar]
- Kim TH, Weiner CP, Thompson LP. Effect of pregnancy on contraction and endothelium-mediated relaxation of renal and mesenteric arteries. Am J Physiol Heart Circ Physiol. 1994;267:H41–H47. doi: 10.1152/ajpheart.1994.267.1.H41. [DOI] [PubMed] [Google Scholar]
- Lang NN, Luksha L, Newby DE, Kublickiene K. Connexin 43 mediates endothelium-derived hyperpolarizing factor-induced vasodilatation in subcutaneous resistance arteries from healthy pregnant women. Am J Physiol Heart Circ Physiol. 2007;292:H1026–H1032. doi: 10.1152/ajpheart.00797.2006. [DOI] [PubMed] [Google Scholar]
- Luksha L, Nisell H, Kublickiene K. The mechanism of EDHF-mediated responses in subcutaneous small arteries from healthy pregnant women. Am J Physiol Regul Integr Comp Physiol. 2004;286:R1102–R1109. doi: 10.1152/ajpregu.00550.2003. [DOI] [PubMed] [Google Scholar]
- Mackey K, Meyer MC, Stirewalt WS, Starcher BC, McLaughlin MK. Composition and mechanics of mesenteric resistance arteries from pregnant rats. Am J Physiol Regul Integr Comp Physiol. 1992;263:R2–R8. doi: 10.1152/ajpregu.1992.263.1.R2. [DOI] [PubMed] [Google Scholar]
- Magness RR, Sullivan JA, Li Y, Phernetton TM, Bird IM. Endothelial vasodilator production by uterine and systemic arteries. VI. Ovarian and pregnancy effects on eNOS and NO(x) Am J Physiol Heart Circ Physiol. 2001;280:H1692–H1698. doi: 10.1152/ajpheart.2001.280.4.H1692. [DOI] [PubMed] [Google Scholar]
- Mazzuca MQ, Wlodek ME, Dragomir NM, Parkington HC, Tare M. Uteroplacental insufficiency programs regional vascular dysfunction and alters arterial stiffness in female offspring. J Physiol. 2010;588:1997–2010. doi: 10.1113/jphysiol.2010.187849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz K, Cuffe J, Wilson L, Dickinson H, Wlodek M, Simmons D, Denton KM. Sex specific programming: a critical role for the renal renin-angiotensin system. Placenta. 2010;31 Suppl:S40–46. doi: 10.1016/j.placenta.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Moritz KM, Mazzuca MQ, Siebel AL, Mibus A, Arena D, Tare M, Owens JA, Wlodek ME. Uteroplacental insufficiency causes a nephron deficit, modest renal insufficiency but no hypertension with ageing in female rats. J Physiol. 2009;587:2635–2646. doi: 10.1113/jphysiol.2009.170407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musha Y, Itoh S, Miyakawa M, Ohtsuji M, Hanson MA, Kinoshita K, Takeda S. Vascular, renal and placental effects on pregnant offspring of protein-restricted rat dams. J Obstet Gynaecol Res. 2011;37:343–351. doi: 10.1111/j.1447-0756.2010.01351.x. [DOI] [PubMed] [Google Scholar]
- Nelson SH, Steinsland OS, Suresh MS, Lee NM. Pregnancy augments nitric oxide-dependent dilator response to acetylcholine in the human uterine artery. Hum Reprod. 1998;13:1361–1367. doi: 10.1093/humrep/13.5.1361. [DOI] [PubMed] [Google Scholar]
- Nelson SH, Steinsland OS, Wang Y, Yallampalli C, Dong YL, Sanchez JM. Increased nitric oxide synthase activity and expression in the human uterine artery during pregnancy. Circ Res. 2000;87:406–411. doi: 10.1161/01.res.87.5.406. [DOI] [PubMed] [Google Scholar]
- Ni Y, Meyer M, Osol G. Gestation increases nitric oxide-mediated vasodilation in rat uterine arteries. Am J Obstet Gynecol. 1997;176:856–864. doi: 10.1016/s0002-9378(97)70611-2. [DOI] [PubMed] [Google Scholar]
- O’Dowd R, Kent JC, Moseley JM, Wlodek ME. Effects of uteroplacental insufficiency and reducing litter size on maternal mammary function and postnatal offspring growth. Am J Physiol Regul Integr Comp Physiol. 2008;294:R539–R548. doi: 10.1152/ajpregu.00628.2007. [DOI] [PubMed] [Google Scholar]
- Osol G, Mandala M. Maternal uterine vascular remodeling during pregnancy. Physiology. 2009;24:58–71. doi: 10.1152/physiol.00033.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoal IF, Umans JG. Effect of pregnancy on mechanisms of relaxation in human omental microvessels. Hypertension. 1996;28:183–187. doi: 10.1161/01.hyp.28.2.183. [DOI] [PubMed] [Google Scholar]
- Poston L, McCarthy AL, Ritter JM. Control of vascular resistance in the maternal and feto-placental arterial beds. Pharmacol Ther. 1995;65:215–239. doi: 10.1016/0163-7258(94)00064-a. [DOI] [PubMed] [Google Scholar]
- Reslan OM, Khalil RA. Molecular and vascular targets in the pathogenesis and management of the hypertension associated with preeclampsia. Cardiovasc Hematol Agents Med Chem. 2010;8:204–226. doi: 10.2174/187152510792481234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Louis J, Pare H, Sicotte B, Brochu M. Increased reactivity of rat uterine arcuate artery throughout gestation and postpartum. Am J Physiol Heart Circ Physiol. 1997;273:H1148–H1153. doi: 10.1152/ajpheart.1997.273.3.H1148. [DOI] [PubMed] [Google Scholar]
- St Louis J, Sicotte B, Bedard S, Brochu M. Blockade of angiotensin receptor subtypes in arcuate uterine artery of pregnant and postpartum rats. Hypertens. 2001;38:1017–1023. doi: 10.1161/hy1101.095008. [DOI] [PubMed] [Google Scholar]
- Thornburg KL, Jacobson SL, Giraud GD, Morton MJ. Hemodynamic changes in pregnancy. Semin Perinatol. 2000;24:11–14. doi: 10.1016/s0146-0005(00)80047-6. [DOI] [PubMed] [Google Scholar]
- Torrens C, Brawley L, Barker AC, Itoh S, Poston L, Hanson MA. Maternal protein restriction in the rat impairs resistance artery but not conduit artery function in pregnant offspring. J Physiol. 2003;547:77–84. doi: 10.1113/jphysiol.2002.026120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte PM, Tang EH. Endothelium-dependent contractions: when a good guy turns bad! J Physiol. 2008;586:5295–5304. doi: 10.1113/jphysiol.2008.161430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigg SJ, Tare M, Tonta MA, O’Brien RC, Meredith IT, Parkington HC. Comparison of effects of diabetes mellitus on an EDHF-dependent and an EDHF-independent artery. Am J Physiol Heart Circ Physiol. 2001;281:H232–H240. doi: 10.1152/ajpheart.2001.281.1.H232. [DOI] [PubMed] [Google Scholar]
- Wlodek ME, Mibus A, Tan A, Siebel AL, Owens JA, Moritz KM. Normal lactational environment restores nephron endowment and prevents hypertension after placental restriction in the rat. J Am Soc Nephrol. 2007;18:1688–1696. doi: 10.1681/ASN.2007010015. [DOI] [PubMed] [Google Scholar]
- Wlodek ME, Westcott K, Siebel AL, Owens JA, Moritz KM. Growth restriction before or after birth reduces nephron number and increases blood pressure in male rats. Kidney Int. 2008;74:187–195. doi: 10.1038/ki.2008.153. [DOI] [PubMed] [Google Scholar]
- Wlodek ME, Westcott KT, O’Dowd R, Serruto A, Wassef L, Moritz KM, Moseley JM. Uteroplacental restriction in the rat impairs fetal growth in association with alterations in placental growth factors including PTHrP. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1620–R1627. doi: 10.1152/ajpregu.00789.2004. [DOI] [PubMed] [Google Scholar]
- Xiao D, Zhang L. Adaptation of uterine artery thick- and thin-filament regulatory pathways to pregnancy. Am J Physiol Heart Circ Physiol. 2005;288:H142–H148. doi: 10.1152/ajpheart.00655.2004. [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Lindheimer MD, Umans JG. Effects of pregnancy on femoral microvascular responses in the rat. Am J Obstet Gynecol. 1996;175:730–736. doi: 10.1053/ob.1996.v175.a73870. [DOI] [PubMed] [Google Scholar]
- Zwart AS, Davis EA, Widdop RE. Modulation of AT1 receptor-mediated contraction of rat uterine artery by AT2 receptors. Br J Pharmacol. 1998;125:1429–1436. doi: 10.1038/sj.bjp.0702210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.