

Abstract
Nascent polypeptides entering the endoplasmic reticulum (ER) are covalently modified with pre-assembled oligosaccharides. The terminal glucose and mannose residues are immediately removed after transfer of the oligosaccharide onto newly synthesized polypeptides. This processing determines whether the polypeptide will be retained in the ER, transported along the secretory pathway, or dislocated across the ER membrane for destruction. New avenues of research and some issues of controversy have recently been opened by the discovery that lectin–oligosaccharide interactions stabilize supramolecular complexes between regulators of ER-associated degradation (ERAD). In this Opinion article, we propose a unified model that depicts carbohydrates acting both as flags signaling the fitness of a maturing protein and as docking sites that regulate the assembly and stability of the ERAD machinery.
Keywords: secretory pathway, carbohydrates, endoplasmic reticulum
N-Linked glycans
Proteins that pass through the eukaryotic secretory pathway are glycosylated on Asn residues (at Asn-Xxx-Ser/Thr or more rarely Asn-Xxx-Cys, Asn-Xxx-Val, or Asn-Gly sequons) on emergence into the ER [1]. Pre-assembled N-acetylglucosamine2-mannose9-glucose3 moieties are transferred en bloc from a lipid donor onto nascent polypeptide chains by the oligosaccharyltransferase complex. N-Linked oligosaccharides are hydrophilic and therefore they increase the solubility and inhibit the aggregation of unstructured nascent chains [2]. The processing of these added glycans determines the fate of the associated cargo protein by influencing which folding, quality control, or degradation factors will be engaged. These factors are present in the ER at delicately balanced and highly controlled concentrations. How N-glycan modifications retain immature or aberrant cargo proteins in the folding environment, control the incorporation of native glycoproteins into secretory vesicles, or select terminally misfolded proteins for ERAD (see Glossary) has been widely covered in recent reviews 3, 4, 5, 6. By contrast, the focus of this Opinion article is discussion of a newly discovered role of N-glycans in cellular proteostasis: the involvement of oligosaccharides on ER-resident members of quality control and ERAD machineries in promoting the assembly of supramolecular complexes. These complexes play crucial roles in inspecting the content of the ER lumen to aid in the clearance of defective gene products, orphan subunits of oligomeric complexes, and metabolically regulated proteins. The intraluminal concentration of misfolded polypeptides might modulate their formation and stability in a substrate-dependent feedback mechanism that eventually determines cellular ERAD activity.
Glossary.
EDEM1–3 [Htm1p (Mnl1p) and Htm2p]: ER-degradation-enhancing mannosidase-like proteins. Their intraluminal concentration is regulated by ERAD tuning and by UPR. They determine the rate of glycoproteins disposal. The catalytic residues in EDEM1, EDEM2, and EDEM3 are conserved.
ERAD: endoplasmic reticulum-associated protein degradation. Clearance of proteins from the ER by their dislocation to the cytoplasm and subsequent degradation using the ubiquitin–proteasome system.
ERAD tuning: selective removal of ERAD factors from the ER that sets the level of ERAD activity. When defective, ERAD activity is enhanced to levels that might interfere with protein biogenesis [45].
ER mannosidase I (Mns Ip): an ER α1,2-exomannosidase that cleaves the first mannose from the B-branch of the glycan to generate Man8B glycoforms, regardless of the conformation of the polypeptide [2].
HRD1 (Hrd1p): ER membrane-localized, RING finger-containing E3 ubiquitin ligase that participates in dislocation across the ER membrane and polyubiquitylation of ERAD substrates [56].
LC3-I (Atg8p): cytoplasmic form of the autophagic marker LC3. LC3-I is also non-covalently associated with membranes of ERAD tuning vesicles (via direct or indirect interaction with the type I membrane protein SEL1L) and with coronavirus replication and transcription platform vesicles [45].
Mannosidase-like domain: structural domain of EDEM proteins conserved among all members of the glycosyl hydrolase 47 family of α1,2 exomannosidases comprising ER mannosidase I and several Golgi mannosidases [57].
MRH domain: mannose 6-phosphate receptor homology domain, which is conserved in few sugar-binding and -modifying proteins. It consists of a flattened β-barrel containing three conserved disulfide bonds. The substrate recognition loop between the fifth and sixth cysteines is well conserved in sequence and precisely conserved in length [58].
OS-9 (Yos9p): ERAD lectin that contains a single MRH domain for which a variety of splice variants are expressed in the mammalian ER. OS-9 recognizes proteins possessing oligosaccharides that expose a terminal α1,6-mannose residue 19, 29. The intraluminal concentration of OS-9 is regulated by ERAD tuning and can be substantially increased on UPR activation 37, 49.
SEL1L (Hrd3p): type I membrane glycoprotein with dual activity in cellular proteostasis as both a component of the dislocation machinery built around the membrane-embedded E3 ubiquitin ligase HRD1 and, at least in mammalian cells, a component of an ERAD tuning receptor that segregates luminal ERAD factors in ERAD tuning vesicles, thereby regulating their constitutive clearance from the ER 38, 51.
UPR: unfolded protein response. An evolutionarily conserved response to ER stress that reduces global protein translation and enhances biosynthesis of membrane lipids, folding chaperones, and ERAD factors. It can eventually result in cell death [59].
XTP3-B/erlectin: mammalian ERAD lectin containing two MRH domains that is expressed as two splice variants [41].
N-Glycans on cargo proteins: maturation, quality control, and sorting signals
Cycles of glucose removal and re-addition, which begin shortly after a glycan is transferred to a nascent chain, or delayed trimming of mannose residues determine whether associated polypeptides will be retained by the folding machinery or selected for ERAD, respectively.
The role of glucose processing in folding promotion
The A-branch or glucose-containing arm of N-linked glycans (Figure 1 ) recruits molecular chaperones that assist in efficient folding of glycoproteins 2, 7. Terminal glucose residue n of the transferred triglucosylated glycan is rapidly removed by α-glucosidase I, which is associated with the translocon complex. Malectin, a Glc2-binding protein, might then intervene to retain immature and/or misfolded polypeptides in the ER 8, 9. Removal of glucose m by α-glucosidase II, a luminal heterodimeric enzyme, supports co- or post-translational association of folding polypeptides with the ER lectins calnexin and calreticulin 2, 7. The oxidoreductase ERp57 and the peptidyl-prolyl isomerase cyclophilin B are associated with calnexin and calreticulin, and they catalyze rate-limiting folding reactions to facilitate attainment of the native structure 10, 11. Removal of innermost glucose residue l by α-glucosidase II dissociates the polypeptide from the calnexin–calreticulin chaperone system. At this stage, UDP-glucose: glycoprotein glucosyltransferase 1 (UGT1) inspects the structure of the released proteins. Non-native polypeptides are re-glucosylated by UGT1, which initiates rebinding to the lectin chaperones and their associated folding factors. Native polypeptides are not recognized by UGT1 and are secreted [12]. Depending on the glycoprotein substrate, one or more bind-and-release events might be required to attain the correct conformation 13, 14, 15.
Figure 1.
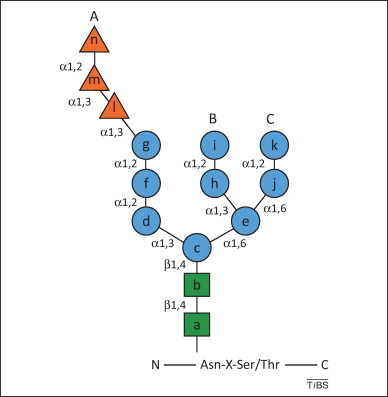
N-Linked glycan structure. The Asn-linked glycan is composed of three glucoses (orange triangles), nine mannoses (blue circles) and two N-acetylglucosamines (green squares). The types of linkage between sugars are denoted and each residue is assigned a letter.
Mannose processing and the mannose timer model of glycoprotein quality control
Studies in Saccharomyces cerevisiae revealed that Mns1p (mannosidase 1) removes mannose residue i from the B-branch of protein-linked oligosaccharides (Figure 1) [5]. For properly folded secretory proteins, this allows binding of cargo lectin sorting receptors, which regulate the export of native conformers from the ER [16]. For folding-defective polypeptides, removal of mannose i is followed by removal of at least one additional α1,2-linked mannose residue (mannose k, branch C) by Htm1p, which forms a functional complex with the oxidoreductase Pdi1p [17]. This trimming event exposes α1,6-linked mannose j, which is recognized by the mannose-6-phosphate receptor homology (MRH) domain of the ERAD lectin Yos9p 17, 18, 19.
In mammalian cells, de-mannosylation of folding-defective polypeptides might be more extensive, with removal of up to four α1,2-linked mannose residues. As in budding yeast, ER mannosidase I (Mns1p ortholog) catalyzes removal of mannose residue i. If misfolding persists, N-linked glycans are further processed by EDEM1 and EDEM3 (ER-degradation enhancing mannosidase-like proteins, Htm1p and Htm2p orthologs) or by other resident α1,2 mannosidase (Figure 2 , steps 1 and 2, respectively). Demonstration of mannosidase activity with purified EDEM1 and EDEM3 is still lacking, and the role of a third EDEM variant, EDEM2, in ERAD is even less well characterized 20, 21, 22, 23, 24, 25. It should therefore be considered that proteins of the EDEM family could operate as mannose-binding lectins that associate with de-mannosylated oligosaccharides generated by extensive processing in a sub-region of the ER that is populated by high ER mannosidase I concentrations (i.e., the ER quality control compartment or ERQC 3, 5).
Figure 2.
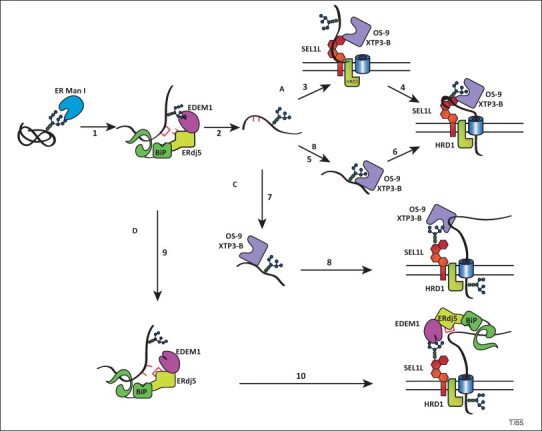
Models of glycoprotein endoplasmic reticulum-associated degradation (ERAD). Various models of the delivery of ERAD substrates to an ER membrane ERAD complex are depicted. Letters A–D designate the different routes for a misfolded substrate. The glycan attached to an ERAD substrate (black sinuous line) is trimmed of a mannose residue by ER mannosidase I (ER Man I, step 1). This is followed by ER-degradation enhancing mannosidase-like protein (EDEM)1 recognition. EDEM1 is present in a complex with ERdj5 and immunoglobulin binding protein (BiP). In the mannose timer model, EDEM1 acts as a mannosidase to trim additional mannose residues (step 2). In route A, ERAD substrates are directly recognized by type I membrane glycoprotein (SEL1L) (step 3). OS-9 and XTP3-B associate with SEL1L to act as ERAD gatekeepers by querying the ERAD substrate for exposed α1,6-linked mannose residues. If the protein possesses the proper glycans, it is passed along the ERAD pathway (step 4). In route B, alternatively, trimmed ERAD substrates are recognized by OS-9 and XTP3-B (step 5) and delivered to the ERAD membrane complex (step 6). Routes C and D depict glycan docking models for OS-9–XTP3-B and EDEM1, respectively. In route C, OS-9 and XTP3-B recognize the misfolded substrates through protein–protein interactions (step 7). This can involve associated co-factors such as GRP94. OS-9 and XTP3-B then deliver the ERAD substrate to the ER membrane ERAD complex by binding to the glycans on the adapter SEL1L (step 8) or on a glycosylated SEL1L associated protein. In route D, the EDEM1 complex associates with ERAD substrates (step 9). EDEM1 employs its mannosidase-like domain to deliver the ERAD substrate to the ER membrane ERAD complex (step 10). The models depicted are not necessarily mutually exclusive. Hybrid models that use multiple pathways are possible. Note that although glycans trimmed to Man7–5 can signal for ERAD in metazoans, the Man6 composition is displayed for simplicity.
In the mannose timer model of glycoprotein quality control [26], extensive substrate de-mannosylation provides two signals that flag misfolded proteins for disposal. Removal of mannose g, the acceptor for the re-glucosylating activity of UGT1 (which is absent in budding yeast), irreversibly extracts the folding-defective polypeptide from the calnexin–calreticulin cycle to mark the end of the folding phase for maturing nascent chains (Figure 2, step 2; note that mannose f can also eventually be removed) [22]. Removal of mannose k exposes the α1,6-linked mannose j, thereby recruiting the ERAD MRH-containing lectins OS-9 and XTP3-B, which are expressed in several splice variants in the mammalian ER (Figure 2, steps 2–6) 27, 28, 29, 30.
Accumulating evidence reveals that de-mannosylating and mannose-binding ERAD factors reside in complexes with other luminal ER-resident proteins. For example, EDEM1 associates with BiP (immunoglobulin binding protein), which possibly contributes to the recognition of non-native polypeptides, and with ERdj5, which might facilitate ERAD by reducing inter- and intramolecular disulfides of ERAD candidates (Figure 2) [31]. Interactions between OS-9 or XTP3-B and BiP or GRP94, respectively, have also been reported 27, 32. Participation in such multi-protein complexes might explain the findings that EDEM proteins and OS-9 and XTP3-B variants also associate or regulate ER retention and disposal of non-glycosylated folding-defective polypeptides 22, 24, 25, 27, 28, 33.
EDEM1, EDEM3, OS-9, and XTP3-B also interact with the type I membrane glycoprotein SEL1L (Hrd3p in yeast) 24, 27, 34, 35, 36, 37. SEL1L nucleates interactions between these luminal ERAD components and a supramolecular membrane complex containing the E3 ubiquitin ligase HRD1, which regulates dislocation across the ER membrane and polyubiquitylation of ERAD substrates (Figure 2, steps 3, 6, 8, and 10) 24, 27, 34, 35, 36, 37, 38. In the next section, we explain how the glycan-mediated interactions between EDEM1, EDEM3, OS-9, or XTP3-B and SEL1 regulate cellular proteostasis in metazoans by modulating clearance of misfolded polypeptides (ERAD) and removal of ERAD factors from the ER lumen (ERAD tuning).
N-Glycans on ER-resident proteins: regulating ERAD and ERAD tuning
Glycosylation of ER-resident quality control factors
Although the proteins that act early in the secretory pathway tend to be non-glycosylated (e.g., the nascent chain-interacting proteins Sec61α, BiP, PDI, malectin, calnexin, and ERp57), several factors that function post-translationally for quality control and ERAD (e.g., EDEM proteins, OS-9, XTP3-B, and SEL1L) are glycosylated. Whether glycosylation plays a role in the segregation or organization of ER factors into specialized complexes or subregions within the ER is unknown. With the exception of EDEM1, for which N-glycans mediate the transient association with calnexin required for efficient maturation [39], the role of N-linked oligosaccharides in the biogenesis of ER-resident chaperones has not been studied. By contrast, several recent studies have revealed the crucial role of oligosaccharides displayed on ER-resident quality control factors in the formation of functional supramolecular complexes 24, 27, 34, 35, 36, 37.
Glycans as docking signals in ERAD
Affinity-tagged ERAD factors have been used as bait to characterize interacting components of multimeric ERAD complexes 38, 40. Christianson et al. used this approach to confirm that a prominent interactor of the type I membrane protein SEL1L is the membrane-embedded E3 ubiquitin ligase HRD1 [27]. These studies also revealed an association between SEL1L and the luminal MRH-containing proteins OS-9 and XTP3-B. Additional studies have revealed that the mannosidase-like domain-containing proteins EDEM1 and EDEM3 also interact with SEL1L 24, 25, 27, 35, 37. Disruption of the EDEM1, EDEM3, OS-9, or XTP3-B oligosaccharide-binding motifs, or cell treatment with kifunensine (an inhibitor of de-mannosylation of protein-bound oligosaccharides), decreases the association of these ERAD factors with the penta-glycosylated protein SEL1L 24, 27, 29, 30, 34, 35, 41, 42. Taken together, these data reveal that EDEM1, EDEM3, OS-9, and XTP3-B activity is not restricted to oligosaccharides displayed on folding-defective polypeptides as described in the mannose timer model of glycoprotein quality control (Figure 2, routes A and B). Rather, these luminal ERAD factors might also engage SEL1L oligosaccharides to dock at the HRD1 dislocation complex (Figure 2, routes C and D; docking model). This facilitates delivery of ERAD substrates to dislocation sites and their transport across the ER membrane for their eventual proteasomal degradation. Several studies also revealed oligosaccharide-independent association of EDEM proteins, OS-9, and XTP3-B with misfolded proteins (Figure 2, routes C and D) 22, 24, 25, 27, 28, 29, 33, 43, 44, or with SEL1L (Figure 2, routes A and B) [36]. This shows that the mode of binding of these ER lectins might be affected by the presence of associated partners such as BiP and GRP94.
Glycans as docking signals for ERAD tuning
Control of the luminal concentration and activity of ERAD factors is important for maintenance of cellular proteostasis. Misregulated ERAD might select on-pathway folding intermediates for degradation before they are given a sufficient opportunity to reach their native structure [45]. For example, aberrantly high levels of the ER E3 ligases HRD1 and gp78 cause the inappropriate degradation of individual gene products and are associated with rheumatoid arthritis and highly metastatic sarcoma, respectively 46, 47. The concept of ERAD tuning [45] is based on emerging evidence showing that an increasing number of ERAD factors, including ER mannosidase I, HERP, gp78, JAMP, EDEM1, OS-9, and SEL1L, are constitutively removed from the unstressed ER 3, 37, 48, 49, 50, 51, 52, 53, 54. Luminal expression of misfolded proteins might inactivate constitutive removal of select ERAD factors from the ER and stabilize complexes between ERAD regulatory components. This might enhance ERAD in the absence of unfolded protein response (UPR) activation. This implies the existence of ERAD substrate-dependent feedback mechanisms that rapidly adapt ERAD activity to the misfolded protein load [37].
Association of EDEM1 and OS-9 with SEL1L regulates delivery of ERAD substrates to the HRD1 dislocon (Figure 2). However, in the absence of ERAD substrates, the ERAD tuning pathways are activated. SEL1L is disengaged from inactive dislocons. Under these circumstances, the cytosolic tail of SEL1L (or a SEL1L-associated protein) recruits the ubiquitin-like protein LC3-I to build a membrane-bound receptor that regulates selective and constitutive clearance of SEL1L-associated OS-9 and EDEM1 from the unstressed ER (Figure 3 ) [37]. These receptor:ligand complexes enter so-called ERAD tuning vesicles, which lack a coatomer coat and are larger than the COPII vesicles that ferry cargo proteins from the ER to the Golgi 37, 48, 49, 55. ERAD tuning vesicles contain up to 80% of the cellular EDEM1 in unstressed cells 33, 48, 55, but much less in cells expressing ERAD substrates, which substantially inhibit removal of ERAD factors from the ER [37]. They eventually deliver OS-9, EDEM1, possibly the short-living ER mannosidase I, and other SEL1L-interacting ERAD factors such as XTP3-B and EDEM3 to an ill-defined endolysosomal degradative organelle. In summary, the ER load of misfolded polypeptides might determine whether the lectin–glycan association with SEL1L drives ERAD substrate progression downstream through the ERAD pathway or directs the removal of ERAD lectins from the ER.
Figure 3.
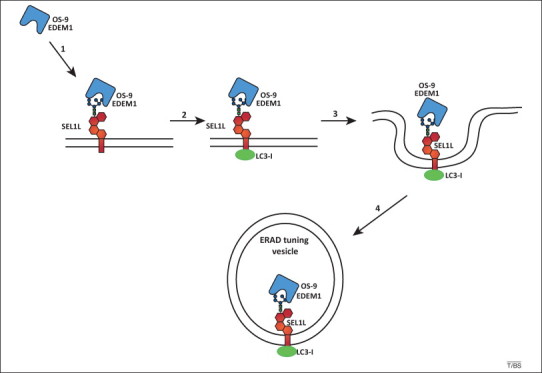
Endoplasmic reticulum-associated degradation (ERAD) tuning model. The receptor-mediated removal of ER-degradation enhancing mannosidase-like protein (EDEM1) and OS-9 from the ER lumen is shown. In the absence of misfolded proteins, type I membrane glycoprotein (SEL1L) is disengaged from the HRD1 dislocation machinery. The penta-glycosylated ectodomain of SEL1L associates with EDEM1 and OS-9 (step 1). The cytosolic tail of SEL1L or a SEL1L-associated protein binds the cytosolic ubiquitin-like protein LC3-I. The complex is released from the ER in vesicles (steps 3 and 4) that may eventually fuse with an ill-defined degradative compartment. ERAD tuning vesicles are co-opted by coronaviruses as replication and transcription platform vesicles.
Concluding remarks
The role of oligosaccharides as folding and quality control tags that regulate protein biogenesis in the ER of eukaryotic cells has been studied extensively over the past two decades. The glycosidases and glycosyltransferases that process protein-bound oligosaccharides, the lectins engaged by the different oligosaccharide structures, and the role of each trimming intermediate in protein quality control have been characterized. In this Opinion article, we propose a new unified model for the role of glycans in protein quality control that combines components of the mannose timer and docking models.
The mannose timer model shows how N-linked glycans can act as flags that signal terminally misfolded polypeptides for degradation by recruiting ERAD lectins. N-Linked glycans can also support lectin–oligosaccharides interactions between components of the ERAD machinery, as demonstrated in the docking model 24, 27, 34, 35, 36, 37. These associations drive progression of substrates along the ERAD pathway or promote removal of ERAD factors from the ER lumen for ERAD tuning in the absence of misfolded polypeptides. On accumulation of terminally misfolded polypeptides and under stress conditions in which the UPR is activated, the formation and stability of active dislocation machineries would be favored to ensure rapid turnover of aberrant cargo. This implies a model of N-glycoprotein processing in the ER that includes regulatory tuning of ERAD components by a substrate-dependent feedback mechanism. A substrate-dependent feedback mechanism has been proposed in yeast, in which the activity of the EDEM1 ortholog Htm1p is enhanced on misfolded polypeptide-induced formation of a functional complex between the mannosidase and Pdi1p [17].
A unified carbohydrate quality control model explains the recognition of oligosaccharides as flags and docking sites by ERAD lectins. With the exception of XTP3-B (which has two separate MRH domains), all other ERAD lectins or lectin-like proteins have only one oligosaccharide-binding domain and therefore cannot simultaneously recognize the flag that indicates the folding state of cargo and the glycosylated docking site on a protein such as SEL1L. It will be important to determine the quaternary structure of the ERAD lectin complexes. If organized in oligomers, the presence of multiple glycan-binding sites would support the recognition of oligosaccharide flags on misfolded polypeptides and of docking oligosaccharides on the dislocation machinery at the ER membrane by individual ERAD lectin complexes. Resolution of this issue and other open questions (Box 1 ) might aid in the design of approaches to support interventions in protein misfolding diseases and in the optimization of the production of ectopic proteins used in clinics or industry.
Box 1. Outstanding questions.
-
•
Are the processes of protein folding, quality control, and degradation segregated into separate domains within the ER? If so, how are folding, quality control, and ERAD factors segregated and retained in sub-compartments (e.g., the ER quality control compartment, ERQC)? How are cargo proteins delivered to functionally distinct regions within the ER?
-
•
How are non-native on-pathway folding intermediates that should be retained in the folding environment distinguished from terminally misfolded proteins that should be degraded?
-
•
How many different ERAD pathways exist and what are the misfolded determinants of cargo proteins that elicit intervention of components of specific ERAD pathways?
-
•
How are misfolded proteins delivered to and dislocated across the ER membrane? For example, is unfolding of ERAD substrates (e.g., reduction of disulfides, or cis to trans isomerization of peptidyl–prolyl bonds) required? What is the nature of the dislocation channel?
-
•
Do members of the EDEM family of proteins act as mannosidases or lectins? How do they recognize substrates?
-
•
Are EDEM2 and EDEM3 also found in complexes with chaperones?
-
•
What is the composition of the individual glycans of ERAD factors such as SEL1L/Hrd3p, EDEMs, OS-9/Yos9p, and XTP3-B? Is their glycan composition regulated?
-
•
How are ERAD tuning pathways regulated at the molecular level?
-
•
How are ERAD tuning pathways hijacked by pathogens?
-
•
What is the complete composition of ERAD tuning vesicles?
-
•
Is there crosstalk between ERAD and autophagy, the two major cellular degradative pathways?
Acknowledgments
This work was supported by US Public Health grants GM086874 and GM094848 to D.N.H. M.M. is supported by grants from the Foundation for Research on Neurodegenerative Diseases, Fondazione San Salvatore, the Swiss National Science Foundation, Association Française contre les Myopathies, Novartis Stiftung für medizinisch-biologische Forschung, and the Gabriele Foundation. Members of the D.N.H and M.M. groups are acknowledged for critical reading of the manuscript.
References
- 1.Zielinska D.F. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell. 2010;141:897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Helenius A., Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 3.Lederkremer G.Z. Glycoprotein folding, quality control and ER-associated degradation. Curr. Opin. Struct. Biol. 2009;19:515–523. doi: 10.1016/j.sbi.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Hebert D.N. ERAD substrates: which way out? Semin. Cell Dev. Biol. 2010;21:526–532. doi: 10.1016/j.semcdb.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 5.Aebi M. N-Glycan structures: recognition and processing in the ER. Trends Biochem. Sci. 2010;35:74–82. doi: 10.1016/j.tibs.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Smith M.H. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearse B.R., Hebert D.N. Lectin chaperones help direct the maturation of glycoproteins in the endoplasmic reticulum. Biochim. Biophys. Acta. 2010;1803:684–693. doi: 10.1016/j.bbamcr.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schallus T. Malectin: a novel carbohydrate-binding protein of the endoplasmic reticulum and a candidate player in the early steps of protein N-glycosylation. Mol. Biol. Cell. 2008;19:3404–3414. doi: 10.1091/mbc.E08-04-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galli C. Malectin participates in a backup glycoprotein quality control pathway in the mammalian ER. PLoS ONE. 2011;6:e16304. doi: 10.1371/journal.pone.0016304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frickel E.M. ERp57 is a multifunctional thiol-disulfide oxidoreductase. J. Biol. Chem. 2004;279:18277–18287. doi: 10.1074/jbc.M314089200. [DOI] [PubMed] [Google Scholar]
- 11.Kozlov G. Structural basis of cyclophilin B binding by the calnexin/calreticulin P-domain. J. Biol. Chem. 2010;285:35551–35557. doi: 10.1074/jbc.M110.160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caramelo J.J., Parodi A.J. Getting in and out from calnexin/calreticulin cycles. J. Biol. Chem. 2008;283:10221–10225. doi: 10.1074/jbc.R700048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solda T. Substrate-specific requirements for UGT1-dependent release from calnexin. Mol. Cell. 2007;27:238–249. doi: 10.1016/j.molcel.2007.05.032. [DOI] [PubMed] [Google Scholar]
- 14.Pearse B.R. A cell-based reglucosylation assay demonstrates the role of GT1 in the quality control of a maturing glycoprotein. J. Cell Biol. 2008;181:309–320. doi: 10.1083/jcb.200712068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pearse B.R. The role of UDP-Glc:glycoprotein glucosyltransferase 1 in the maturation of an obligate substrate prosaposin. J. Cell Biol. 2010;189:829–841. doi: 10.1083/jcb.200912105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Appenzeller C. The lectin ERGIC-53 is a cargo transport receptor for glycoproteins. Nat. Cell Biol. 1999;1:330–334. doi: 10.1038/14020. [DOI] [PubMed] [Google Scholar]
- 17.Gauss R. A complex of pdi1p and the mannosidase htm1p initiates clearance of unfolded glycoproteins from the endoplasmic reticulum. Mol. Cell. 2011;42:782–793. doi: 10.1016/j.molcel.2011.04.027. [DOI] [PubMed] [Google Scholar]
- 18.Clerc S. Htm1 protein generates the N-glycan signal for glycoprotein degradation in the endoplasmic reticulum. J. Cell Biol. 2009;184:159–172. doi: 10.1083/jcb.200809198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quan E.M. Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol. Cell. 2008;32:870–877. doi: 10.1016/j.molcel.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mast S.W. Human EDEM2, a novel homolog of family 47 glycosidases, is involved in ER-associated degradation of glycoproteins. Glycobiology. 2005;15:421–436. doi: 10.1093/glycob/cwi014. [DOI] [PubMed] [Google Scholar]
- 21.Olivari S. A novel stress-induced EDEM variant regulating endoplasmic reticulum-associated glycoprotein degradation. J. Biol. Chem. 2005;280:2424–2428. doi: 10.1074/jbc.C400534200. [DOI] [PubMed] [Google Scholar]
- 22.Olivari S. EDEM1 regulates ER-associated degradation by accelerating de-mannosylation of folding-defective polypeptides and by inhibiting their covalent aggregation. Biochem. Biophys. Res. Commun. 2006;349:1278–1284. doi: 10.1016/j.bbrc.2006.08.186. [DOI] [PubMed] [Google Scholar]
- 23.Hirao K. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 2006;281:9650–9658. doi: 10.1074/jbc.M512191200. [DOI] [PubMed] [Google Scholar]
- 24.Cormier J.H. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol. Cell. 2009;34:627–633. doi: 10.1016/j.molcel.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hosokawa N. EDEM1 accelerates the trimming of alpha1,2-linked mannose on the C branch of N-glycans. Glycobiology. 2010;20:567–575. doi: 10.1093/glycob/cwq001. [DOI] [PubMed] [Google Scholar]
- 26.Helenius A. How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol. Biol. Cell. 1994;5:253–265. doi: 10.1091/mbc.5.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christianson J.C. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1:SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernasconi R. A dual task for the Xbp1-responsive OS-9 variants in the mammalian endoplasmic reticulum: inhibiting secretion of misfolded protein conformers and enhancing their disposal. J. Biol. Chem. 2008;283:16446–16454. doi: 10.1074/jbc.M802272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hosokawa N. Human OS-9, a lectin required for glycoprotein ERAD, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 2009;284:17061–17068. doi: 10.1074/jbc.M809725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamaguchi D. Human XTP3-B binds to alpha1-antitrypsin variant null (Hong Kong) via the C-terminal MRH domain in a glycan-dependent manner. Glycobiology. 2010;20:348–355. doi: 10.1093/glycob/cwp182. [DOI] [PubMed] [Google Scholar]
- 31.Ushioda R. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science. 2008;321:569–572. doi: 10.1126/science.1159293. [DOI] [PubMed] [Google Scholar]
- 32.Christianson J.C. Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 2012;14:93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ron E. Bypass of glycan-dependent glycoprotein delivery to ERAD by up-regulated EDEM1. Mol. Biol. Cell. 2011;22:3945–3954. doi: 10.1091/mbc.E10-12-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Satoh T. Structural basis for oligosaccharide recognition of misfolded glycoproteins by OS-9 in ER-associated degradation. Mol. Cell. 2010;40:905–916. doi: 10.1016/j.molcel.2010.11.017. [DOI] [PubMed] [Google Scholar]
- 35.Saeed M. Role of the endoplasmic reticulum-associated degradation (ERAD) pathway in degradation of hepatitis C virus envelope proteins and production of virus particles. J. Biol. Chem. 2011;286:37264–37273. doi: 10.1074/jbc.M111.259085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanna J. Structural and biochemical basis of Yos9 protein dimerization and possible contribution to self-association of 3-hydroxy-3-methylglutaryl-coenzyme A reductase degradation ubiquitin–ligase complex. J. Biol. Chem. 2012;287:8633–8640. doi: 10.1074/jbc.M111.317644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bernasconi R. Role of the SEL1L:LC3-I complex as an ERAD tuning receptor in the mammalian ER. Mol. Cell. 2012;46:809–819. doi: 10.1016/j.molcel.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 38.Mueller B. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. U.S.A. 2008;105:12325–12330. doi: 10.1073/pnas.0805371105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tamura T. Characterization of early EDEM1 protein maturation events and their functional implications. J. Biol. Chem. 2011;286:24906–24915. doi: 10.1074/jbc.M111.243998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denic V. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- 41.Cruciat C.M. The MRH protein erlectin is a member of the endoplasmic reticulum synexpression group and functions in N-glycan recognition. J. Biol. Chem. 2006;281:12986–12993. doi: 10.1074/jbc.M511872200. [DOI] [PubMed] [Google Scholar]
- 42.Mikami K. The sugar-binding ability of human OS-9 and its involvement in ER-associated degradation. Glycobiology. 2010;20:310–321. doi: 10.1093/glycob/cwp175. [DOI] [PubMed] [Google Scholar]
- 43.Kosmaoglou M. A dual role for EDEM1 in the processing of rod opsin. J. Cell Sci. 2009;122:4465–4472. doi: 10.1242/jcs.055228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jaenicke L.A. Yos9p assists in the degradation of certain nonglycosylated proteins from the endoplasmic reticulum. Mol. Biol. Cell. 2011;22:2937–2945. doi: 10.1091/mbc.E10-10-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bernasconi R., Molinari M. ERAD and ERAD tuning: disposal of cargo and of ERAD regulators from the mammalian ER. Curr. Opin. Cell Biol. 2011;23:176–183. doi: 10.1016/j.ceb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsai Y.C. The ubiquitin ligase gp78 promotes sarcoma metastasis by targeting KAI1 for degradation. Nat. Med. 2007;13:1504–1509. doi: 10.1038/nm1686. [DOI] [PubMed] [Google Scholar]
- 47.Yamasaki S. Rheumatoid arthritis as a hyper-endoplasmic-reticulum-associated degradation disease. Arthritis Res. Ther. 2005;7:181–186. doi: 10.1186/ar1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cali T. Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem. Biophys. Res. Commun. 2008;371:405–410. doi: 10.1016/j.bbrc.2008.04.098. [DOI] [PubMed] [Google Scholar]
- 49.Reggiori F. Coronaviruses hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe. 2010;7:500–508. doi: 10.1016/j.chom.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hori O. Role of Herp in the endoplasmic reticulum stress response. Genes Cells. 2004;9:457–469. doi: 10.1111/j.1356-9597.2004.00735.x. [DOI] [PubMed] [Google Scholar]
- 51.Mueller B. SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J. Cell Biol. 2006;175:261–270. doi: 10.1083/jcb.200605196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu Y. Human endoplasmic reticulum mannosidase I is subject to regulated proteolysis. J. Biol. Chem. 2007;282:4841–4849. doi: 10.1074/jbc.M607156200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shmueli A. Targeting of gp78 for ubiquitin-mediated proteasomal degradation by Hrd1: cross-talk between E3s in the endoplasmic reticulum. Biochem. Biophys. Res. Commun. 2009;390:758–762. doi: 10.1016/j.bbrc.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tcherpakov M. JAMP optimizes ERAD to protect cells from unfolded proteins. Mol. Biol. Cell. 2008;19:5019–5028. doi: 10.1091/mbc.E08-08-0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zuber C. EDEM1 reveals a quality control vesicular transport pathway out of the endoplasmic reticulum not involving the COPII exit sites. Proc. Natl. Acad. Sci. U.S.A. 2007;104:4407–4412. doi: 10.1073/pnas.0700154104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hampton R.Y. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell. 1996;7:2029–2044. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moremen K.W., Molinari M. N-linked glycan recognition and processing: the molecular basis of endoplasmic reticulum quality control. Curr. Opin. Struct. Biol. 2006;16:592–599. doi: 10.1016/j.sbi.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Munro S. The MRH domain suggests a shared ancestry for the mannose 6-phosphate receptors and other N-glycan-recognising proteins. Cur. Biol. 2001;11:R499–R501. doi: 10.1016/s0960-9822(01)00302-5. [DOI] [PubMed] [Google Scholar]
- 59.Walter P., Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]