

Abstract
Liver ischemia-reperfusion injury (IRI) remains a challenging problem in clinical settings. The expression of fibronectin (FN) by endothelial cells is a prominent feature of the hepatic response to injury. Here we investigate the effects of the connecting segment-1 (CS-1) peptide therapy, which blocks fibronectin (FN)-α4β1 integrin leukocyte interactions, in a well-established model of 24-hour cold liver IRI. CS-1 peptides significantly inhibited leukocyte recruitment and local release of proinflammatory mediators (COX-2, iNOS, and TNF-α), ameliorating liver IRI and improving recipient survival rate. CS1 therapy inhibited the phosphorylation of p38 MAPK, a kinase linked to inflammatory processes. Moreover, in addition to downregulating the expression of matrix metalloproteinase-9 (MMP-9) in hepatic IRI, CS-1 peptide therapy depressed the expression of membrane type 1-matrix metalloproteinase (MT1-MMP/MMP-14) by macrophages, a membrane-tethered MMP important for focal matrix proteolysis. Inhibition of p38 MAPK activity, with its pharmacological antagonist SB203580, downregulated MMP-9 and MT1-MMP/MMP-14 expressions by fibronectin-stimulated macrophages, suggesting that p38 MAPK kinase pathway controls fibronectin mediated inductions of MMP-9 and MT1-MMP/MMP-14. Hence, this study provides new insights on the role of fibronectin in liver injury, which can potentially be applied to the development of new pharmacological strategies for the successful protection against hepatic IRI.
Keywords: Leukocyte migration, fibronectin, matrix metalloproteinases, liver cold ischemia, reperfusion injury
INTRODUCTION
Ischemia-reperfusion injury (IRI) represents a major problem in orthotopic liver transplantation (OLT). IRI is a multifactorial antigen-independent inflammatory process that can lead to early graft failure and to a higher incidence of both acute and chronic organ dysfunction after transplantation 1, 2.
The migration of leukocytes into tissues is a central event in inflammatory processes 3, including in acute inflammatory liver injury 4. Leukocyte transmigration across endothelial and extracellular matrix (ECM) protein barriers is dependent on complex series of adhesion and focal matrix degradation events 5, 6. Fibronectin (FN) is a ECM glycoprotein implicated in a variety of pathological conditions associated with cell turnover and migration such as tumor metastasis 7, rheumatoid arthritis 8, multiple sclerosis 9, and organ transplantation 10. Moreover, clinical trials using humanized antibodies against the α4 integrin, a receptor for the connecting segment-1 (CS-1) region of fibronectin 11, have been effective in controlling inflammatory conditions like multiple sclerosis (MS) 12 and inflammatory bowel disease 13. The role of fibronectin in leukocyte adhesion, migration and activation has been extensively reported 14. Indeed, it was recently demonstrated that adhesion of leukocytes from MS patients to brain microvascular endothelial cells under flow conditions is preferentially mediated by the a4 integrin/FN-CS1 interactions 15.
We have previously shown that CS-1 peptides, which are FN-specific peptides that interact with the α4β1-integrin and inhibit its binding to FN 16, profoundly depressed leukocyte recruitment and improved liver function of steatotic liver transplants in a model of ex vivo 4-hour cold ischemia followed by isotransplantation 17, 18. In the present study, we evaluated the effects of the CS-1 peptide therapy in a well-established rat liver model of prolonged cold hepatic IRI, in which normal livers are cold stored for 24 hours prior to being transplanted in syngeneic recipients. Our results show a beneficial role for CS-1 peptides in ameliorating prolonged cold hepatic IRI. Moreover, they provide evidence that FN regulates the expression of both matrix metalloproteinase-9 (MMP-9) and membrane type 1-matrix metalloproteinase (MT1-MMP/MMP-14) through activation of the p38 MAP kinase cell signaling pathway.
MATERIALS AND METHODS
Animals, grafting techniques and CS-1 peptide therapy
Male Sprague Dawley rats (250–300 g) were obtained from Harlan Sprague Dawley, Inc. (Indianapolis, IN). Syngeneic OLTs were performed at the University of California, Los Angeles under a protocol approved by the UCLA-Animal Research Committee (ARC-2000-177). Briefly, after skeletonization of the liver, the portal vein, bile duct, and inferior vena cava were cannulated, and the liver was flushed with University of Wisconsin (UW) solution. Livers were stored for 24 hours at 4°C in UW solution before being transplanted in syngeneic recipients with revascularization without hepatic artery reconstruction with an anhepatic phase of 16–20 min 19, 20. Cellular FN was significantly up-regulated in the liver vasculature after 24h of cold storage followed by OLT (Fig. 1). Based on these observations and in our previous studies 17, 18, CS-1 peptides (500 μg/rat) were dissolved in saline and administered ex vivo via portal vein to livers before cold storage and immediately prior to reperfusion; OLT recipients received an additional peptide dose (1 mg/rat i.p.) 1h post-transplantation. Controls received vehicle and were subjected to the same surgical procedures. Rat recipients of liver transplants were sacrificed at 6 hours (n=6/group) and 24 hours (n=5–7/group) after OLT or followed for survival studies (n=8/group).
Figure 1.

Representative cellular fibronectin staining in 24h cold liver IRI. Cellular fibronectin was virtually absent in naïve livers (A) and it was abundantly upregulated in livers after 24h of cold storage followed by 6h (B) and 24h (C) of reperfusion (n=4/group).
Assessment of liver Damage
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured in blood samples with an auto analyzer by ANTECH Diagnostics (Los Angeles, CA). Liver paraffin sections were stained with H&E and the histological severity was graded from 0 to 4 as described 17. No necrosis, congestion, or centrilobular ballooning was given a score of 0, while severe congestion, ballooning degeneration and >60% lobular necrosis was given a value of 4.
Immunohistochemistry
Immunostaining was performed in cryostat sections, as described 18, 21. Appropriate primary antibodies against rat T-Cells (R73), NK-cells (CD161), monocyte/macrophages (ED1) (AbD Serotec), cellular FN (IST-9) (Accurate Chemical), MMP-9 (gelatinase-B) (EMDchemicals) and MMP-14 (H-72) (Santa Cruz Biotech) were added at optimal dilutions. Sections were evaluated blindly by counting the labeled cells in triplicates; results are presented as number of cells/10 high-power fields. Dual Staining was achieved by immunofluorescence with Alexa Fluor 594-red anti-rabbit IgG (H+L) and Alexa Fluor 488-green IgG (H+L) antibodies (Molecular probes, Carlsbad, CA). Slides were analyzed using a Leica Confocal Microscope (UCLA, Confocal Microscope Core Facility).
Myeloperoxidase (MPO) Assay
Myeloperoxidase activity was evaluated in frozen tissue homogenized in an iced solution of 0.5% hexadecyltrimethyl-ammonium and 50 mmol/L of potassium phosphate buffer solution 22. After centrifugation, the supernatants were mixed in a solution of hydrogen peroxide-sodium acetate and tetramethyl benzidine (Sigma). The quantity of enzyme degrading 1 μmol/L of peroxide/minute at 25°C per gram of tissue was defined as 1U of MPO activity.
Western Blot and Zymography Analyses
Western blots and Zymography were performed as described 18. Proteins (40 μg/sample) in sodium dodecyl sulfate (SDS)-loading buffer were electrophoresed through 10%–12% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to PVDF membranes (Thermo Scientific). Membranes were incubated with specific primary antibodies against MMP-9 (Millipore, Billerica, MA), MT1-MMP (Sigma), phospho-p38, phospho-p44/42, p38, p44/42 (Cell Signaling Technology), COX-2 (Cayman), and iNOS (Santa Cruz). After development, membranes were striped and re-blotted with an antibody against β-actin (Abcam). Gelatinolytic activity was detected in liver extracts (100μg) by 10% SDS-PAGE contained 1mg/ml of gelatin (Invitrogen), under non-reducing conditions. After incubation in development buffer (50 mmol/L Tris-HCl, 5 mmol/L CaCl2, and 0.02% NaN3, pH 7.5), gels were stained with Coomassie brilliant blue R-250 (Bio-Rad), and destained with methanol/acetic acid/water (20:10:70). Prestained molecular weight markers (Fermentas) and MMP-9 (BIOMOL International) served as standards. Relative quantities of protein were determined using a densitometer (Image J, NIH software).
ELISA
TNF-α protein content in liver samples was determined using a commercially available ELISA kit (eBioscience, San Diego, CA) following the manufacturer’s instructions, with final results expressed in picograms of TNF-α per milliliter of serum.
RNA Extraction and Reverse Transcriptase PCR
RNA was extracted from livers with Trizol (Life Technologies) as described 18, 22. Reverse transcription was performed using 5 μg of total RNA in a first-strand cDNA synthesis reaction with SuperScript III RNaseH Reverse Transcriptase (Life Technologies), as recommended by the manufacturer. The cDNA product was amplified by PCR using primers specific for each target cDNA. Densitometric quantification of band intensity was performed using NIH Image J software.
Leukocyte Isolation
Murine neutrophils and macrophages were isolated as previously described 22, 23. Neutrophil isolation: bone marrow was flushed from harvested femurs and tibias with Hanks’ balanced salt solution (HBSS) containing 0.1% (wt/vol) bovine serum albumin (BSA) and 1% (wt/vol) glucose on ice. Cells were pelleted and erythrocytes removed by hypotonic lysis. The bone marrow preparation was resuspended at 5×107 cells/mL in HBSS and layered on a Percoll (Sigma) gradient (55% Percoll, top; 65% Percoll, middle; 80% Percoll, bottom). After centrifugation, mature neutrophils were recovered at the interface of the 65% and 80% fractions and were >90% pure. Macrophage isolation: 1 ml of 3% thioglycollate medium was injected into the peritoneal cavity 72 hours before collecting macrophages. The peritoneal cavities were lavaged with HBSS, and the aspirate was placed on ice. After centrifugation, the pellets were resuspended in DMEM. Cell viability was determined by trypan blue exclusion.
Leukocyte Migration Assay
Transmigration of isolated macrophages and neutrophils through fibronectin was performed using a commercially available in vitro cell migration assay kit (BD Bioscience) and as previously described 21, 23. Leukocytes were resuspended in DMEM without fetal bovine serum at a final concentration of 1.0×106 cells/ml. Transwell inserts with 3-μm pore size coated with fibronectin were placed in the 24-well plates, and then macrophages, or neutrophils, (5×105 cells/well) were added to the upper chambers. Where indicated, cells were pretreated with anti-MT1-MMP antibody (20 μg/ml; LEM-2/63.1) or control IgG (20 μg/ml) for 30m prior addition to the upper chambers, MMP-9 inhibitor-I (10 nmol/L; C27H33N3O5S; Calbiochem) and TNF-α(10 ng/ml; eBioscience) were included in the DMEM medium of the lower chambers. Cells were incubated at 37°C and 5% CO2 for 6 hours, and the leukocytes that had migrated into the lower chambers were collected, stained and counted.
Cell Culture
Isolated macrophages were cultured in medium without fetal bovine serum overnight and pretreated for 30 min with the p38 inhibitor SB203580 (10 μM) prior to being plated on FN-coated plates (Biocoat, BD Biosciences). Controls included combination of cells cultured on polylysine-coated plates, cells stimulated with lipopolysaccharide (10 ng/ml, LPS, Sigma), and cells treated with a specific LPS inhibitor, Polymixin B (10 ng/ml). Cells were cultured on 24-well plates at a concentration of 5×105 cells/well and incubated at 37°C, 5% CO2 for 12 hours. After, incubation, cells and supernatants were collected for RT-PCR and zymography, respectively.
Data Analysis
Data are shown as means +/− SD. Statistical comparisons between groups were performed by Student’s t-test using the statistical package SPSS (SPSS Inc., Chicago, IL, USA). Kaplan-Meier analysis was used to determine statistical significance of the differences in rat survival. P values of <0.05 were considered statistically significant.
RESULTS
CS-1 peptide therapy ameliorates hepatocellular damage and increases recipient survival in 24h cold liver IRI
We examined the effects of the CS-1 peptide therapy in a well-established model of 24-hour liver cold ischemia followed by OLT. Recipients of livers that had been treated with CS-1 peptides were characterized by improved liver function, as shown by the decreased serum transaminase levels (IU/L) at 6h (sAST: 1412±420 vs. 2866±864, p<0.05; sALT: 728 ± 428 vs.1690 ± 211, p<0.05) and 24h (sAST: 1350±142 vs. 4000±1358, p<0.05; sALT: 1261±233 vs. 3051±958, p<0.05) post-OLT (Fig. 2A). CS-1 treated OLTs showed relatively good histological preservation, contrasting with high vascular congestion, extensive necrosis, and significant disruption of lobular architecture observed in control livers, (score: 6h/1.5±0.5 vs. 2.7±0.6, p<0.05; 24h/1.7±0.4 vs. 3.1±0.5, p<0.05) (Fig. 2B). Moreover, CS-1 peptide therapy significantly increased the 14-day survival rate (100% vs. 50%, p<0.05; n=8/group) post-OLT (Fig 2C). Therefore, our results agree with previous observations in a 4h model of steatotic liver IRI 17 and support a broadly beneficial role of FN-α4β1 integrin blockade in cold hepatic IRI.
Figure 2.

Transaminase levels, histology, and recipient survival in 24h cold liver IRI. CS-1peptide therapy significantly improved liver function as evidenced by the lower AST and ALT levels (panel A) in the CS-1 peptide treated recipients at 6h and 24h post-cold liver IRI. Hematoxylin and eosin staining of liver grafts (panel, B) indicated a better histological preservation in the CS-1 peptide treated liver OLTs (c, and d) as compared with respective controls (a, and b), at 6h (a, and c) and 24h (b, and d) post-OLT. Moreover, CS-1 peptide treated OLTs had a significantly prolonged survival rate (panel C) as compared to respective controls at 14-day post-OLT (*p<0.05; ×100 H&E; panels A and B n=5–7 rats/group; panel C n=8 rats/group).
CS-1 peptide therapy disrupts leukocyte infiltration in 24h cold liver IRI
CS-1 peptide therapy significantly depressed leukocyte infiltration (/10HPF) in prolonged cold liver IRI. The numbers of T lymphocytes (31±8 vs. 64±3, p<0.05), NK cells (19±2 vs. 41±3, p<0.05) and ED1 monocyte/macrophages (21±6 vs. 32± 1, p<0.05) were decreased in the CS-1 peptide treated livers at 6h post-transplantation (Fig. 3). T-cell (30±3 vs. 83±15, p<0.05), NK cell (16±3 vs. 30±8, p<0.05) and ED1 macrophage (35±7 vs. 57±16, p<0.05) infiltration was also lower in the CS-1 treated recipients 24h post-OLT (Fig. 3). MPO activity (U/g), an index of neutrophil infiltration, was significantly reduced in the CS-1 treated rats at 6h (1.22 ± 0.48 vs. 2.93 ± 0.57 p<0.05) and 24h (0.48 ± 0.02 vs. 3.18 ± 0.94, p<0.05) post-OLT (Fig. 3E).
Figure 3.

Leukocyte infiltration in 24h cold liver IRI. The infiltration of T lymphocytes (panel A), NK cells (panel B), and monocyte/ macrophages (panel C) was significantly depressed in CS-1 peptide treated OLTs at 6h and 24h post-reperfusion. Panel D illustrates immunoperoxidase staining of T cells (a, and b), NK lymphocytes (c, and d), and macrophages (e, and f) in CS-1 (b, d, and f) and control (a, c, and e) OLTs at 6h post-transplantation. MPO activity (panel E), an index of neutrophil infiltration, was significantly depressed in CS-1 peptide treated OLTs at 6h and 24h post-transplantation as compared to respective controls (*p<0.05; ×200; n=5–6 rats/group).
CS-1 peptide therapy decreases iNOS, COX-2 and proinflammatory cytokine expression in prolonged cold liver IRI
Inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) are two of the most prominent inflammatory mediators 24 and their expressions have been linked to liver IRI 23, 25. As shown in figure 4, CS1 peptide therapy reduced the intragraft mRNA expression of both iNOS (0.06±0.02 vs. 1.40±0.41, p<0.05) and COX-2 (0.80±0.11 vs. 2.20±0.61, p<0.05) at 6h post-OLT (Fig. 4A). Other pro-inflammatory mediators, such as TNF-α(0.03±0.04 vs. 0.83±0.26, p<0.05), IL1β (0.35 ± 0.13 vs. 0.74 ± 0.30, p<0.05), IL-6 (0.45±0.17 vs. 1.10±0.32, p<0.05), and IFN-γ(0.41±0.11 vs. 0.58±0.06, p<0.05) were also markedly depressed in the CS-1 peptide treated OLTs at mRNA level (Fig. 4A). Evaluations of iNOS, COX-2, and TNF-α at protein level confirmed the beneficial effect of the CS-1 peptide therapy in the reduction of these major inflammatory mediators in prolonged cold liver IRI (Fig. 4B and C).
Figure 4.

COX-2, iNOS and proinflammatory cytokine expression in cold liver IRI. The mRNA expressions (panel A) of COX-2 and iNOS, two major tissue injury mediators, as well as of various proinflammatory cytokines were profoundly depressed in the CS-1 treated grafts at 6h post-IRI. The mRNA expressions correlated with the protein expressions (panels B and C), as iNOS, COX-2, and TNF-α were significantly depressed at protein level in the CS-1 peptide treated OLTs at 6h after reperfusion (*p<0.05; panel A n=4 rats/group; panels B and C n=5 rats/group).
CS-1 peptide therapy downregulates MMP-9 and MT1-MMP/MMP-14 expressions in 24h cold liver IRI
Our earlier studies have shown that MMP-9 is induced upon leukocyte attachment to fibronectin in damaged steatotic livers 18 and that MMP-9 mediates leukocyte migration in liver IRI 21. Here, we evaluated whether CS-1 peptide mediated therapy affected the expressions of MMP-9 and MT1-MMP/MMP-14 in the liver model of prolonged cold IRI. CS-1 peptide therapy reduced the intragraft MMP-9 expression at mRNA (0.10±0.12 vs. 0.50±0.35, p<0.05), protein (0.03±0.01 vs. 0.23±0.14, p<0.05), and activity (~2.5-fold decrease, p<0.05) levels at 6h post-OLT (Fig. 5). MMP-9 expression was also depressed at mRNA (0.33±0.19 vs. 1.13±0.22, p<0.05), protein (0.15±0.07 vs. 0.70±0.14, p<0.05), and activity (~4.7-fold decrease, p<0.05) levels in CS-1 peptide treated OLTs at 24h post-IRI, compared to controls. Moreover, while control OLTs were characterized by significant MMP-9+ leukocyte infiltration, CS-1 peptide treated livers showed only very few intragraft MMP-9+ leukocytes, (Fig. 5C). MT1-MMP/MMP-14 was virtually absent from naïve livers and upregulated in livers post-OLT; however, its expression was significantly depressed in CS-1 peptide treated OLTs at mRNA (0.27±0.20 vs. 0.76±0.09; p<0.05) and protein (0.32±0.07 vs. 0.61±0.04; p<0.05) levels at 6h post-OLT, compared to respective controls (Fig. 5E and F). MT1-MMP/MMP-14 expression was similar in CS-1 peptide treated and control OLTs at 24h post-transplantation (data not shown), raising the possibility that the MT1-MMP/MMP-14 expression could perhaps be more relevant during the initial phase of liver IRI. Double immunofluorescence staining was performed in serial liver sections to identify the sources of MT1-MMP/MMP-14, which detected MT1-MMP/MMP-14 staining predominantly in liver infiltrating monocyte/macrophages at 6h post-cold IRI (Fig. 5G).
Figure 5.

MMP-9 and MT1-MMP expressions in 24h cold liver IRI. The expressions of MMP-9 (panels A–D) and MT1-MMP (panels E–G) at mRNA (panel A and E), protein (panel B and F), and activity (panel D) levels were readily detected in control OLTs (lanes 1, and 2) and only slightly detected in CS-1 peptide treated livers at 6h post-IRI (lanes 3, and 4); MMP-9 and MT1-MMP expressions were nearly undetectable in naïve livers (lane 5). Panel C shows MMP-9 + leukocyte infiltration in control (a) and CS-1 peptide treated (b) OLTs at 6h post-IRI. Panel G displays MT1-MMP and Mac-1 immunofluorescence staining in control (a, c, and e) and CS-1 peptide treated (b, d, and f) livers at 6h post-OLT. Mac-1 (a, and b) is stained in green (Alexa Fluor 488) and MT1-MMP (c, and d) is labeled in red (Alexa Fluor 594); cell colocalization of Mac-1/MT1-MMP markers is shown in yellow-orange (e, and f) (*p<0.05; arrows denote positive labeling; × 200; n=5–6 rats/group).
MMP-9 and MT1-MMP/MMP-14 induce distinct patterns of leukocyte migration across fibronectin
We have shown that MMP-9 regulates the migration of neutrophils 21, 23, others have shown that MT1-MMP/MMP-14 controls the migration of macrophages 26 in vitro. Therefore, to complement these earlier studies, we assessed the role of MMP-9 and MT1-MMP/MMP-14 on the migration of both TNF-α-stimulated neutrophils and macrophages across fibronectin. Migration of both macrophages and neutrophils across fibronectin was markedly increased in the presence of TNF-α stimulation. However, MMP-9 inhibition significantly depressed TNF-α-stimulated macrophage (~2.0-fold decrease, p<0.05) and neutrophil (~1.8-fold decrease, p<0.05) migration (Fig. 6). In contrast, MMP/MMP-14 inhibition, while significantly reducing TNF-α-stimulated macrophage migration (~1.7-fold decrease, p<0.05), it was virtually ineffective in preventing stimulated neutrophils to migrate across fibronectin. Therefore, these data suggest a rather selective role for MT1-MMP/MMP-14 on macrophage migration (Fig. 6).
Figure 6.

Regulation of neutrophil and macrophage migration by MMP-9 and MT1-MMP/MMP-14. Migration of macrophages across fibronectin (A) was markedly increased in the presence of TNF-α; MMP-9 inhibition as well as MT1-MMP/MMP-14 inhibition significantly reduced macrophage migration to levels almost comparable with those observed in the absence of TNF-α stimulation. Migration of neutrophils across fibronectin (B) was also markedly increased in the presence of TNF-α stimulation; however, while MMP-9 inhibition markedly reduced neutrophil infiltration, MT1-MMP/MMP-14 inhibition was virtually ineffective in depressing the migration of these cells. The cell migration index was measured as the ratio of cells that migrated into the lower wells in the experimental groups to those in the control groups without TNF-α stimulation. MMP indexes are expressed as mean ± SD of four experiments (&p<0.05, relative to controls without TNF-α stimulation; *p<0.05 relative to TNF-α-stimulated controls).
CS-1 peptide therapy inhibits the phosphorylation of p38 mitogen-activated protein kinase in 24h cold liver IRI
Mitogen-activated protein kinases (MAPKs) are a family of serine/threonine protein kinases implied in the regulation of cellular responses to the environment 27. In an attempt to elucidate the significance of CS-1 peptide therapy upon MAPK cell signaling, we evaluated the activation of p38 MAPK and p44/42 MAPK signaling pathways in our rat liver model of ex vivo 24h cold ischemia followed by transplantation. The phosphorylation of the p38 MAPK threonine and tyrosine residues (Thr180/Tyr182), which was virtually undetected in naïve livers, was slightly upregulated in CS-1 treated livers (0.06±0.01 vs. 0.27±0.13; p<0.05) at 6h post-reperfusion, contrasting with the strong p38 phosphorylation levels detected in the respective control OLTs, (Fig. 7). On the other hand, significant differences in p44/42 MAPK threonine and tyrosine phosphorylation (Thr202/Tyr204) were not detected between CS-1 peptide treated and control OLTs (0.60±0.28 vs. 0.57±0.27) at 6h post-reperfusion, (Fig. 7). Thus, our data suggest that CS-1 peptide therapy preferentially inhibits the p38 MAPK signaling pathway in cold liver IRI.
Figure 7.

Mitogen-activated protein kinase pathways in 24h cold liver IRI. Levels of p38 MAPK and p44/42 MAPK phosphorylation (panel A) in control (lanes 1, and 2) and in CS-1 peptide (lanes 3, and 4) treated livers at 6h post-IRI. Densitometric analysis (panel B) revealed a marked decrease in p38 MAPK activation in CS-1 peptide treated OLTs as compared to controls; there were no significant differences on p44/42 MAPK phosphorylation between CS-1 peptide treated and control OLTs (*p<0.05; n=6/group).
Induction of MMP-9 and MT1-MMP/MMP-14 by fibronectin is mediated by p38 mitogen-activated protein kinase
We have previously shown that fibronectin-leukocyte interactions regulate MMP-9 expression in a macrophage cell line 18. Here, we tested whether fibronectin was able to upregulate the expressions of both MMP-9 and MT1-MMP/MMP-14 in isolated macrophages. Indeed, in addition to MMP-9 upregulation (0.80±0.24 vs. 0.36±0.11; p<0.05), we observed that FN is also capable of upregulating MT1-MMP/MMP-14 expression in cultured macrophages (0.97±0.16 vs. 0.48±0.11; p<0.05) (Fig 8A). It has been shown that human monocytes stimulated with LPS express MT1-MMP/MMP-14 28; polymyxin B was added to some cultures to eliminate the effect of potential endotoxin contamination. We further determined the role of the p38 MAP kinase pathway in fibronectin-stimulated MMP-9 and MT1-MMP/MMP-14 expressions by culturing isolated macrophages in fibronectin in the absence or presence of the pharmacological inhibitor of p38 MAPK SB203580. As shown in figure 8, SB203580 significantly inhibited fibronectin-stimulated MMP-9 (0.36±0.07 vs. 0.80±0.24; p<0.05) and MT1-MMP/MMP-14 (0.62±0.04 vs. 0.97±0.16; p<0.05) mRNA expressions. Moreover, gelatin zymography carried out on protein extracts from the cultures of fibronectin-stimulated macrophages confirmed a significant decrease in MMP-9 activity (p<0.05) in the presence of SB203580. Thus, our data suggest that fibronectin-stimulated MMP-9 and MT1-MMP/MMP-14 expressions are p38 MAPK signaling pathway-dependent.
Figure 8.
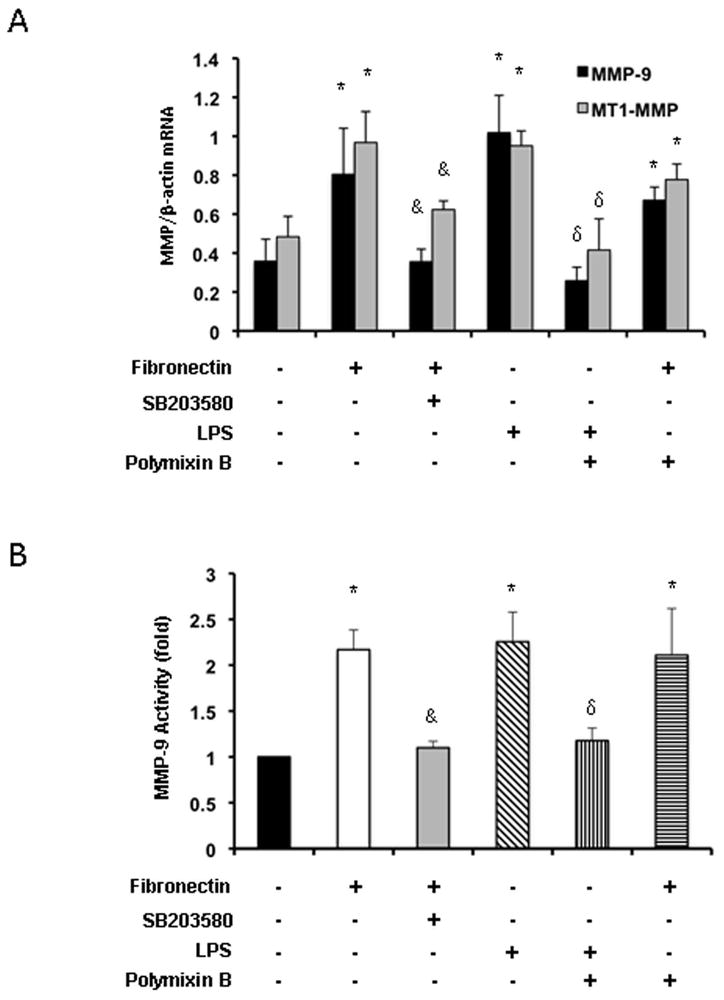
Fibronectin-mediated MMP-9 and MT1-MMP expressions are dependent on p38 MAPK activation. Attenuation of FN-mediated MMP-9 and MT1-MMP mRNA expressions (panel A) by inhibition of p38 MAPK in isolated macrophages treated with SB-203580 (10 μg/ml). While polymyxin B (10 ng/ml) profoundly depressed the expression of MMP-9 and MT1-MMP in LPS-stimulated macrophages, it didn’t significantly affect the production of MMP-9 or MT1-MMP induced by fibronectin in isolated macrophages. Conditioned media obtained from cultured macrophages was subjected to a gelatin zymography assay (panel B); inhibition of p38 MAPK with SB-203580 significantly depressed the MMP-9 activity in fibronectin-stimulated macrophages (graph represents fold increases in enzymatic activity over unstimulated macrophages). MMP levels expressed as mean ± SD of four experiments (*p<0.05, relative to unstimulated controls; &p<0.05, relative to fibronectin stimulated controls; δp<0.05, relative to LPS stimulated controls).
DISCUSSION
It is generally accepted that hepatic IRI associated with leukocyte recruitment and release of cytokines and free radicals plays a major role in liver dysfunction after OLT 29; nevertheless, the goal of improving therapies for liver IRI has been hindered by the need to develop a thorough understanding about which factors drive leukocyte recruitment and production of inflammatory cytokines. This study provides new insights on the role of fibronectin in the pathophysiology of hepatic IRI. FN can exist in two forms, plasma, which lacks the EDA sequence, and cellular FN; plasma FN circulates in the blood in a closed (allegedly) non-active form, while cellular FN, which exists as part of the extracellular matrix, has been linked to most of the FN activities in the body 30, 31. EDA-containing cellular fibronectin is virtually absent in normal adult tissues and is expressed by endothelial cells very early after injury 10, 32. Its expression has been associated to both physiologic wound healing and pathologic tissue fibrosis. While EDA+ FN expression by sinusoidal endothelial cells mediates TGF-β induced wound repair 33, its absence results in abnormal wound healing 34. The dysregulation of tissue-repair processes can lead to fibrosis 35; cellular FN, which stimulates the conversion of hepatic stellate cells into fibrogenic myofibroblasts 36, may play an important role in the development of this condition. Indeed, EDA-null mice fail to significantly develop pulmonary fibrosis. 37.
We have previously shown that cellular fibronectin is upregulated in the liver vasculature preceding leukocyte recruitment and that CS-1 peptide facilitate blockade of α4β1-FN interactions disrupted leukocyte infiltration and ameliorated steatotic hepatic IRI 17. Comparable beneficial effects of the CS-1 peptide therapy, on both leukocyte recruitment and recipient survival rate, were observed in prolonged cold liver IRI. In 24-hour cold liver IRI, CS-1 peptides significantly improved liver histological preservation and increased recipient survival. CS-1 peptide therapy disrupted the recruitment of T cells, NK cells, macrophages, and neutrophils, which are leukocytes associated with the development of liver IRI 38, 39. Moreover, the CS-1 peptide therapy suppressed the release of several pro-inflammatory mediators, such as TNF-α, IFN-γ, COX-2 and iNOS. TNF-α as well as IFN-γ are critical mediators of hepatic IRI 38. COX-2 and iNOS have been shown by us 23, 25 and by others 40, 41 to have deleterious effects on liver IRI.
Leukocyte transmigration across vascular barriers is dependent on both adhesive and matrix degradation mechanisms. Whereas adhesion molecules are important to leukocyte transmigration by providing leukocyte attachment to the vascular endothelium, matrix metalloproteinases are critical for facilitating leukocyte movement across vascular barriers. Among different MMPs, MMP-9, an inducible gelatinase expressed by leukocytes during hepatic IRI, is emerging as an important mediator of leukocyte traffic to inflamed liver 5. Fibronectin-α4β1 interactions are capable of upregulating MMP-9 expression by infiltrating leukocytes 18, which is a critical mediator of leukocyte recruitment in liver IRI 21. There is growing evidence supporting a complex spatiotemporal regulation of the proteolytic activity is involved in focal matrix degradation during extravasation 26. In addition to downregulating the expression of MMP-9, CS-1 peptide therapy also depressed the expression of MT1-MMP/MMP-14 in hepatic IRI. The membrane-anchored MT1-MMP/MMP-14 is a MMP involved in the breakdown of several adhesion molecules, including fibronectin 42. Unlike soluble MMPs, MT1-MMP/MMP-14 has a stretch of hydrophobic amino acids that anchors the enzyme to the plasma membrane and restricts its activity to the cell surface 43, 44. We show for the first time that MT1-MMP/MMP-14 expression, which was undetectable in naïve livers, was upregulated by infiltrating monocyte/macrophages after prolonged liver IRI, suggesting a potential role for MT1-MMP/MMP-14 in cold liver IRI. MT1-MMP/MMP-14 has been associated to focalized ECM degradation and to migration of a variety of cell types, including macrophages, endothelial, and tumor cells 26, 45. Indeed, it has been recently demonstrated that MT1-MMP/MMP-14 is needed to increase the migration of cancer cells in mammary tumors 38. In our settings, MT1-MMP/MMP-14 appears to preferentially regulate macrophage migration as its inhibition didn’t affect neutrophil migration across fibronectin. All together, these observations support the view that MT1-MMP/MMP-14 may act as an amplifier in the recruitment of macrophages, important regulators of inflammation and fibrosis 46, in liver IRI; additional experimentation is warranted to further unveil the role of MT1-MMP/MMP-14 in inflamed livers.
Integrins transmit information from the ECM to the cell resulting in activation of cell signaling pathways important for regulating different cell functions, including adhesion, migration, and proliferation 47. The p38 MAPK signaling transduction pathway, which is activated through extracellular stimuli, plays an essential role in regulating inflammatory processes 48, including hepatic IRI 49. Moreover, it has been suggested that the main biological response of p38 MAPK activation has been linked to initiation of leukocyte recruitment and activation 49. Indeed, the results of this study show that CS-1 mediated blockade of the FN-α4β1 interactions, which disrupted leukocyte recruitment, markedly depressed the phosphorylation of p38 MAPK. We next investigated whether treatment with the pharmacological inhibitor of p38 MAPK SB203580 would affect the expressions of MMP-9 and MT1-MMP/MMP-14 in fibronectin-stimulated macrophages. Macrophages are major sources of MMP-9 in cold liver IRI 18 and, as we report here, of MT1-MMP/MMP-14 as well. Indeed, inhibition of p38 MAPK activity with the antagonist SB203580 depressed the expressions of MMP-9 and MT1-MMP/MMP-14 by fibronectin-stimulated macrophages in culture, suggesting that the p38 MAPK kinase pathway controls fibronectin MMP-9 and MT1-MMP/MMP-14 inductions. In this regard, activation of p38 MAPK has been implied in TNF-α mediated MMP-9 induction 50 and in promoting cancer cell invasion via regulation of MMP mRNA stability 51. In fact, inhibition of the p38 MAPK pathway significantly depressed TNF-α stimulated leukocyte migration across fibronectin (data not shown).
In summary, the findings we report here further emphasize an important role for the FN-α4β1 integrin interactions in cold hepatic IRI. Our results show that CS-1 peptide therapy down-regulated the expressions of MMP-9 and MT1-MMP/MMP-14, disrupted leukocyte recruitment, and decreased the release of pro-inflammatory mediators, resulting in protection against prolonged cold liver IRI and increased OLT recipient survival. Additionally, the CS-1 peptide facilitated blockade of the FN-α4β1integrin interactions depressed the phosphorylation of p38 MAPK, which is considered to be an attractive target for pharmacologic intervention 52, and therefore implying a regulatory role for fibronectin on the activation of p38 MAPK in hepatic IRI. Furthermore, we provide evidence that p38 MAPK kinase pathway controls the fibronectin mediated induction of both MMP-9 and MT1-MMP/MMP-14 by macrophages.
Acknowledgments
This work was supported by the following grants from the National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases (NIAID) R01AI057832 and the Pfleger Foundation (to AJC). S.D. was supported in part by a doctoral fellowship from the Fundação para a Ciência e Tecnologia (FCT), Portugal. We thank Drs. L. Messersmith for supplying the peptides, A.G. Arroyo for providing the antibodies against MT1-MMP/MMP-14 used in the in vitro. studies, and C. Moore for her valuable assistance with specimen collection and processing.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CS-1
connecting segment-1
- COX-2
cyclooxygenase-2
- FN
fibronectin
- ECM
extracellular matrix
- iNOS
inducible nitric oxide synthase
- IRI
ischemia-reperfusion injury
- MMP
matrix metalloproteinase
- MT1-MMP
membrane type 1-matrix metalloproteinase
- MAPK
mitogen-activated protein kinase
- MPO
myeloperoxidase
- OLT
orthotopic liver transplantation
Footnotes
DISCLOSURE: The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
References
- 1.Selzner N, Rudiger H, Graf R, Clavien PA. Protective strategies against ischemic injury of the liver. Gastroenterology. 2003;125:917–936. doi: 10.1016/s0016-5085(03)01048-5. [DOI] [PubMed] [Google Scholar]
- 2.Busuttil RW, Tanaka K. The utility of marginal donors in liver transplantation. Liver Transpl. 2003;9:651–663. doi: 10.1053/jlts.2003.50105. [DOI] [PubMed] [Google Scholar]
- 3.Carman CV, Springer TA. Trans-cellular migration: cell-cell contacts get intimate. Curr Opin Cell Biol. 2008;20:533–540. doi: 10.1016/j.ceb.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaeschke H, Hasegawa T. Role of neutrophils in acute inflammatory liver injury. Liver Int. 2006;26:912–919. doi: 10.1111/j.1478-3231.2006.01327.x. [DOI] [PubMed] [Google Scholar]
- 5.Coito AJ. Leukocyte transmigration across endothelial and extracellular matrix protein barriers in liver ischemia/reperfusion injury. Curr Opin Organ Transplant. 2011;16:34–40. doi: 10.1097/MOT.0b013e328342542e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bianchi E, Bender JR, Blasi F, Pardi R. Through and beyond the wall: late steps in leukocyte transendothelial migration. Immunol Today. 1997;18:586–591. doi: 10.1016/s0167-5699(97)01162-6. [DOI] [PubMed] [Google Scholar]
- 7.Garmy-Susini B, Avraamides CJ, Schmid MC, Foubert P, Ellies LG, Barnes L, Feral C, Papayannopoulou T, Lowy A, Blair SL, Cheresh D, Ginsberg M, Varner JA. Integrin alpha4beta1 signaling is required for lymphangiogenesis and tumor metastasis. Cancer Res. 2010;70:3042–3051. doi: 10.1158/0008-5472.CAN-09-3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lefebvre JS, Levesque T, Picard S, Pare G, Gravel A, Flamand L, Borgeat P. Extra domain A of fibronectin primes leukotriene biosynthesis and stimulates neutrophil migration through activation of Toll-like receptor 4. Arthritis Rheum. 2011;63:1527–1533. doi: 10.1002/art.30308. [DOI] [PubMed] [Google Scholar]
- 9.van Horssen J, Dijkstra CD, de Vries HE. The extracellular matrix in multiple sclerosis pathology. J Neurochem. 2007;103:1293–1301. doi: 10.1111/j.1471-4159.2007.04897.x. [DOI] [PubMed] [Google Scholar]
- 10.Coito AJ, Brown LF, Peters JH, Kupiec-Weglinski JW, van de Water L. Expression of fibronectin splicing variants in organ transplantation: a differential pattern between rat cardiac allografts and isografts. Am J Pathol. 1997;150:1757–1772. [PMC free article] [PubMed] [Google Scholar]
- 11.Pulido R, Elices MJ, Campanero MR, Osborn L, Schiffer S, Garcia-Pardo A, Lobb R, Hemler ME, Sanchez-Madrid F. Functional evidence for three distinct and independently inhibitable adhesion activities mediated by the human integrin VLA-4. Correlation with distinct alpha 4 epitopes. J Biol Chem. 1991;266:10241–10245. [PubMed] [Google Scholar]
- 12.O’Connor P. Natalizumab and the role of alpha 4-integrin antagonism in the treatment of multiple sclerosis. Expert Opin Biol Ther. 2007;7:123–136. doi: 10.1517/14712598.7.1.123. [DOI] [PubMed] [Google Scholar]
- 13.Fiorino G, Correale C, Fries W, Repici A, Malesci A, Danese S. Leukocyte traffic control: a novel therapeutic strategy for inflammatory bowel disease. Expert Rev Clin Immunol. 2010;6:567–572. doi: 10.1586/eci.10.40. [DOI] [PubMed] [Google Scholar]
- 14.Coito AJ, de Sousa M, Kupiec-Weglinski JW. Fibronectin in immune responses in organ transplant recipients. Dev Immunol. 2000;7:239–248. doi: 10.1155/2000/98187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Man S, Tucky B, Bagheri N, Li X, Kochar R, Ransohoff RM. alpha4 Integrin/FN-CS1 mediated leukocyte adhesion to brain microvascular endothelial cells under flow conditions. J Neuroimmunol. 2009;210:92–99. doi: 10.1016/j.jneuroim.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perez VA, Ali Z, Alastalo TP, Ikeno F, Sawada H, Lai YJ, Kleisli T, Spiekerkoetter E, Qu X, Rubinos LH, Ashley E, Amieva M, Dedhar S, Rabinovitch M. BMP promotes motility and represses growth of smooth muscle cells by activation of tandem Wnt pathways. J Cell Biol. 2011;192:171–188. doi: 10.1083/jcb.201008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amersi F, Shen XD, Moore C, Melinek J, Busuttil RW, Kupiec-Weglinski JW, Coito AJ. Fibronectin-alpha 4 beta 1 integrin-mediated blockade protects genetically fat Zucker rat livers from ischemia/reperfusion injury. Am J Pathol. 2003;162:1229–1239. doi: 10.1016/s0002-9440(10)63919-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore C, Shen XD, Gao F, Busuttil RW, Coito AJ. Fibronectin-alpha4beta1 integrin interactions regulate metalloproteinase-9 expression in steatotic liver ischemia and reperfusion injury. Am J Pathol. 2007;170:567–577. doi: 10.2353/ajpath.2007.060456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamada N, Davies HS, Wight D, Culank L, Roser B. Liver transplantation in the rat. Biochemical and histological evidence of complete tolerance induction in non-rejector strains. Transplantation. 1983;35:304–311. [PubMed] [Google Scholar]
- 20.Fondevila C, Shen XD, Tsuchiyashi S, Yamashita K, Csizmadia E, Lassman C, Busuttil RW, Kupiec-Weglinski JW, Bach FH. Biliverdin therapy protects rat livers from ischemia and reperfusion injury. Hepatology. 2004;40:1333–1341. doi: 10.1002/hep.20480. [DOI] [PubMed] [Google Scholar]
- 21.Hamada T, Fondevila C, Busuttil RW, Coito AJ. Metalloproteinase-9 deficiency protects against hepatic ischemia/reperfusion injury. Hepatology. 2008;47:186–198. doi: 10.1002/hep.21922. [DOI] [PubMed] [Google Scholar]
- 22.Kuriyama N, Duarte S, Hamada T, Busuttil RW, Coito AJ. Tenascin-C: a novel mediator of hepatic ischemia and reperfusion injury. Hepatology. 2011;54:2125–2136. doi: 10.1002/hep.24639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamada T, Duarte S, Tsuchihashi S, Busuttil RW, Coito AJ. Inducible nitric oxide synthase deficiency impairs matrix metalloproteinase-9 activity and disrupts leukocyte migration in hepatic ischemia/reperfusion injury. Am J Pathol. 2009;174:2265–2277. doi: 10.2353/ajpath.2009.080872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310:1966–1970. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 25.Hamada T, Tsuchihashi S, Avanesyan A, Duarte S, Moore C, Busuttil RW, Coito AJ. Cyclooxygenase-2 deficiency enhances Th2 immune responses and impairs neutrophil recruitment in hepatic ischemia/reperfusion injury. J Immunol. 2008;180:1843–1853. doi: 10.4049/jimmunol.180.3.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matias-Roman S, Galvez BG, Genis L, Yanez-Mo M, de la Rosa G, Sanchez-Mateos P, Sanchez-Madrid F, Arroyo AG. Membrane type 1-matrix metalloproteinase is involved in migration of human monocytes and is regulated through their interaction with fibronectin or endothelium. Blood. 2005;105:3956–3964. doi: 10.1182/blood-2004-06-2382. [DOI] [PubMed] [Google Scholar]
- 27.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 28.Shankavaram UT, Lai WC, Netzel-Arnett S, Mangan PR, Ardans JA, Caterina N, Stetler-Stevenson WG, Birkedal-Hansen H, Wahl LM. Monocyte membrane type 1-matrix metalloproteinase. Prostaglandin-dependent regulation and role in metalloproteinase-2 activation. J Biol Chem. 2001;276:19027–19032. doi: 10.1074/jbc.M009562200. [DOI] [PubMed] [Google Scholar]
- 29.Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, Pina E, Geller DA. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J Surg Res. 2008;147:153–159. doi: 10.1016/j.jss.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115:3861–3863. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- 31.Wierzbicka-Patynowski I, Schwarzbauer JE. The ins and outs of fibronectin matrix assembly. J Cell Sci. 2003;116:3269–3276. doi: 10.1242/jcs.00670. [DOI] [PubMed] [Google Scholar]
- 32.Jarnagin WR, Rockey DC, Koteliansky VE, Wang SS, Bissell DM. Expression of variant fibronectins in wound healing: cellular source and biological activity of the EIIIA segment in rat hepatic fibrogenesis. J Cell Biol. 1994;127:2037–2048. doi: 10.1083/jcb.127.6.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.George J, Wang SS, Sevcsik AM, Sanicola M, Cate RL, Koteliansky VE, Bissell DM. Transforming growth factor-beta initiates wound repair in rat liver through induction of the EIIIA-fibronectin splice isoform. Am J Pathol. 2000;156:115–124. doi: 10.1016/s0002-9440(10)64711-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muro AF, Chauhan AK, Gajovic S, Iaconcig A, Porro F, Stanta G, Baralle FE. Regulated splicing of the fibronectin EDA exon is essential for proper skin wound healing and normal lifespan. J Cell Biol. 2003;162:149–160. doi: 10.1083/jcb.200212079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bissell DM. Hepatic fibrosis as wound repair: a progress report. J Gastroenterol. 1998;33:295–302. doi: 10.1007/s005350050087. [DOI] [PubMed] [Google Scholar]
- 37.Muro AF, Moretti FA, Moore BB, Yan M, Atrasz RG, Wilke CA, Flaherty KR, Martinez FJ, Tsui JL, Sheppard D, Baralle FE, Toews GB, White ES. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177:638–645. doi: 10.1164/rccm.200708-1291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le Moine O, Louis H, Demols A, Desalle F, Demoor F, Quertinmont E, Goldman M, Deviere J. Cold liver ischemia-reperfusion injury critically depends on liver T cells and is improved by donor pretreatment with interleukin 10 in mice. Hepatology. 2000;31:1266–1274. doi: 10.1053/jhep.2000.7881. [DOI] [PubMed] [Google Scholar]
- 39.Gao B, Jeong WI, Tian Z. Liver: An organ with predominant innate immunity. Hepatology. 2008;47:729–736. doi: 10.1002/hep.22034. [DOI] [PubMed] [Google Scholar]
- 40.Ozturk H, Gezici A. The effect of celecoxib, a selective COX-2 inhibitor, on liver ischemia/reperfusion-induced oxidative stress in rats. Hepatol Res. 2006;34:76–83. doi: 10.1016/j.hepres.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 41.Moon KH, Hood BL, Mukhopadhyay P, Rajesh M, Abdelmegeed MA, Kwon YI, Conrads TP, Veenstra TD, Song BJ, Pacher P. Oxidative inactivation of key mitochondrial proteins leads to dysfunction and injury in hepatic ischemia reperfusion. Gastroenterology. 2008;135:1344–1357. doi: 10.1053/j.gastro.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perentes JY, Kirkpatrick ND, Nagano S, Smith EY, Shaver CM, Sgroi D, Garkavtsev I, Munn LL, Jain RK, Boucher Y. Cancer cell-associated MT1-MMP promotes blood vessel invasion and distant metastasis in triple-negative mammary tumors. Cancer Res. 2011;71:4527–4538. doi: 10.1158/0008-5472.CAN-10-4376. [DOI] [PubMed] [Google Scholar]
- 44.Seiki M. Membrane-type 1 matrix metalloproteinase: a key enzyme for tumor invasion. Cancer Lett. 2003;194:1–11. doi: 10.1016/s0304-3835(02)00699-7. [DOI] [PubMed] [Google Scholar]
- 45.Moss NM, Wu YI, Liu Y, Munshi HG, Stack MS. Modulation of the membrane type 1 matrix metalloproteinase cytoplasmic tail enhances tumor cell invasion and proliferation in three-dimensional collagen matrices. J Biol Chem. 2009;284:19791–19799. doi: 10.1074/jbc.M109.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mainiero F, Gismondi A, Soriani A, Cippitelli M, Palmieri G, Jacobelli J, Piccoli M, Frati L, Santoni A. Integrin-mediated ras-extracellular regulated kinase (ERK) signaling regulates interferon gamma production in human natural killer cells. J Exp Med. 1998;188:1267–1275. doi: 10.1084/jem.188.7.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 49.King LA, Toledo AH, Rivera-Chavez FA, Toledo-Pereyra LH. Role of p38 and JNK in liver ischemia and reperfusion. J Hepatobiliary Pancreat Surg. 2009;16:763–770. doi: 10.1007/s00534-009-0155-x. [DOI] [PubMed] [Google Scholar]
- 50.Li H, Mittal A, Paul PK, Kumar M, Srivastava DS, Tyagi SC, Kumar A. Tumor necrosis factor-related weak inducer of apoptosis augments matrix metalloproteinase 9 (MMP-9) production in skeletal muscle through the activation of nuclear factor-kappaB-inducing kinase and p38 mitogen-activated protein kinase. a potential role of MMP-9 in myopathy. J Biol Chem. 2009;284:4439–4450. doi: 10.1074/jbc.M805546200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar B, Koul S, Petersen J, Khandrika L, Hwa JS, Meacham RB, Wilson S, Koul HK. p38 mitogen-activated protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer Res. 2010;70:832–841. doi: 10.1158/0008-5472.CAN-09-2918. [DOI] [PubMed] [Google Scholar]
- 52.Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, Braunwart J, Glascock BJ, Klevitsky R, Kimball TF, Hewett TE, Molkentin JD. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest. 2003;111:1475–1486. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]