

Abstract
Recent genomic studies have identified risk SNPs in or near eight genes associated with testicular germ cell tumors (TGCT). Mouse models suggest a role for Dnd1 epigenetics in TGCT susceptibility, and we have recently reported that transgenerational inheritance of epigenetic events may be associated with familial TGCT risk. We now investigate whether aberrant promoter methylation of selected candidate genes is associated with familial TGCT risk. Pyrosequencing assays were designed to evaluate CpG methylation in the promoters of selected genes in peripheral blood DNA from 153 TGCT affecteds and 116 healthy male relatives from 101 multiple-case families. Wilcoxon rank-sum tests and logistic regression models were used to investigate associations between promoter methylation and TGCT. We also quantified gene product expression of these genes, using quantitative PCR. We observed increased PDE11A, SPRY4 and BAK1 promoter methylation, and decreased KITLG promoter methylation, in familial TGCT cases versus healthy male family controls. A significant upward risk trend was observed for PDE11A when comparing the middle and highest tertiles of methylation to the lowest [odds ratio (OR) =1.55, 95% confidence intervals (CI) 0.82-2.93, and 1.94, 95% CI 1.03-3.66], respectively; P trend=0.042). A significant inverse association was observed for KITLG when comparing the middle and lowest tertiles to the highest (OR=2.15, 95% CI 1.12-4.11, and 2.15, 95% CI 1.12-4.14, respectively; P trend=0.031). There was a weak inverse correlation between promoter methylation and KITLG expression. Our results suggest that familial TGCT susceptibility may be associated with promoter methylation of previously-identified TGCT risk-modifying genes. Larger studies are warranted.
Keywords: Promoter methylation, testicular germ cell tumors, familial testicular cancer, epidemiology, candidate gene
Introduction
Testicular germ cell tumor (TGCT) is the most commonly-diagnosed cancer among young men in industrialized countries. Until recently, little was known of its genetic susceptibility, despite numerous epidemiological studies suggesting a strong genetic basis [1-4]. Genome-wide linkage analysis in familial TGCT found no significant evidence of genetic linkage, and suggested that multiple common, low-penetrance loci likely contributed to TGCT susceptibility [5]. Candidate gene studies identified two loci of interest, the Y-chromosome gr/gr deletion [6] and PDE11A gene variants [7]. In addition, recent genomewide association studies (GWAS) have uncovered candidate TGCT susceptibility loci in six gene regions: KITLG, SPRY4, BAK1, TERT-CLPTM1L, ATF7IP, and DMRT1 [8-10]. A candidate gene study of single nucleotide polymorphisms (SNPs) in BAK1, DMRT1, TERT-CLPTM1L, and KITLG gene regions showed that these risk variants predispose to both familial and bilateral TGCT [11]. The GWAS risk SNPs together only accounted for 11% of the risk to brothers and 16% of the risk to sons of TGCT cases [10], suggesting that the majority of TGCT risk loci are still unidentified.
Notably, most of the identified TGCT susceptibility loci are located in genes involved in biologically-plausible pathways. Three loci are involved in the KITLG-KIT signaling pathway that regulates the survival, proliferation and migration of germ cells [12]: KITLG encodes the ligand that activates the receptor tyrosine kinase KIT; SPRY4 encodes an inhibitor of the mitogen-activated protein kinase pathway, which is activated by the KITLG-KIT pathway [13]; and BAK1 is a pro-apoptotic protein, expression of which is repressed by the KITLG-KIT pathway [14]. PDE11A is highly-expressed in endocrine steroidogenic tissues, especially the testis, and its loss causes infertility (a known human TGCT risk factor [15]) in mice [16,17]. The most recently-identified loci [10] in the TERT-CLPTM1L, ATF7IP, and DMRT1 gene regions are involved in telomere maintenance (TERT-CLPTM1L and ATF7IP) and sex determination (DMRT1).
DNA methylation is an epigenetic modification that can be heritable without changing the DNA sequence. DNA methylation regulates gene transcription by targeting CpG islands found in the 5’ regulatory and promoter regions of genes [18]. Hypermethylation of these islands has been described in carcinogenesis, and is a well-documented mechanism by which tumor suppressor genes are silenced [19]. Our previous study of global LINE-1 methylation among multiple-case testicular cancer family members suggested that inheritance of LINE-1 methylation levels may be associated with familial TGCT risk [20]. Interestingly, transgenerational epigenetic interactions have been shown to control susceptibility to TGCTs in mice with a Dead-end homologue 1 (Dnd1) mutation [21]. Dnd1 is expressed in the fetal murine testis during the critical period when TGCTs are believed to develop. Although germline mutations in DND1 have been excluded as contributing significantly to human TGCT risk [22], the epigenetics of DND1 have not been described.
Aberrant DNA methylation may provide an alternate genetic mechanism for familial TGCT susceptibility. The promoter regions of candidate genes may be regulated through epigenetic silencing, and could show a unique profile in high-risk families. Motivated by our prior work [20], the exciting results of recent GWAS studies [8-11], and our inability to confirm a classical Mendelian mode of inheritance for familial TGCT [15], we undertook an exploratory, pilot study aimed at investigating the association between promoter methylation of 5 candidate genes involved in normal testicular embryologic development and/or male infertility (PDE11A, KITLG, SPRY4, and BAK1, as well as the DND1 candidate gene from the mouse model) and TGCT risk among individuals from 101 multiple-case testicular cancer families. These experiments were designed prior to the identification of TERT-CLPTM1L, ATF7IP, and DMRT1 as genomic regions of interest in TGCT, hence their omission from the current analysis. In addition, we examined expression of the protein products of these candidate genes to determine if promoter methylation levels correlated with gene expression.
Materials and methods
Study population
Families with two or more cases of TGCT or a single family member with bilateral TGCT were recruited as part of the NCI Clinical Genetics Branch Familial TGCT study; details are described elsewhere [23]. Family members ≥age 12 years were invited to participate in the study if they were first-degree relatives of a case, spouses of a case who had participating children, non-first-degree blood relatives who provided a genetic link between cases, or blood relatives with cancer. All participants completed detailed questionnaires and provided blood samples; a subset of participants underwent a detailed medical evaluation at the NIH Clinical Center. Blood was collected between 2003 and 2006 and stored at the NCI/CGB Biorepository. The majority of patients underwent blood collection 5 or more years after they were diagnosed with TGCT (median time between diagnosis and blood draw was 5 years). The parent study was reviewed and approved by the NCI Institutional Review Board (NCI Protocol 02-C-0178; NCT00039598), and all participants provided written informed consent.
DNA extraction and bisulfite treatment
Genomic DNA was extracted from fresh whole blood by standard methods. For bisulfite conversion, 500ng of DNA was treated using the EZ DNA Methylation-Gold™ kit (Zymo Research Corp., Orange, CA) according to the manufacturer's recommendations. The final elution was in 20 μl of M-Elution Buffer.
PCR amplification and pyrosequencing
The genomic sequence (using published Gen-Bank sequences) of each gene was searched with an online search engine (www.cpgislands.com) to identify CpG islands in the promoter region (Supplementary Table S1). The transcription start site was based on Ensembl transcripts for each gene (Supplementary Table S1). DNA methylation (%5-methylcytosine [5-mC]) of consecutive CpG sites was quantified using pyrosequencing of the bisulfite-treated DNA by EpigenDx Laboratory Service (Worcester, MA). Gene-specific primers, assay details, and cycling conditions are given in Supplementary Table S1. Each of the newly-designed primer sets was tested for PCR bias in a mixing experiment using in vitro methylated and non-methylated DNA. For each gene, PCR was performed using 10X PCR buffer, 3.0 mM MgCl2, 200 μM of each dNTP, 0.2 μM each of forward and reverse primers, HotStar DNA polymerase (Qiagen, Valencia, CA) 1.25 U, and 10 ng of bisulfite converted DNA per 50 μl reaction. The PCR was performed with one of the primers biotinylated to convert the PCR product to single-stranded DNA templates. The PCR products (each 10 μl) were sequenced by Pyrosequencing PSQ96 HS System (PSQ H96A, Qiagen Pyrosequencing) following the manufacturer’s instructions, according to the established method [24]. Built-in controls were used to verify bisulphite conversion efficiency. The methylation status of each CpG site in the promoter region of each gene was individually analyzed as a T/C SNP using QCpG software (PSQ H96A, Qiagen Pyrosequencing). The relative 5-mC content was expressed as percentage of methylated cytosines divided by the sum of methylated and unmethylated cytosines (5-mC/[5-mC + unmethylated cytosine] = %5-mC). The median coefficient of variation (CV) among 28 blinded replicate samples for each gene was below 12%.
Gene expression
RNA was extracted from cryopreserved lymphocytes, and cDNA was created using the High-capacity cDNA Reverse Transcription Kit from Applied Biosystems (PN 4368814), following the manufacturer’s protocol with 20ng of total RNA per reaction. The following TaqMan Gene Expression assay IDs from Applied Biosystems were used for each gene: Hs00241497_m1 for KITLG; Hs00184027_m1 for PDE11A; Hs00832876_g1 for BAK1; Hs01935412_s1 for SPRY4; and Hs00832091_s1 for DND1. The Pre-Amplification master mix from Applied Biosystems (PN 4391128) was used to pre-amplify all of the assays. A 0.2X dilution pool of all of the 20X assays from Applied Biosystems was created, and 1.25μL of the cDNA was mixed with 2.5μL of pre-amp master mix, and 1.25μL of the 0.2x assay pool. The following cycling conditions were used: 95°C for 10 min, then 14 cycles of 95°C for 15 sec and 60°C for 4 min. Samples were diluted 1:5 with 1X TE and loaded into the 48x48 dynamic array from Fluidigm (PN BMK-M-48.48) according to the manufacturer’s protocol. A delta delta Ct analysis was performed using GAPDH as the endogenous control. To increase precision, each sample was tested 4 times, and the 4-test mean was used in statistical analyses (average fold change). The median CV for the fold change in expression among replicate samples was 5.8%.
Statistical analysis
The percent methylation between TGCT patients and controls (unaffected male relatives) was analyzed as a continuous variable using a nonparametric Wilcoxon rank-sum test and linear regression models. We used unconditional logistic regression models to estimate the odds ratio (OR) and 95% confidence intervals (CI) for the strength of the association between TGCT risk and promoter methylation, adjusting for potential confounders (e.g., age). For this analysis, methylation levels were categorized in tertiles or dichotomized at the median, based on the distribution in the healthy male family controls. Analyses were done for each individual CpG site in each gene’s promoter region and for the average methylation across the entire promoter CpG island (combined methylation level). We computed the variance of the OR estimates using a robust variance estimator to adjust for the correlations between participants from the same family. Spearman rank correlations and general linear models were used to investigate the strength of the associations between %5-mC and age and TGCT type, and between %5-mC and gene expression (average fold change in expression). Statistical significance refers to a P ≤ 0.05 or an OR with 95% CI that excludes 1.0. All analyses were carried out using SAS software version 9.1 (SAS Institute, Cary, NC). The Basic Local Alignment Search Tool (BLAST) was used to compare the SPRY4 nucleotide sequence to mammalian sequence databases (http://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastHome). The 1000 Genomes data (June 2011 release; http://www.1000genomes.org/home) was used to determine minor allele frequencies (MAF).
Results
Table 1 shows the characteristics of the 116 healthy male family controls and 153 TGCT cases. The mean age of the healthy male family controls was 45 (standard deviation [SD] 19.8; range 13 to 94) and the mean age of the TGCT cases was 41 (SD 11; range 17 to 79). There were no significant correlations between age or TGCT histology and promoter methylation levels for any of the 5 genes (Table 1).
Table 1.
Characteristics of study subjects and the average promoter methylation level for each candidate gene.
| Variables | No. (%) | KITLG | PDE11A | SPRY4 | BAK1 | DND1 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Mean level† | SD | Mean level | SD | Mean level | SD | Mean level | SD | Mean level | SD | ||
| Family, healthy | 116 (43.1) | 2.68 | 1.0 | 13.53 | 3.9 | 1.94 | 0.7 | 1.73 | 0.7 | 82.95 | 2.6 |
| TGCT patients | 153 (56.9) | 2.39 | 0.8 | 14.41 | 4.7 | 2.09 | 0.8 | 1.94 | 1.0 | 82.74 | 2.4 |
|
| |||||||||||
| P | 0.028 | 0.082 | 0.24 | 0.12 | 0.16 | ||||||
|
| |||||||||||
| Age group* | |||||||||||
| 13-35 | 90 (33.5) | 2.8 | 1.4 | 13.36 | 3.6 | 1.93 | 0.6 | 1.68 | 0.8 | 82.66 | 2.5 |
| 36-50 | 93 (34.6) | 2.33 | 0.6 | 13.0 | 3.6 | 1.74 | 0.5 | 1.87 | 0.7 | 83.19 | 2.6 |
| 51-94 | 86 (31.9) | 2.71 | 0.9 | 13.87 | 4.2 | 2.04 | 0.8 | 1.76 | 0.7 | 83.2 | 2.6 |
|
| |||||||||||
| P | 0.26 | 0.69 | 0.23 | 0.64 | 0.57 | ||||||
|
| |||||||||||
| TGCT histological type‡ | |||||||||||
| Seminoma | 74 (48.4) | 2.29 | 0.7 | 13.99 | 3.7 | 2.07 | 0.8 | 1.93 | 1.0 | 82.38 | 2.6 |
| Non-seminoma | 75 (49.0) | 2.47 | 0.7 | 14.77 | 5.6 | 2.1 | 0.7 | 1.78 | 0.7 | 83.03 | 2.2 |
|
| |||||||||||
| P | 0.37 | 0.71 | 0.49 | 0.37 | 0.17 | ||||||
No, number of subjects (percent of subjects); SD, standard deviation;
the average % methylation across the entire promoter CpG island;
% methylation for each age group is estimated for controls only;
4 cases were not otherwise specified.
Promoter methylation associations with TGCT
KITLG
There were 24 CpG sites in the promoter region of KITLG. We observed lower methylation levels at most individual CpG sites in the promoter region of KITLG in the familial TGCT cases (average % methylation: 2.4) compared with the healthy male family controls (average % methylation: 2.7). There were significant differences between methylation in TGCT cases and controls at 4 CpG sites and for the average methylation across the entire promoter region (Figure 1 and Supplementary Table S2). With methylation categorized into tertiles based on the control distribution, low methylation (first tertile of methylation) was associated with an increased risk of TGCT at each of these 4 CpG sites and across the promoter region (ORs ranged from 1.8 to 2.2; Figure 2 and Supplementary Table S3).
Figure 1.
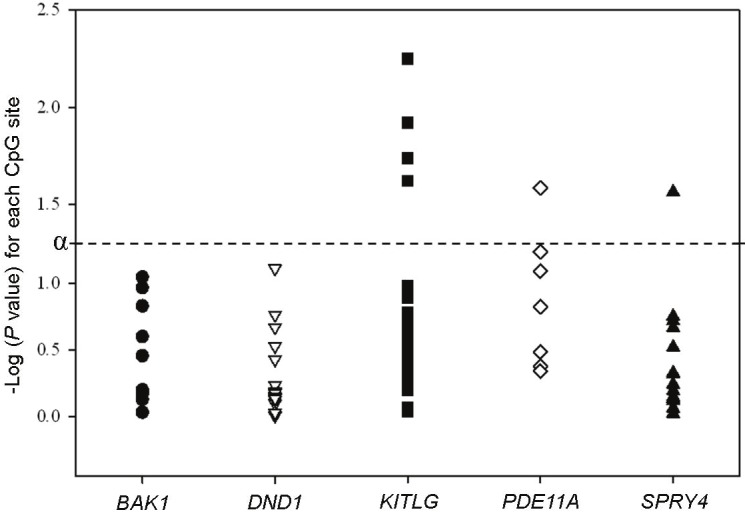
Plot of Wilcoxon rank sum P values for each individual CpG site in the promoter region of the five genes. P values are from a Wilcoxon rank sum test comparing the % methylation at each individual CpG site in the promoter CpG island of each gene among TGCT cases and their healthy male relatives. α = 0.05; dashed line represents an extension of α.
Figure 2.

Graphs illustrating the number of individuals in each methylation tertile (low, middle, high methylation) andodds ratios (OR) for the association between TGCT risk and promoter methylation levels for KITLG (A.), PDE11A (B.),and SPRY4 (C.). Only the average methylation across the promoter region and the CpG sites that were significantlyassociated with risk of TGCT are shown. For PDE11A and SPRY4, ORs are for the middle and highest tertiles of methylationcompared to low methylation; and, for KITLG, ORs are for having middle or low methylation compared tohaving high methylation. Logistic regression models were adjusted for age. Methylation levels for each promoter region,and for each individual CpG site, were categorized into tertiles, based on the distribution in the controls. SignificantORs (P < 0.05) are bolded.
PDE11A
There were 7 CpG sites in the promoter region of PDE11A. Methylation levels were higher in the TGCT cases (average % methylation: 14.4) compared with the male family controls (average % methylation: 13.5) at all CpG sites; levels were significantly different at one CpG site (Figure 1 and Supplementary Table S2). High methylation (top tertile of methylation) was significantly associated with an increased risk of TGCT at 2 CpG sites in the promoter region (ORs = 1.9, 95% CI 1.02-3.7, and 2.3, 95% CI 1.2-4.3; Figure 2 and Supplementary Table S3). There was an increased risk of TGCT associated with high methylation (top tertile of methylation) for the average methylation across the promoter region (OR 1.94, 95% CI 1.03-3.65; P trend= 0.04).
The sequence and variant status of PDE11A in 90 of our TGCT cases has been previously published [7]. We have incorporated this sequence information to determine if TGCT cases without a known PDE11A inactivating variant [7] have increased promoter methylation. There were no statistically significant differences between promoter methylation levels in cases with and without a sequence variant (P values >0.2). However, if we exclude the 20 cases with a known sequence variant, the remaining cases did have a slightly higher TGCT risk associated with high methylation (top tertile of methylation) at most CpG sites and for the average methylation across the promoter region (OR 2.01, 95% CI 1.04-3.90) compared to the results including all cases (OR 1.94, 95% CI 1.03-3.65), and an additional CpG site became significantly associated with risk.
SPRY4
There were 16 CpG sites in the promoter region of SPRY4. Methylation levels were slightly higher in TGCT cases (average % methylation: 2.1) compared with the male family controls (average % methylation: 1.9) at most promoter CpG sites. There was a significant difference between methylation levels in TGCT cases and controls at 1 CpG site (Figure 1; Supplementary Table S2), with high methylation (top tertile of methylation) associated with an increased risk of TGCT (OR 1.99, 95% CI 1.06-3.76; P trend= 0.03; Figure 2; Supplementary Table S3).
In addition, we found a single nucleotide polymorphism (SNP) in the SPRY4 promoter region of one multiple-case family, a heterozygous A>G change (genome build 37.2, Chr 5: 141,704,544) downstream of CpG number 6 (genome build 37.2, Chr 5: 141,704,546). This SPRY4 variant was observed in 3 brothers: 2 were TGCT cases and one was unaffected. The unaffected father of these brothers was also included in our study and did not carry this SPRY4 variant. This SNP has been previously identified in the 1000 Genomes study, and reported in dbSNP (rs146162871 in the 5’ UTR). Using data from 1000 Genomes for 1094 individuals (June 2011 release), the MAF of the variant G allele was 0.7% in all populations, 0% in persons of European ancestry; only 8 individuals of African ancestry carried the variant G allele (7/8 were AG heterozygotes). Using BLAST, the A allele was highly-conserved among other mammalian species (rhesus macaque, chimpanzee, mouse, rat, rabbit, and cat).
BAK1
There were 10 CpG sites in the promoter of BAK1. Most CpG sites in the promoter region had slightly higher methylation levels in TGCT cases (average % methylation: 1.9) compared with the male family controls (average % methylation: 1.7), but none of these differences were statistically significant (Figure 1; Supplementary Table S2). However, high methylation (top tertile of methylation) for the combined methylation level across the promoter region was associated with an increased risk of TGCT (OR 1.86, 95% CI 1.01-3.42; P trend= 0.04; Supplementary Table S3).
DND1
There were 15 CpG sites in the promoter of DND1. Across the promoter region, there were no significant differences between methylation levels in TGCT cases (average % methylation: 82.7) and male family controls (average % methylation: 82.9; Figure 1 and Supplementary Table S2).
Combining the effects of average promoter methylation in these candidate genes considered 2 or 3 at a time did not have an additive effect on TGCT risk, with most risk estimates increasing only slightly (data not shown). The strongest combined effect was observed among those having high methylation (top tertile) of PDE11A, SPRY4, and BAK1 compared with having low methylation of these genes (OR 4.77, 95% CI 1.5-15.8), and there was a strong upward risk trend with increasing number of these genes with high methylation (P trend= 0.007). Stratified analyses by tumor type showed similar associations between combined methylation level across the promoter region and TGCT risk for both the seminoma and non-seminoma tumors (Supplemental Table S4). The associations were only significant for cases with seminoma tumors and lower promoter methylation of KITLG and higher promoter methylation of BAK1. There was also a significant association between lower promoter methylation of DND1 and risk of seminoma tumors. The strong biologically plausibility of DND1 being a TGCT risk modifier warrants a careful look at this gene. However, since we have not corrected our exploratory analyses for false discovery due to multiple testing, the statistical significance here must be regarded with caution.
Gene expression and promoter methylation
There were 109 TGCT cases and 10 healthy male family members with cryopreserved lymphocytes available for RNA extraction and expression analysis. For KITLG, there was an inverse correlation between gene expression and average methylation across the promoter region in our TGCT cases (r = -0.21, P = 0.080; Figure 3). For the 4 individual CpG sites that had methylation levels significantly associated with TGCT, methylation levels at 3 were inversely correlated with gene expression in TGCT cases, but none were individually statistically significant (P >0.1). Methylation levels at 3 other CpG sites in the promoter region of KITLG showed a significant inverse correlation with gene expression (r = -0.26 to -0.28; P = 0.02).
Figure 3.
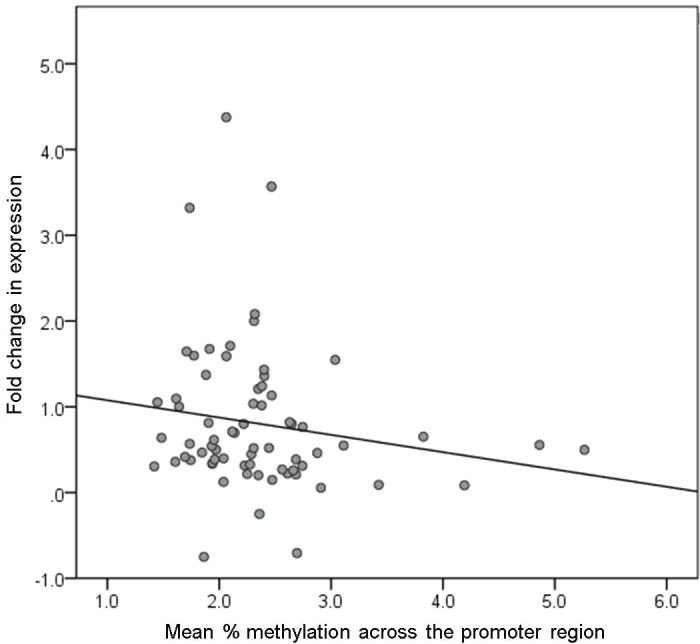
Gene expression and mean % promoter methylation of KITLG. The scatterplot shows the fold change in expression of KITLG and the mean % methylation across the promoter region in TGCT cases only.
PDE11A failed to amplify in any of the samples tested, suggesting very low or no expression in our samples. For SPRY4, there was no correlation between the individual CpG site significantly associated with TGCT risk and gene expression (r = -0.017, P = 0.88). Methylation levels across the promoter regions of BAK1 and DND1 were not associated with gene expression (r = 0.02, P = 0.81; and r = -0.11, P = 0.24, respectively).
Discussion
Our data suggest that increased promoter methylation of PDE11A, SPRY4 and BAK1, and decreased promoter methylation of KITLG in primary lymphocytes, are associated with risk of familial TGCT. PDE11A, SPRY4 and BAK1 genes may be inactivated and KITLG potentially activated by these changes in promoter methylation in TGCT cases. Although, the absolute differences in methylation levels between cases and controls were small, they were statistically significant. The strongest associations were observed between KITLG (low methylation) and PDE11A (high methylation) promoter methylation changes and increased TGCT risk. These differences are consistent with the known activating and inactivating effects, respectively, of the cancer risk alleles in these two genes (see below). In general, however, we did not observe strong associations between promoter methylation and gene expression in lymphocytes; there may be a tissue-specific effect of aberrant DNA methylation, with blood cell assays underestimating this tissue-specific effect.
Our methylation findings fit with prior observations and hypotheses regarding how each gene/pathway may modify TGCT risk. The KIT pathway has been suggested to be constitutively activated in human TGCTs as a result of gain-of-function mutations in the KIT oncogene and/or overexpression of KIT [25]. In mice, germline heterozygous deletions of KITLG cause an increase in TGCT incidence [25]. The decreased KITLG promoter methylation we observe in TGCT cases may also activate the KIT pathway. In addition, the significant association between lower promoter methylation and only seminoma tumors fits with previous studies that have observed over-expression of KIT predominantly in seminoma tumors [26,27]. The weak inverse correlation we observed between promoter methylation and KITLG expression suggests that decreased promoter methylation may increase KITLG expression in lymphocytes only modestly. The GWAS SNPs in KITLG previously associated with TGCTs were not correlated with expression changes in lymphoblastoid cell lines [8].
Inactivating PDE11A sequence variants have been associated with risk of familial and bilateral TGCTs [7]; we observed increased PDE11A promoter methylation in TGCT cases, an alteration that is potentially inactivating. Incorporating PDE11A sequence variant data [7] into our analysis showed that TGCT cases without a PDE11A variant had a slightly higher risk of TGCT associated with high promoter methylation, suggesting there may be increased PDE11A inactivation in these cases without a known inactivating variant. Our failure to detect PDE11A expression in lymphocytes may be due to either very low expression levels that were below the qPCR detection threshold or true absence of expression in our samples, since the limited number of control lymphocytes compromised our ability to make an accurate case-control comparison. Thus, we cannot definitively conclude that PDE11A expression in our TGCT cases has been lost. Others have observed very low or no expression of PDE11A in leukocytes [28,29]. The PDE11A mutations previously associated with familial TGCT risk were associated with significantly decreased PDE11A expression in affected tumor tissues [7]; however, we were only able to examine expression in lymphocytes.
SPRY4 and BAK1 are also involved in the KITLG-KIT pathway. The associations observed in SPRY4 and BAK1 were weaker than those just described, but are consistent with potential inactivation of these genes through increased promoter methylation. SPRY4 inhibits the protein kinase pathway that is activated by KITLG-KIT signaling, and BAK1 expression in testicular germ cells has been shown to be repressed by KITLG-KIT signaling [13,14]. Rapley et al. [8] found that the BAK1 allele associated with an increased risk of testis cancer in their GWAS was also associated with a lower expression of BAK1 in lymphoblastoid cell lines. The increased promoter methylation we observed may also reduce the expression of BAK1 in TGCT cases, although we did not observe a change in BAK1 expression in our primary lymphocyte samples.
SPRY4 SNPs previously associated with TGCT risk were not correlated with expression changes [8]. The SPRY4 promoter SNP, rs146162871, has not been previously associated with disease, to our knowledge. It was found in one TGCT multi-case family: the proband and his affected brother both carried this promoter G variant allele, as did their unaffected brother who has not developed a TGCT. The common A allele is highly-conserved among mammalian species, and the 0% MAF observed in individuals of European ancestry, similar to our FTC families, are consistent with the possibility that the G variant allele may be a very rare TGCT risk allele.
Others have found that aberrant constitutional DNA methylation of the BRCA1 [30], MLH1 [31,32] and MSH2 [33,34] cancer predisposition genes accounts for a significant proportion of patients with features of inherited cancers [35]. Recently, BRCA1 promoter methylation in peripheral blood was strongly associated with breast cancer and also with the development of BRCA1 methylated tumors with features similar to BRCA1 mutated tumors [30]. In addition, they observed a mosaic pattern of BRCA1 methylation, with levels ranging from 0.1-17%; methylation in normal tissues can be either complete or in a mosaic form [36]. The low levels of methylation in KITLG, SPRY4, and BAK1 may be suggesting methylation mosaicism in these genes.
Conclusions
We have found that differences in promoter methylation of KITLG, PDE11A, SPRY4 and BAK1 in primary lymphocytes are associated with familial TGCT. Promoter methylation of these candidate genes may modify familial TGCT risk in a manner that is compatible with the influence that variants in these genes exert on TGCT risk. Finally, our data suggest an inverse correlation between KITLG promoter methylation and lymphocyte expression of KITLG. Although we observed only a relatively small effect of methylation on lymphocyte protein expression, there may be stronger tissue-specific effects in the testis that could more plausibly influence gene expression and possibly carcinogenesis. This is the first study to investigate promoter methylation of these genes, and our data provide new insight into the complexity of TGCT genetics, to which epigenetic effects may be contributing. However, this pilot study must be viewed as an hypothesis-generating experiment, the results of which must be confirmed in larger studies, perhaps including sporadic TGCT, before definitive conclusions can be reached.
Acknowledgements
We thank Drs Marston Linehan and Jennifer Loud, Susan Pfeiffer, and June Peters for their contributions to the CGB Familial Testicular Cancer study. We are grateful to the patients and families who participated in this study. We thank Charles Chung at the NCI Advanced Technology Center for assistance with 1000 Genomes data. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Division of Cancer Epidemiology and Genetics, and by support services contracts NO2-CP-11019-50 and NO2-CP-65504-50 with Westat Inc.
Supporting Information
References
- 1.Hemminki K, Li X. Familial risk in testicular cancer as a clue to a heritable and environmental aetiology. Br J Cancer. 2004;90:1765–1770. doi: 10.1038/sj.bjc.6601714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dong C, Hemminki K. Modification of cancer risks in offspring by sibling and parental cancers from 2,112,616 nuclear families. Int J Cancer. 2001;92:144–150. [PubMed] [Google Scholar]
- 3.Fosså S, Chen J, Schonfeld S, McGlynn K, McMaster M, Gail M, Travis L. Risk of contralateral testicular cancer: a population-based study of 29,515 U. S. men. J Natl Cancer Inst. 2005;97:1056–1066. doi: 10.1093/jnci/dji185. [DOI] [PubMed] [Google Scholar]
- 4.Swerdlow A, De Stavola B, Swanwick M, Maconochie N. Risks of breast and testicular cancers in young adult twins in England and Wales: evidence on prenatal and genetic aetiology. Lancet. 1997;350:1723–1728. doi: 10.1016/s0140-6736(97)05526-8. [DOI] [PubMed] [Google Scholar]
- 5.Crockford G, Linger R, Hockley S, Dudakia D, Johnson L, Huddart R, Tucker K, Friedlander M, Phillips K, Hogg D, Jewett M, Lohynska R, Daugaard G, Richard S, Chompret A, Bonaiti-Pellie C, Heidenreich A, Albers P, Olah E, Geczi L, Bodrogi I, Ormiston WJ, Daly PA, Guilford P, Fossa SD, Heimdal K, Tjulandin SA, Liubchenko L, Stoll H, Weber W, Forman D, Oliver T, Einhorn L, McMaster M, Kramer J, Greene MH, Weber BL, Nathanson KL, Cortessis V, Easton DF, Bishop DT, Stratton MR, Rapley EA. Genomewide linkage screen for testicular germ cell tumour susceptibility loci. Hum Mol Genet. 2006;15:443–451. doi: 10.1093/hmg/ddi459. [DOI] [PubMed] [Google Scholar]
- 6.Nathanson KL, Kanetsky PA, Hawes R, Vaughn DJ, Letrero R, Tucker K, Friedlander M, Phillips K-A, Hogg D, Jewett MAS, Lohynska R, Daugaard G, Richard S, Chompret A, Bonaïti-Pellié C, Heidenreich A, Olah E, Geczi L, Bodrogi I, Ormiston WJ, Daly PA, Oosterhuis JW, Gillis AJM, Looijenga LHJ, Guilford P, Fosså SD, Heimdal K, Tjulandin SA, Liubchenko L, Stoll H, Weber W, Rudd M, Huddart R, Crockford GP, Forman D, Oliver DT, Einhorn L, Weber BL, Kramer J, McMaster M, Greene MH, Pike M, Cortessis V, Chen C, Schwartz SM, Bishop DT, Easton DF, Stratton MR, Rapley EA. The Y deletion gr/gr and susceptibility to testicular germ cell tumor. Am J Hum Genet. 2005;77:1034–1043. doi: 10.1086/498455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horvath A, Korde L, Greene MH, Libe R, Osorio P, Faucz FR, Raffin-Sanson ML, Tsang KM, Drori-Herishanu L, Patronas Y, Remmers EF, Nikita M-E, Moran J, Greene J, Nesterova M, Merino M, Bertherat J, Stratakis C. Functional phosphodiesterase 11A mutations may modify the risk of familial and bilateral testicular germ cell tumors. Cancer Res. 2009;69:5301–5306. doi: 10.1158/0008-5472.CAN-09-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rapley EA, Turnbull C, Al Olama AA, Dermitzakis ET, Linger R, Huddart RA, Renwick A, Hughes D, Hines S, Seal S, Morrison J, Nsengimana J, Deloukas P, Rahman N, Bishop DT, Easton DF, Stratton MR. A genome-wide association study of testicular germ cell tumor. Nat Genet. 2009;41:807–810. doi: 10.1038/ng.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanetsky PA, Mitra N, Vardhanabhuti S, Li M, Vaughn DJ, Letrero R, Ciosek SL, Doody DR, Smith LM, Weaver J, Albano A, Chen C, Starr JR, Rader DJ, Godwin AK, Reilly MP, Hakonarson H, Schwartz SM, Nathanson KL. Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. Nat Genet. 2009;41:811–815. doi: 10.1038/ng.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turnbull C, Rapley E, Seal S, Pernet D, Renwick A, Hughes D, Ricketts M, Linger R, Nsengimana J, Deloukas P, Huddart R, Bishop D, Easton D, Stratton M, Rahman N UK Testicular Cancer Collaboration. Testicular Cancer Collaboration: Variants near DMRT1, TERT and ATF7IP are associated with testicular germ cell cancer. Nat Genet. 2010;42:604–607. doi: 10.1038/ng.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kratz C, Han S, Rosenberg P, Berndt S, Burdett L, Yeager M, Korde L, Mai P, Pfeiffer R, Greene M. Variants in or near KITLG, BAK1, DMRT1, and TERT-CLPTM1L predispose to familial testicular germ cell tumour. J Med Genet. 2011;48:473–476. doi: 10.1136/jmedgenet-2011-100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boldajipour B, Raz E. What is left behind-quality control in germ cell migration. Sci STKE. 2007;383:pe16. doi: 10.1126/stke.3832007pe16. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki A, Taketomi T, Kato R, Saeki K, Nonami A, Sasaki M, Kuriyama M, Saito N, Shibuya M, Yoshimura A. Mammalian Sprouty4 suppresses Ras-independent ERK activation by binding to Raf1. Cell Cycle. 2003;2:281–282. [PubMed] [Google Scholar]
- 14.Yan W, Samson M, Jegou B, Toppari J. Bcl-w forms complexes with Bax and Bak, and elevated ratios of Bax/Bcl-w and Bak/Bcl-w correspond to spermatogonial and spermatocyte apoptosis in the testis. Mol Endocrinol. 2000;14:682–699. doi: 10.1210/mend.14.5.0443. [DOI] [PubMed] [Google Scholar]
- 15.Greene M, Kratz C, Mai P, Mueller C, Peters J, Bratslavsky G, Ling A, Choyke P, Premkumar A, Bracci J, Watkins R, McMaster M, Korde L. Familial testicular germ cell tumors in adults: 2010 summary of genetic risk factors and clinical phenotype. Endocr Relat Cancer. 2010;17:R109–R121. doi: 10.1677/ERC-09-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wayman C, Phillips S, Lunny C, Webb T, Fawcett L, Baxendale R, Burgess G. Phosphodiesterase 11 (PDE11) regulation of spermatozoa physiology. Int J Impot Res. 2005;17:216–223. doi: 10.1038/sj.ijir.3901307. [DOI] [PubMed] [Google Scholar]
- 17.Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, Griffin KJ, Stein E, Levine E, Delimpasi G, Hsiao HP, Keil M, Heyerdahl S, Matyakhina L, Libè R, Fratticci A, Kirschner LS, Cramer K, Gaillard RC, Bertagna X, Carney JA, Bertherat J, Bossis I, Stratakis CA. A genomewide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet. 2006;38:794–800. doi: 10.1038/ng1809. [DOI] [PubMed] [Google Scholar]
- 18.Antequera F, Bird A. CpG islands. EXS. 1993;64:169–185. doi: 10.1007/978-3-0348-9118-9_8. [DOI] [PubMed] [Google Scholar]
- 19.Lettini A, Guidoboni M, Fonsatti E, Anzalone L, Cortini E, Maio M. Review: Epigenetic remodeling of DNA in cancer. Histol Histopathol. 2007;22:1413–1424. doi: 10.14670/HH-22.1413. [DOI] [PubMed] [Google Scholar]
- 20.Mirabello L, Savage S, Korde L, Gadalla S, Greene M. LINE-1 methylation is inherited in familial testicular cancer kindreds. BMC Med Genet. 2010;11:77. doi: 10.1186/1471-2350-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lam M-YJ, Heaney JD, Youngren KK, Kawasoe JH, Nadeau JH. Trans-generational epistasis between Dnd1Ter and other modifier genes controls susceptibility to testicular germ cell tumors. Hum Mol Genet. 2007;16:2233–2240. doi: 10.1093/hmg/ddm175. [DOI] [PubMed] [Google Scholar]
- 22.Linger R, Dudakia D, Huddart R, Tucker K, Friedlander M, Phillips K, Hogg D, Jewett M, Lohynska R, Daugaard G, Richard S, Chompret A, Stoppa-Lyonnet D, Bonaïti-Pellié C, Heidenreich A, Albers P, Olah E, Geczi L, Bodrogi I, Daly P, Guilford P, Fosså S, Heimdal K, Tjulandin S, Liubchenko L, Stoll H, Weber W, Einhorn L, McMaster M, Korde L, Greene M, Nathanson K, Cortessis V, Easton D, Bishop D, Stratton M, Rapley E. Analysis of the DND1 gene in men with sporadic and familial testicular germ cell tumors. Genes, Chromosomes and Cancer. 2008;47:247–252. doi: 10.1002/gcc.20526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korde LA, Premkumar A, Mueller C, Rosenberg P, Soho C, Bratslavsky G, Greene MH. Increased prevalence of testicular microlithiasis in men with familial testicular cancer and their relatives. Br J Cancer. 2008;99:1748–1753. doi: 10.1038/sj.bjc.6604704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dupont J, Tost J, Jammes H, Gut I. De novo quantitative bisulfite sequencing using the pyrosequencing technology. Anal Biochem. 2004;333:119–127. doi: 10.1016/j.ab.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 25.Heaney J, Lam M, Michelson M, Nadeau J. Loss of the transmembrane but not the soluble kit ligand isoform increases testicular germ cell tumor susceptibility in mice. Cancer Res. 2008;68:5193–5197. doi: 10.1158/0008-5472.CAN-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikolaou M, Valavanis C, Aravantinos G, Fountzilas G, Tamvakis N, Lekka I, Arapantoni-Dadioti P, Zizi A, Ghiconti I, Economopoulos T, Pectasides D. Kit expression in male germ cell tumors. Anticancer Res. 2007;27:1685–1688. [PubMed] [Google Scholar]
- 27.Biermann K, Heukamp L, Steger K, Zhou H, Franke F, Sonnack V, Brehm R, Berg J, Bastian P, Muller S, Wang-Eckert L, Buettner R. Genome-wide expression profiling reveals new insights into pathogenesis and progression of testicular germ cell tumors. Cancer Genomics Proteomics. 2007;4:359–367. [PubMed] [Google Scholar]
- 28.Yuasa K, Kotera J, Fujishige K, Michibata H, Sasaki T, Omori K. Isolation and characterization of two novel phosphodiesterase PDE11A variants showing unique structure and tissue-specific expression. J Biol Chem. 2000;275:31469–31479. doi: 10.1074/jbc.M003041200. [DOI] [PubMed] [Google Scholar]
- 29.Fawcett L, Baxendale R, Stacey P, McGrouther C, Harrow I, Soderling S, Hetman J, Beavo JA, Phillips SC. Molecular cloning and characterization of a distinct human phosphodiesterase gene family: PDE11A. Proc Natl Acad Sci USA. 2000;97:3702–3707. doi: 10.1073/pnas.050585197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong EM, Southey MC, Fox SB, Brown MA, Dowty JG, Jenkins MA, Giles GG, Hopper JL, Dobrovic A. Constitutional methylation of the BRCA1 promoter is specifically associated with BRCA1 mutation-associated pathology in early-onset breast cancer. Cancer Prev Res. 2011;4:23–33. doi: 10.1158/1940-6207.CAPR-10-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suter C, Martin D, Ward R. Germline epimutation of MLH1 in individuals with multiple cancers. Nat Genet. 2004;36:497–501. doi: 10.1038/ng1342. [DOI] [PubMed] [Google Scholar]
- 32.Hitchins M, Ward R. Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary nonpolyposis colorectal cancer. J Med Genet. 2009;46:793–802. doi: 10.1136/jmg.2009.068122. [DOI] [PubMed] [Google Scholar]
- 33.Chan TL, Yuen ST, Kong CK, Chan YW, Chan AS, Ng WF, Tsui WY, Lo MW, Tam WY, Li VS, Leung SY. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat Genet. 2006;38:1178–1183. doi: 10.1038/ng1866. [DOI] [PubMed] [Google Scholar]
- 34.Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, Lee TY, Bodmer D, Hoenselaar E, Hendriks-Cornelissen SJ, Tsui WY, Kong CK, Brunner HG, van Kessel AG, Yuen ST, van Krieken JH, Leung SY, Hoogerbrugge N. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’ exons of TACSTD1. Nat Genet. 2009;41:112–117. doi: 10.1038/ng.283. [DOI] [PubMed] [Google Scholar]
- 35.Issa J, Garber J. Time to think outside the (genetic) box. Cancer Prev Res (Phila) 2011;4:6–8. doi: 10.1158/1940-6207.CAPR-10-0348. [DOI] [PubMed] [Google Scholar]
- 36.Dobrovic A, Kristensen L. DNA methylation, epimutations and cancer predisposition. Int J Biochem Cell Biol. 2009;41:34–39. doi: 10.1016/j.biocel.2008.09.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.