

Abstract
Tyrosine deprotonation in peptides yields preferential electron detachment upon NETD or UVPD, resulting in prominent N – Cα bond cleavage N-terminal to the tyrosine residue. UVPD of iodo-tyrosine modified peptides was used to generate localized radicals on neutral tyrosine side chains by homolytic cleavage of the C – I bond. Subsequent collisional activation of the radical species yielded the same preferential cleavage of the adjacent N-terminal N – Cα bond. LC-MS/MS analysis of a tryptic digest of BSA demonstrated that these cleavages are regularly observed for peptides when using high pH mobile phases.
Bottom-up workflows for qualitative and quantitative protein identification utilizing liquid chromatography tandem mass spectrometry (LC-MS/MS) have emerged as one of the top technologies of choice for proteome analyses.1 The success of these strategies hinges upon the ability to accurately predict fragmentation patterns of theoretical peptide sequences generated in silico from protein sequence databases.2,3 The bench-mark for peptide cation dissociation is collision induced dissociation (CID),4,5 in addition to electron capture dissociation (ECD)6,7 and electron transfer dissociation (ETD)8,9 which afford complementary information to CID and enhanced identification of labile post-translational modifications (PTMs).10 The dissociation mechanisms and characteristic product ion types of these methods have been investigated in depth and are fairly well understood. A number of preferential cleavage types have been reported for peptide cations. For example, cleavages at protonated histidine and N-terminal to proline residues are commonly observed upon CID of protonated peptides, and in the absence of a mobile proton, enhanced cleavage also occurs at the amide bond immediately C-terminal to acidic residues.11,12 Disulfide bond cleavage in multiply charged peptide and protein cations upon ECD and ETD is readily observed and for lower charge states is more favored than peptide/protein backbone cleavage.13–15
Exploration of the negative ion mode for protein identification affords an attractive alternative due to the fact that a large portion of biologically significant PTMs, such as phosphorylation, sulfonation, nitration, and glycosylation with acidic glycans, as well as many naturally occurring peptides and proteins, are acidic and thus readily form anions. Negative ion mode analyses16,17 provide complementary information to that obtained in the positive ion mode, and the combined data sets allow more comprehensive proteomics analyses by tandem mass spectrometry. However, CID of peptide anions is generally ineffective due to extensive and uninformative neutral losses.18,19 Recently, a number of dissociation methods including electron detachment dissociation (EDD),20,21 negative electron transfer dissociation (NETD),22,23 ultraviolet photo-dissociation (UVPD)24 at 193 nm, negative-ion electron capture dissociation (niECD)25, and electron photodetachment dissociation (EPD)26–28 have been shown to be viable alternatives to CID for peptide anion characterization. Higher pH conditions using basic mobile phases for LCMS enhance deprotonation of peptide sites not routinely deprotonated (e.g. tyrosine side chain with pKa of ~10). Deprotonation of alternative sites may promote alternative fragmentation pathways as described herein.
We report preferential cleavage N-terminal to deprotonated tyrosine residues in peptide anions upon UVPD at 193 nm or upon negative electron transfer dissociation (NETD). The dominant backbone cleavages of peptide anions upon NETD21-22 typically result in a•- and x-type product ions and upon UPVD23 a- and x-type with lower levels of b, c, y, Y, and z ions. Conversely, NETD (Figure 1A), NETD with simultaneous infrared photoactivation (a process termed AI-NETD) (Figure 1B), and UVPD (Figure 1C) of triply deprotonated DRVYIHPFHLVIHN yield abundant c3 and z11• ions which arise uniquely from cleavage N-terminal to the tyrosine residue. This process is likewise notable upon NETD and UVPD of the 3- charge states of peptides NEKYAQAYPNVS, Ac-DRVYIHPFHLVIHN, DRVYIHPFHLLVYS and KTMTESSFYSNMLA (not shown). The formation of high abundance c and z-type productions has not been reported previously for NETD or UVPD at 193 nm; however, the highly selective nature of the fragmentation suggests that a favorable radical-directed cleavage process may occur upon electron detachment from tyrosine-containing peptides.
Figure 1.

NETD (A), NETD with simultaneous infrared photoactivation (B) and UVPD (C) spectra of triply deprotonated DRVYIHPFHLVIHN. * and ’ represent the precursor and loss of water, respectively.
The electron hole recombination energy upon electron transfer from peptide anions to fluoranthene radical cations has been previously estimated to be 2.5 - 4.5 eV based on the difference in ionization energy (IE) of fluoranthene (IE = 7.9 eV) and the electron affinities (EA) of carboxylate (EA = 3.4 eV) and phosphate (EA = 5.4 eV) groups in phosphopeptides.23 The energy of a 193 nm photon is 6.4 eV. In a recent top-down EDD study, Breuker et al. tabulated the electron affinities (EA) of radical functional groups in peptides and proteins.29 The EA of each radical functional group is equivalent to the ionization energy of the corresponding deprotonated functional group, and all are less than 3.5 eV. In contrast, ionization energies of neutral amino acids are between 7 and 10 eV.30–32 Therefore, electron detachment, a key step in the selective backbone cleavage showcased in this report, likely occurs at sites of deprotonation.
The likely sites of deprotonation in DRVYIHPFHLVIHN include the side-chains of aspartic acid and tyrosine, and the C-terminus, which are all expected to be deprotonated in the triply charged species. Delocalization of the negative charges over backbone amides may also occur as a means of charge solvation.21 The IEs of deprotonated aspartic acid and the C-terminus have been estimated to be 3.34 eV, and the IE of deprotonated tyrosine as 2.17 eV.29 Due to the lower IE and greater absorption cross section of deprotonated tyrosine, electron detachment is most favored at the deprotonated phenol of tyrosine over the carboxylates of aspartic acid or the C-terminus. In the doubly deprotonated species a majority of the ions are expected to be deprotonated at the carboxylates of aspartic acid and the C-terminus, not the tyrosine side-chain. In fact, NETD and UVPD of doubly deprotonated DRVIHPFHLVIHN (SI Figure 1A and B) yielded very low abundance c3 and z11• product ions. This outcome is expected if it is tyrosine deprotonation that specifically promotes the preferential cleavage.
Breuker et al. proposed a mechanism for the formation of c and z ions N-terminal to tyrosine residues upon EDD (see Ref 29, Scheme S2), and it is reasonable to consider a similar pathway during NETD and 193 nm UVPD. In fact, deuterium labeling of the beta carbon atom of tyrosine in our experiments yields results entirely consistent with the previously proposed mechanism (see SI Fig 2). The substantially increased abundances of the c3 and z11• ions upon NETD with simultaneous IR photoactivation (Figure 1B) compared to NETD alone (Figure 1A) suggests that the additional vibrational energy provided by IR photoactivation allows a greater portion of tyrosyl radical ions to undergo the radical migration/N – Cα backbone cleavage process.
CID of the charge reduced precursor ([M - 3H]2-•) produced by NETD (Figure 2A) or by UVPD (Figure 2B) of triply deprotonated DRVYIHPFHLVIHN yields the same abundant c3 and z11• ions along with neutral losses of water, carbon dioxide and tyrosine side chain. This indicates that a portion of the [M - 3H]2-• ions formed upon electron detachment from the triply deprotonated peptide precursors contain stable radicals localized on the tyrosine side chain or c and z ions held together through non-covalent interactions. Subsequent collisional activation of these species provides enough internal energy to surpass the activation barriers for radical migration or non-covalent bond cleavage.
Figure 2.
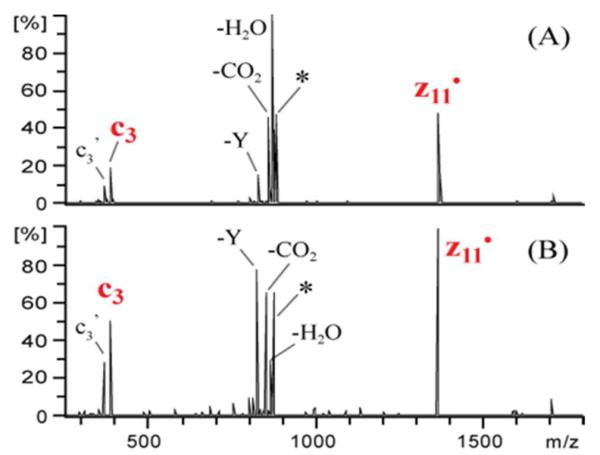
MS3 spectrum upon CID of the charge-reduced precursor ([M - 3H]2-.) produced by (A) NETD and (B) UVPD of triply deprotonated DRVYIHPFHLVIHN. * and ’ represent the precursor and loss of water, respectively.
Previous work has shown that 266 nm UVPD of iodotyrosine-containing peptide and protein cations results in homolytic cleavage of the C – I bond, thus yielding a localized radical on the tyrosine residue.33,34 Subsequent CID of these peptide/protein radical cations led to dominant backbone fragmentation in the form of a ions C-terminal to the tyrosine residue.33,34 This process is illustrated in Equation 1 below.
| (1) |
In addition to selective a ion formation adjacent to the modified tyrosine residue, abundant secondary cleavages producing complementary a and y ions were observed N-terminal to proline residues in the vicinity of the modified tyrosine residue. These experiments were recently extended to peptide anions,35 thus providing a convenient method for creating radicals on tyrosine side chains in peptide anions in the present study. The resulting ions allow determination of whether the selective formation of c and z ions N-terminal to tyrosine upon NETD and 193 nm UVPD proceeds through a tyrosyl radical.
UVPD at 193 nm (Figure 3A) of doubly deprotonated DRVYIIHPFHLVIHN (where YI represents iodo-tyrosine) yields predominantly an intact charge reduction product ([M – 2H + I]2-.) via electron photodetachment and a wide array of low abundance a, x, c and z ions as expected. Homolytic cleavage of the C – I bond (i.e. [M - 2H]2-•) also occurs at approximately 10% relative abundance. Very low abundance c3 and [z11 + I]• ions are observed and are expected based on the results from the non-iodinated peptide (i.e. in the 2- charge state YI is not expected to be deprotonated, and thus Y-directed cleavage is not favored.). Based on previously observed results with iodinated peptides, collisional activation of [M - 2H]2-• is expected to yield selective a ion formation C-terminal to the iodinated tyrosine residue. Instead, CID of [M - 2H]2-• (Figure 3B) yields dominant c3 and z11• ions corresponding to cleavage N-terminal to the tyrosine residue. In fact, the a4 and x10 ions which arise from cleavage C-terminal to the tyrosine residue are observed at only 1% relative abundance. In addition, y7 ions are observed at 90% relative abundance which corresponds to cleavage C-terminal to proline. The UVPD/CID experiments of other iodo-tyrosine peptides similarly reproduced the same tyrosine selective cleavages that occur from non-iodinated peptides containing a deprotonated tyrosine. However, the abundant y7 ion formation C-terminal to proline is not observed upon NETD or UVPD of the triply deprotonated non-iodinated peptide or upon CID of [M - 3H]2-• ion produced by NETD or UVPD of triply charged DRVYIHPFHLVIHN. CID of the doubly deprotonated even electron species (SI Figure 3) also fails to yield y7 ions, a results which indicates the y7 ions are produced by radical directed dissociation (RDD). This is consistent with the analogous results reported previously showing that y ion formation N-terminal to proline proceeds through a radical mechanism.33,35 The fact that the tyrosine residue is not deprotonated in the 2- charge state of the iodo-tyrosine-containing peptide and is deprotonated in the 3- charge state of the non-iodinated peptide likely accounts for the observation of the abundant y7 ion. UVPD of triply deprotonated DRVYIIHPFHLVIHN (Figure 3C) shows that the presence of iodine on the tyrosine side chain does not impede the selective cleavage N-terminal to the deprotonated tyrosine residue. Dominant c3 and [z11+I]• ions are produced. The absence of y7 ions upon UVPD of the 3- charge state of the iodinated peptide supports our hypothesis that the phenol oxygen needs to be protonated for y ion formation. Collectively, these results suggest that the formation of tyrosyl radicals, whether promoted by NETD or UVPD of peptides containing a deprotonated tyrosine residue, is a key step that precedes the selective cleavage N-terminal to the tyrosine residue.
Figure 3.

193 nm UVPD (A) of doubly deprotonated DRVYIIHPFHLVIHN (YI represents iodo-tyrosine) and (B) subsequent CID of [M - 2H]2-•. (C) 193 nm UVPD of [M - 3H + I]3-. *, ’ and I represent the precursor, loss of water or iodine, respectively.
To gauge the frequency with which the preferential cleavage N-terminal to tyrosine might occur in a bottom-up proteomics experiment, we performed LC-MS/NETD and LC-MS/UVPD of a bovine serum albumin (BSA) tryptic digest using 0.05% ammonium hydroxide mobile phases (~pH 10.5). LC-MS/MS spectra were interpreted using the MassMatrix database search engine36,37 which was adapted to search the appropriate ion series for each dissociation method. Tyrosine-containing peptide matches in which the charge state exceeded the number of acidic functionalities (i.e. E, D and C-terminus) were manually interpreted to confirm preferential cleavage N-terminal to deprotonated tyrosine residues. The peptide sequence, charge state, and the percentage of total sequence ion peak area for Y-selective c and z ions for those peptides that produced preferential cleavage upon NETD are shown in Table 1. Figure S4 shows the NETD spectrum of the 4- charge state of RHPYFYAPELLYYANK from BSA. For peptides with multiple tyrosine residues, c and z ion formation was observed N-terminal to all tyrosine residues. Of the 16 tyrosine-containing peptides identified by each method, six yielded preferential cleavage N-terminal to tyrosine residues. However, three of the 16 peptides contained N-terminal tyrosine residues, thus c and z ion formation N-terminal to the tyrosine residue is not possible. The seven remaining tyrosine-containing peptides did not produce charge states consistent with tyrosine deprotonation, explaining why preferential cleavage was not observed upon NETD or UVPD.
Table 1.
BSA tryptic peptides exhibiting preferential cleavage N-terminal to tyrosine upon NETD.
| Peptide Sequence | Charge State1 |
|---|---|
| HPYFYAPELLYYANK | 3-, 4- (39, 56%) |
| RHPYFYAPELLYYANK | 3-, 4- (69, 88%) |
| GLVLIAFSQYLQQCPFDEHVK | 4- (20%) |
| RHPEYAVSVLLR | 3- (63%) |
| LGEYGFQNALIVR | 3- (23%) |
| LGEYGFQNALIVRYTR | 4- (53%) |
The percentage of sequence ion peak area due to the c and z ions from Y-selective cleavage is given in parenthesis for each charge state.
In summary, tyrosine deprotonation during negative ion mode analyses yields selective and enhanced c and z ion formation N-terminal to the tyrosine residue upon NETD and 193 nm UVPD. The lower ionization energy and greater absorption cross section of deprotonated tyrosine phenol compared to the carboxylates of acidic residues and the C-terminus favors preferential electron detachment upon NETD or UVPD. UVPD/CID experiments utilizing iodo-tyrosine derivatives confirmed that selective and enhanced c and z ion formation proceeds through a tyrosyl radical. LC-MS/MS experiments showed that this cleavage specificity can be expected to occur frequently during bottom-up proteomics experiments and could be readily incorporated into database search algorithms to facilitate peptide anion identification. This fragmentation pathway may prove to be a valuable diagnostic for the characterization of post-translational modifications of tyrosine, such as nitration which has been shown to lower the pKa of the phenol hydroxyl function.38
Supplementary Material
Acknowledgments
Funding Sources Funding from NIH (R21GM099028) and the Robert A. Welch Foundation (F1155) is gratefully acknowledged.
Footnotes
Supporting Information Experimental details and additional spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Mann M, Kelleher NL. PNAS. 2008;105:18132–18138. doi: 10.1073/pnas.0800788105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Eng JK, McCormack AL, Yates JR., III J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- (3).Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. ELECTROPHORESIS. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- (4).McLuckey SJ. Am. Soc. Mass Spectrom. 1992;3:599–614. doi: 10.1016/1044-0305(92)85001-Z. [DOI] [PubMed] [Google Scholar]
- (5).Hunt DF, Buko AM, Ballard JM, Shabanowitz J, Giordani AB. Biol. Mass Spectrom. 1981;8:397–408. doi: 10.1002/bms.1200080909. [DOI] [PubMed] [Google Scholar]
- (6).Zubarev RA, Kelleher NL, McLafferty FW. J. Am. Chem. Soc. 1998;120:3265–3266. [Google Scholar]
- (7).Breuker K, Oh H, Lin C, Carpenter BK, McLafferty FW. PNAS. 2004;101:14011–14016. doi: 10.1073/pnas.0406095101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. PNAS. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Good DM, Wirtala M, McAlister GC, Coon JJ. Molecu lar & Cellular Proteomics. 2007;6:1942–1951. doi: 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- (10).Zhou Y, Dong J, Vachet RW. Current Pharmaceutical Biotechnology. 2011;12:1558–1567. doi: 10.2174/138920111798357230. [DOI] [PubMed] [Google Scholar]
- (11).Vaisar T, Urban JJ. Mass Spectrom. 1996;31:1185–1187. doi: 10.1002/(SICI)1096-9888(199610)31:10<1185::AID-JMS396>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- (12).Gu C, Tsaprailis G, Breci L, Wysocki VH. Anal. Chem. 2000;72:5804–5813. doi: 10.1021/ac000555c. [DOI] [PubMed] [Google Scholar]
- (13).Zubarev RA, Kruger NA, Fridriksson EK, Lewis MA, Horn DM, Carpenter BK, McLafferty FW. J. Am. Chem. Soc. 1999;121:2857–2862. [Google Scholar]
- (14).Chrisman PA, Pitteri SJ, Hogan JM, McLuckey SA. J. Am. Soc. Mass Spectrom. 2005;16:1020–1030. doi: 10.1016/j.jasms.2005.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Simons J. Chem. Phys. Lett. 2010;484:81–95. [Google Scholar]
- (16).Ewing N, Cassady CJ. Am. Soc. Mass Spectrom. 2001;12:105–116. doi: 10.1016/S1044-0305(00)00195-1. [DOI] [PubMed] [Google Scholar]
- (17).Janek K, Wenschuh H, Bienert M, Krause E. Rapid Com mun. Mass Spectrom. 2001;15:1593–1599. doi: 10.1002/rcm.417. [DOI] [PubMed] [Google Scholar]
- (18).Bowie JH, Brinkworth CS, Dua S. Mass Spectrom. Rev. 2002;21:87–107. doi: 10.1002/mas.10022. [DOI] [PubMed] [Google Scholar]
- (19).Clipston NL, Jai-nhuknan J, Cassady CJ. Int. J. Mass Spectrom. 2003;222:363–381. [Google Scholar]
- (20).Budnik BA, Haselmann KF, Zubarev RA. Chem. Phys. Lett. 2001;342:299–302. [Google Scholar]
- (21).Kjeldsen F, Silivra OA, Ivonin IA, Haselmann KF, Gorshkov M, Zubarev RA. Chem. Eur. J. 2005;11:1803–1812. doi: 10.1002/chem.200400806. [DOI] [PubMed] [Google Scholar]
- (22).Coon JJ, Shabanowitz J, Hunt DF, Syka JEP. J. Am. Soc. Mass Spectrom. 2005;16:880–882. doi: 10.1016/j.jasms.2005.01.015. [DOI] [PubMed] [Google Scholar]
- (23).Huzarska M, Ugalde I, Kaplan DA, Hartmer R, Easterling ML, Polfer NC. Anal. Chem. 2010;82:2873–2878. doi: 10.1021/ac9028592. [DOI] [PubMed] [Google Scholar]
- (24).Madsen JA, Kaoud TS, Dalby KN, Brodbelt JS. Prote omics. 2011;11:1329–1334. doi: 10.1002/pmic.201000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Yoo HJ, Wang N, Zhuang S, Song H, Håkansson K. J. Am. Chem. Soc. 2011;133:16790–16793. doi: 10.1021/ja207736y. [DOI] [PubMed] [Google Scholar]
- (26).Antoine R, Joly L, Tabarin T, Broyer M, Dugourd P, Lemoine J. Rapid Commun. Mass Spectrom. 2007;21:265–268. doi: 10.1002/rcm.2810. [DOI] [PubMed] [Google Scholar]
- (27).Larraillet V, Antoine R, Dugourd P, Lemoine J. Anal. Chem. 2009;81:8410–8416. doi: 10.1021/ac901304d. [DOI] [PubMed] [Google Scholar]
- (28).Larraillet V, Vorobyev A, Brunet C, Lemoine J, Tsybin YO, Antoine R, Dugourd P. J. Am. Soc. Mass Spectrom. 2010;21:670–680. doi: 10.1016/j.jasms.2010.01.015. [DOI] [PubMed] [Google Scholar]
- (29).Ganisl B, Valovka T, Hartl M, Taucher M, Bister K, Breuker K. Chem. Eur. J. 2011;17:4460–4469. doi: 10.1002/chem.201003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Slifkin MA, Allison AC. Nature. 1967;215:949–950. [Google Scholar]
- (31).Klasinc LJ. Electron. Spectrosc. Relat. Phenom. 1976;8:161–164. [Google Scholar]
- (32).Dehareng D, Dive G. Int. J. Mol. Sci. 2004;5:301–332. [Google Scholar]
- (33).Ly T, Julian RR. J. Am. Chem. Soc. 2008;130:351–358. doi: 10.1021/ja076535a. [DOI] [PubMed] [Google Scholar]
- (34).Liu Z, Julian RR. J. Am. Soc. Mass Spectrom. 2009;20:965–971. doi: 10.1016/j.jasms.2008.12.014. [DOI] [PubMed] [Google Scholar]
- (35).Moore B, Sun Q, Hsu JC, Lee AH, Yoo GC, Ly T, Julian RR. J. Am. Soc. Mass Spectrom. 2011;23:460–468. doi: 10.1007/s13361-011-0318-2. [DOI] [PubMed] [Google Scholar]
- (36).Xu H, Freitas M. BMC Bioinformatics. 2007;8:133. doi: 10.1186/1471-2105-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Xu H, Freitas MA. Proteomics. 2009;9:1548–1555. doi: 10.1002/pmic.200700322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sokolovsky M, Riordan JF, Vallee BL. Biochem. Biophys. Res. Commun. 1967;27:20–25. doi: 10.1016/s0006-291x(67)80033-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.