

Abstract
The dynamic interaction between nuclear proteins and chromatin leads to the functional plasticity necessary to mount adequate responses to regulatory signals. Here, we review the factors regulating the chromatin interactions of the high mobility group proteins (HMGs), an abundant and ubiquitous superfamily of chromatin-binding proteins in living cells. HMGs are highly mobile and interact with the chromatin fiber in a highly dynamic fashion, as part of a protein network. The major factors that affect the binding of HMGs to chromatin are operative at the level of the single nucleosome. These factors include structural features of the HMGs, competition with other chromatin-binding proteins for nucleosome binding sites, complex formation with protein partners, and post-translational modifications in the protein or in the chromatin-binding sites. The versatile modulation of the interaction between HMG proteins and chromatin plays a role in processes that establish the cellular phenotype.
Keywords: chromatin, HMG proteins, protein–DNA interactions
Introduction
The dynamic and continuous interaction of nuclear regulatory factors and structural proteins with the chromatin fiber plays a key role in regulating the fidelity of gene expression and in establishing the cellular phenotype. Therefore, studies of the factors that regulate the interaction of chromatin-binding proteins with their targets provide insights into the molecular mechanisms that regulate chromatin function. One of the most perplexing aspects of chromatin biology is the mechanism of action and the cellular function of a group of nuclear proteins, known as architectural nucleosome-binding proteins. This class of proteins, which includes variants of the highly abundant linker his-tone H1 family (Kasinsky et al. 2001) and members of the high mobility group (HMG) protein superfamily (Bustin 1999; Bianchi and Agresti 2005), are structural proteins that bind to nucleosomes without any known specificity for the underlying DNA sequence. The interactions of architectural proteins with nucleosomes induce local and global changes in chromatin structure that affect various DNA-dependent activities, including transcription, recombination, and repair. These proteins may also play a role in epigenetic regulation, since their interaction with chromatin affects the levels of histone modifications (Lim et al. 2004, 2005; Fan et al. 2005).
Most of the information on the binding of the architectural proteins to chromatin was obtained from in vitro experiments with purified components or with reconstituted chromatin. However, in vitro studies cannot fully reflect the complex binding modes and cross interactions occurring in living cells. Fluorescence microscopy techniques that enable investigations on protein mobility and binding in living cells, in real time, provide new important insights into the interaction of nuclear proteins with chromatin. The results obtained with these approaches indicate that nuclear proteins in living cells are highly mobile, that their interaction with any specific site in chromatin is temporary, and that they move continuously among chromatin-biding sites in a stop-and-go fashion (Lever et al. 2000; Phair et al. 2004). Nevertheless, most of the time, most of the proteins are bound to chromatin because their dwell time on nucleosomes is longer than their transit time among nucleosome. Thus, at any particular binding site, there is a continuous turnover of the proteins, some of which compete with each other for binding to nucleosomes.
In vivo experiments have revealed that H1 and HMGs, and perhaps other nuclear components, form a dynamic network of proteins that continuously move among nucleosomes, constantly molding and reconfiguring their chromatin-binding site (Catez et al. 2004; Bustin et al. 2005). The chromatin interactions of members of this network are interdependent and are affected by structural and biochemical changes in the binding sites, as well as by changes in the levels and properties of the various members of the network. This network of continuous and dynamic interactions between architectural proteins and their nucleosomal binding sites is part of the mechanism that provides functional plasticity to the chromatin fiber.
Linker histone H1 is the major architectural protein family present in metazoan cells, most of which contain sufficient H1 to bind most of the nucleosomes. The major factors affecting the binding of H1 variants to chromatin have been previously summarized (Catez et al. 2006). The HMG protein superfamily, which consists of 3 families, named HMGA, HMGB, and HMGN, is also abundantly and ubiquitously expressed in most eukaryotic cells. Although each HMG family is clearly distinct and contains unique chromatin-binding motifs (Bustin 1999), they all have been shown to affect the properties of the chromatin fiber. The biochemical properties and the biological function of HMG proteins, and their involvement in the etiology of certain diseases, have been described in several reviews (Bustin 1999; Reeves and Beckerbauer 2001; Sgarra et al. 2004; Bianchi and Agresti 2005; Hock et al. 2007). Here, we review the information on the major factors that regulate the interaction of the 3 canonical HMG protein families (for definitions of canonical HMGs, see http://home.ccr.cancer.gov/metabolism/bustin/hmg_proteins.htm) with chromatin, and focus the attention on the more recent results obtained in studies with living cells. The functional motifs characteristic of the 3 HMG families are found in numerous nuclear proteins; therefore, elucidation of the factors that regulate the interaction of HMGs with chromatin may be of general relevance to understanding the molecular mechanisms by which architectural proteins affect chromatin function.
HMGA proteins
AT hooks
HMGA proteins are ubiquitously present in the nuclei of both plants and animals (Bustin and Reeves 1996; Reeves and Beckerbauer 2001). Their expression is related to cellular differentiation and proliferation, and they are involved in the regulation of gene expression, in differentiation processes, and in carcinogenesis (Reeves and Beckerbauer 2001; Sgarra et al. 2004; Fusco and Fedele 2007; Hock et al. 2007). The hallmark of the HMGA protein family is the AT hook, which serves as their major chromatin-binding motif. The canonical HMGA family is encoded by 2 distinct genes: HMGA1 and HMGA2. Alternative splicing of the HMGA1 transcript gives rise to 3 variants: HMGA1a, 1b, and 1c (Reeves and Beckerbauer 2001). The metazoan HMGAs have a molecular mass of ~11 kDa, contain 3 conserved AT hook DNA-binding domains, and have an acidic C-terminal tail (Fig. 1).
Fig. 1.
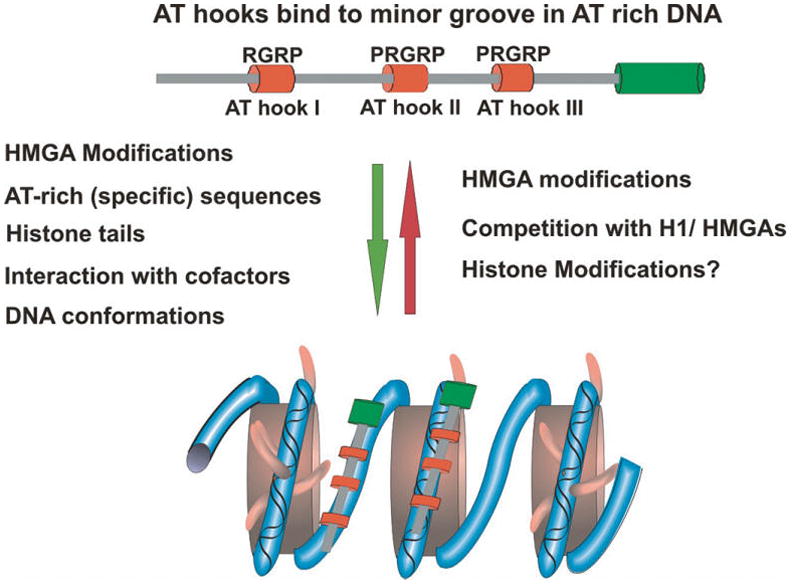
Determinants of HMGA–chromatin interactions. Shown is a schematic diagram of HMGA proteins. The AT hooks appear as red cylinders. The factors listed to the left of the green arrow promote the interaction with HMGA with chromatin, while those listed to the right of the red arrow promote dissociation of HMGA from chromatin.
The AT-hook domain is a small motif containing the consensus PRGRP sequence flanked by additional positively charged residues (Reeves and Nissen 1990; Huth et al. 1997). This consensus sequence is necessary and sufficient for the specific binding of the proteins to DNA (Reeves and Nissen 1990). The central RGR tripeptide penetrates into the minor groove of AT base pairs, where the Arg residues form electrostatic and hydrophobic contacts with the floor of the minor groove DNA (Huth et al. 1997). Early footprinting studies suggested that HMGA proteins bind to any short run of AT-rich DNA, and that the AT hook motifs bind preferentially to AT-rich B-form DNA (Solomon et al. 1986; Reeves and Nissen 1990); however, more recent studies identified 2 consensus sequences for HMGA2 binding, suggesting some degree of sequence specificity for their binding (Cui and Leng 2007). Indeed, in living cells, the proteins display specific effects on the cellular transcription profile and affect distinct cellular processes (Merika and Thanos 2001; Reeves and Beckerbauer 2001; Sgarra et al. 2004; Hock et al. 2007). In addition, the AT-hook motif facilitates the binding of HMGA proteins to DNA substrates with altered structures, including synthetic 4-way junctions (Claus et al. 1994; Hill and Reeves 1997; Hill et al. 1999; Banks et al. 2000), supercoiled plasmids (Nissen and Reeves 1995), and isolated nucleosomes (Reeves and Nissen 1993; Reeves and Wolffe 1996; Li et al. 2006).
Upon binding to their targets, the HMGA proteins undergo conformational changes (Huth et al. 1997), which facilitate their ability to bend DNA (Reeves and Beckerbauer 2001). The enthalpy-entropy compensation associated with the binding of HMGA to DNA is similar to that of minor groove-binding drugs, such as netropesin, distamycin A, and Hoechst 33258 (Cui et al. 2005), which compete with HMGA proteins for chromatin-binding sites both in vitro and in vivo (Reeves and Nissen 1990; Radic et al. 1992). Although a single AT hook is sufficient for the binding of the protein to DNA, the simultaneous binding of 2 or more hooks increases the strength of this interaction (Claus et al. 1994; Maher and Nathans 1996; Yie et al. 1997; Frank et al. 1998). Efficient binding of HMGA proteins to DNA is dependent on the number, the arrangement, and the proper phasing of their AT hooks (Thanos and Maniatis 1995; Kim and Maniatis 1997; Reeves and Beckerbauer 2001).
Photobleaching analyses of living cells revealed that HMGA is included in the pool of chromatin-binding proteins that interact with their targets transiently and constantly move through the cell nucleus (Catez et al. 2004; Harrer et al. 2004). Although the binding of a particular HMGA molecule to any particular site is temporary, most of the HMGA proteins are bound to heterochromatin; in mitotic cells, the protein remains associated with the meta-phase chromatin. Detailed analyses of HMGA point mutants revealed that, in living cells, the Arg residues in the AT hook play a key role in their chromatin interaction. Mutations of a single Arg residue weakened, but did not abolish, the preferential interaction of the protein with heterochromatin. Any double point mutant bearing Arg mutations in 2 different AT hooks did not localize to heterochromatin, and was diffusely distributed throughout the entire nucleus (Harrer et al. 2004). Thus, the AT hooks are the major determinants affecting the interaction of HMGA with chromatin both in vivo and in vitro.
Mutliple post-translational modifications modulate the binding of HMGAs to chromatin
Beyond the AT hooks, the binding of HMGA proteins to chromatin is modulated by a large number of post-translational modifications. HMGA proteins are among the most heavily and extensively modified proteins in the cell nucleus; they are known to be modified by phosphorylation, acetylation, methylation, ribosylation, and sumoylation (Zhang and Wang 2008). The modification patterns change during the cell cycle in response to changes in the physiological state of a cell, and are also cell-type specific (Banks et al. 2000; Diana et al. 2001; Reeves and Beckerbauer 2001; Edberg et al. 2004, 2005; Jiang and Wang 2006; Zou et al. 2007; Cao et al. 2008).
In vitro studies demonstrated that several kinases, including CDC2, PKC, casein kinase 2, and HIPK2, modify distinct residues in HMGA proteins, and that these modifications alter the binding of the proteins to their targets (Schwanbeck et al. 2000; Reeves and Beckerbauer 2001; Zhang and Wang 2007). During mitosis, PKC and CDC2 hyperphosphorylate Ser and Thr residues next to each AT hook, suggesting that some of the modifications are cell-cycle related (Nissen et al. 1991). Photobleaching experiments demonstrated that, in living cells, the Ser and Thr residues that are modified by these kinases do indeed play a role in regulating the interaction of HMGA1 with chromatin. Inhibition of PKC or CDC2 kinase activities increased the HMGA1a mobility preferentially in areas containing compacted chromatin, indicating that suppression of HMGA1 phosphorylation reduces its binding to heterochromatin. Fluorescence recovery after photobleaching (FRAP) studies using point-mutated proteins of mitosis-specific phosphorylation sites support the conclusion that phosphorylation promotes the binding of HMGA1 proteins to mitotic chromosomes and to interphase heterochromatin. Furthermore, the phosphorylation patterns seem to fine tune HMGA1 dynamics and regulate their binding specificity to either DNA or nucleosomes (Harrer et al. 2004).
Another prominent modification found in HMGA proteins is arginine methylation (Banks et al. 2000; Sgarra et al. 2003a, 2003b). Significantly, many of the methyltransferases target the Arg residues found in the AT-hook motifs. The type I protein arginine methyltransferases PMRT1, 3, and 6 have been shown to be involved in most of these methylations. PMRT1 and PMRT3 preferentially methylate Arg 23 and 25, located in the first AT hook of HMGAs. PMRT6 was able to methylate all the Arg residues in all 3 AT hooks, but with some preference for the second AT hook (Miranda et al. 2005; Sgarra et al. 2006; Zou et al. 2007). Some of the methylations were linked to cellular changes, such as apoptosis (Sgarra et al. 2003a, 2003b). Arginine 25 methylation was described in HMGA1a, but not HMGA1b, suggesting functional specificity between these 2 splice variants (Zou and Wang 2007). Given that the Arg residues located in the AT hooks play an important role in stabilizing the interaction of HMGAs with chromatin, it can be expected that methylation of these residues would affect the interaction of the proteins with their chromatin targets sites; however, the effect of this modification on chromatin binding has not been yet examined.
In addition, it is well documented that acetylation of lysine residues affects the binding of HMGA proteins to their target. Elegant studies on the activation of interferon-β revealed that the ordered acetylation of HMGAs on specific lysine residues by PCAF/GCN5 and CBP plays an important role in the activation of endogenous promoters (Munshi et al. 1998, 2001). A correlation between the acetylation of Lys 64 and 70 and the metastasis state of breast cancer cells has also been reported (Edberg et al. 2004). Recent studies suggest that p300 and PCAF acetylate Lys residues at multiple positions with various efficiencies (Zhang et al. 2007). FRAP analyses of cells treated with histone deacetylase inhibitors revealed that hyperacetylation increased significantly the dynamic properties of HMGA1 proteins in both heterochromatin and euchromatin (Harrer et al. 2004). Thus, acetylation of HMGAs and (or) their nucleosomal targets plays an important regulatory role in their chromatin interactions.
Interaction with regulatory proteins
The binding of HMGAs to chromatin is also regulated by their interactions with other chromatin components. One of the best examples is the role of HMGA1 in enhanceosome formation on the promoter of the interferon-β gene, where HMGA1 proteins form a multiprotein complex with specific transcription factors. Enhanceosome formation requires allosteric changes of the DNA, which are induced by HMGA1 binding and followed by protein–protein interactions between HMGA1 nuclear factor κB and ATF2/c-Jun to stabilize the enhanceosome (Yie et al. 1997). Likewise, HMGA proteins target the formation of the origin recognition complex (ORC) via direct interaction with the components Orc1 and Orc6 of this protein complex (Thomae et al. 2008). Significantly, functional AT hooks are required for the targeting of ORC, an indication that the interaction of HMGAs with chromatin plays a major role in this targeting.
Competition with chromatin-binding proteins
In the living nucleus, HMGAs are highly mobile and continuously move among nucleosomes. Photobleaching experiments indicated that, in living cells, HMGA variants compete with histone H1 variants for chromatin-binding sites (Catez et al. 2004). Competition of HMGA proteins and histone H1 for binding sites could involve the conserved SPTXK motifs that play an essential role in the binding of H1 to the minor groove of DNA. This motif, which is repeated several times in the amino and carboxyl tails of histone H1, adopts a β-turn structure, similar to that in Hoechst 33258, and preferentially binds to AT-rich sequences (Churchill and Suzuki 1989), suggesting that it could compete with AT hooks for chromatin binding. Although not yet experimentally proven, it is likely that the HMGA variants compete among themselves, and perhaps also with other nuclear proteins, for chromatin-binding sites. Potentially, changes in these competitors could affect the interaction of the HMGAs with chromatin.
HMGB proteins
The HMG box
The high mobility group box (HMGB) protein family is the most abundant family of HMGs and a major nucleosome-binding constituent of the metazoan nucleus. This protein family has been shown to affect various chromatin-related processes, such as transcription and replication, and plays a role in determining the cellular phenotype (Bustin 1999; Thomas and Travers 2001; Agresti and Bianchi 2003; Bianchi and Agresti 2005; Hock et al. 2007). Interestingly, HMGB proteins also have important functions as extracellular signaling molecules (Lotze and Tracey 2005; Bianchi and Manfredi 2007). The canonical metazoan HMGB protein family consists of 3 major variants, HMGB1, HMGB2, and HMGB3, each encoded by a specific gene. The functional hallmark of this protein family is the HMG-Box, which serves as the major binding site of the proteins to DNA and chromatin. The canonical HMGB proteins have a molecular mass of ~25 kDa, contain 2 DNA-binding domains, called HMG box A and B, and a long acidic tail, which is a continuous array of ~30 acidic amino acids (Thomas and Travers 2001) (Fig. 2).
Fig. 2.
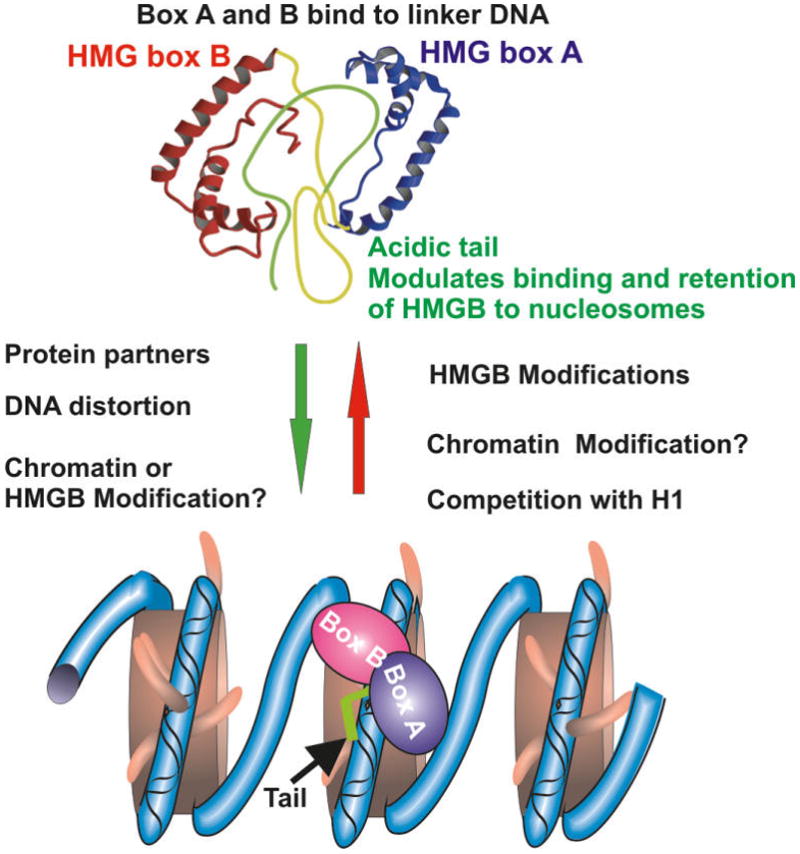
Determinants of HMGB–chromatin interactions. Shown is a schematic diagram of HMGB proteins. The 3 structural domains of HMG box A and HMG box B are indicated. The factors listed to the left of the green arrow promote the interaction of HMGB with chromatin, while those listed to the right of the red arrow promote dissociation of HMGB from chromatin. HMGB structure was adapted from Lee and Thomas (2000).
The HMGB box consists of 3 alpha-helices arranged in an L-like structure (Thomas and Travers 2001). In vitro studies using purified double-stranded DNA revealed that both box A and box B bind to and distort the minor groove of the DNA in a sequence-independent manner. Although either HMG box A or B can bind to DNA by itself, the binding is enhanced by a tandem array of the 2 boxes (Yoshioka et al. 1999). The details of these interactions and the specific features of the various boxes are described elsewhere (Thomas and Travers 2001). The acidic tail of HMGB1 contacts the DNA-binding surfaces of both HMG boxes and reduces the affinity of HMGB1 to double-strand DNA, whereas it enhances the affinity of the protein to distorted DNA (Lee and Thomas 2000; Watson et al. 2007). It seems that the acidic tail modulates the biological functions of HMGBs (Aizawa et al. 1994; Shirakawa et al. 1997); however, its role in the chromatin binding of these proteins in living cells has not been fully clarified.
Beyond the intrinsic structure of the proteins, the in vitro interaction of HMGBs with DNA and chromatin is affected by several factors, including post-translational modifications in the HMGs, interaction with regulatory factors, and DNA conformations (Bustin 1999; Thomas and Travers 2001; Bianchi and Agresti 2005; Assenberg et al. 2008). The organization of HMGB in the nucleus of live cells is highly dynamic and, like other nuclear proteins, HMGBs bind transiently to chromatin (Scaffidi et al. 2002; Phair et al. 2004). Factors known to affect the interaction of HMGBs with DNA in vitro also affected their chromatin binding in living cells. Thus, a mutant HMGB1, bearing mutations in conserved amino acid residues that are important for anchoring HMGB1 to the minor groove of DNA, does not bind to chromatin and mislocalizes to nucleoli (Agresti et al. 2005).
Competition experiments, in which HMGB1 was micro-injected into living cells expressing H1-GFP, indicated that HMGB1 competes with histone H1 for chromatin-binding sites. HMGB1 deletion mutants lacking the acidic C-terminal, which interacted strongly with DNA in vitro, competed efficiently with H1, while deletion mutants that did not interact with DNA in vitro did not compete (Catez et al. 2004). These results suggested that, in vivo, the binding sites for HMGB1 and histone H1 on the chromatin partially overlap, and that the HMGB boxes are the main chromatin-binding domain of this protein family. These results are also in agreement with a previous in vitro experiment, which demonstrated that HMGB1 preferentially binds to the linker DNA rather than to nucleosome core particles (CPs) (Nightingale et al. 1996), and that an HMGB1 truncation mutant lacking the acidic tail can bind to nucleosome cores (Bonaldi et al. 2002). Nevertheless, it has been demonstrated that the acidic tail of HMGB1 interacts with histone H3 and facilitates the proper binding of HMGB1 to the linker DNA between adjacent nucleosome CPs (Ueda et al. 2004).
Post-translational modifications of HMGBs affect their chromatin interactions both in vitro and in living cells. Monomethylation at Lys42 located in HMG box A of HMGB1 weakens its DNA binding, and the protein relocalizes to the cytoplasm (Ito et al. 2007). In monocytes, acetylation of nuclear HMGB1 leads to relocation to the cytoplasm, indicating a weakening in chromatin binding (Bonaldi et al. 2003). Indeed, detailed in vitro analyses revealed that acetylation of specific Lys residues in HMGB1 plays a critical role in its binding to purified DNA (Assenberg et al. 2008). An interesting, albeit puzzling, finding was that HMGB1 binds strongly, almost irreversibly, to the chromatin in apoptotic cells, but not to the chromatin in necrotic cells (Scaffidi et al. 2002). The mechanism responsible for the strong binding of HMGB1 to apoptotic chromatin is not understood. In living cells, the binding of HMGB1 to mitotic chromatin is weaker than that to interphase chromatin (Pallier et al. 2003), a finding that is in full agreement with previous immunofluorescence analyses demonstrating significant depletion of HMGB1 from mitotic chromatin following fixation (Isackson et al. 1980). Thus, both post-translational modifications of HMGBs and the higher-order chromatin conformation affect the chromatin binding of HMGB in living cells.
Numerous studies, including reporter assays in tissue culture cells and in vitro binding assays, indicate that HMGBs interact with many proteins that are known to affect chromatin function (Bustin and Reeves 1996; Bustin 1999; Agresti and Bianchi 2003). Obviously, these interactions may affect the binding of HMGB1 to chromatin in vivo. This possibility has been experimentally demonstrated by analyses of living cells expressing both fluorescently labeled HMGB1 and glucocorticoid receptors. Detailed FRAP and fluorescence resonance energy transfer studies revealed that, in living cells, these 2 proteins interact, but only when bound on chromatin. The chromatin interaction of this complex is more stable than could be predicted from the independent chromatin binding of the 2 proteins, and its disassembly requires energy (Agresti et al. 2005). This elegant study serves as a paradigm of the dynamic cooperative interactions between chromatin-binding proteins and their targets in living cells. However, given the large numbers of proteins that can potentially interact with HMGBs, it is still possible that some of these interactions occur prior to chromatin binding, and that, in some instances, HMGBs bind to chromatin as part of a protein complex.
HMGN proteins
The nucleosome-binding domain
HMGNs are the only nuclear proteins known to specifically bind to the 147 bp nucleosome CP, the building block of the chromatin fiber. This protein family consists of 5 major variants, named HMGN1, HMGN2, HMGN3a, HMGN3b, and HMGN4, all with a molecular mass of ~10 kDa (Bustin 2001; Bianchi and Agresti 2005). A much larger protein, named NSBP1 (or NBD-45), shares chromatin-binding characteristics with the HMGN family (Shirakawa et al. 2000; King and Francomano 2001). The hallmark of this family is the nucleosomal binding domain (NBD), which facilitates the specific binding of these proteins to the CP (Fig. 3). The NBD is a 30 amino acid domain that has been shown to be the minimum peptide able to bind specifically to nucleosomes, both in vitro and in vivo (Crippa et al. 1992; Ueda et al. 2008). Embedded in this domain is an 8 amino acid motif, RRSARLSA, which is encoded by a single exon and is absolutely conserved in all HMGNs. Within this sequence, each of the 4 amino acids in the R-S-RL sequence are absolutely necessary for the specific binding of the HMGN protein to the CP, both in vivo and in vitro (Ueda et al. 2008).
Fig. 3.

Determinants of HMGN–chromatin interactions. Shown is a schematic diagram of HMGN proteins. The R-S-RL listed above the nucleosomal binding domain (NBD) denotes amino acid residues that specify the interaction of HMGN proteins with nucleosome core particles. The factors listed to the left of the green arrow promote the interaction of HMGN with chromatin, and those listed to the right of the red arrow promote dissociation of HMGN from chromatin. NLS, nuclear localization signal, RD, regulatory domain.
While the NBD determines the specificity of the interactions of HMGNs with nucleosomes, the C-terminal region contributes to the binding affinity. The binding of HMGN C-terminal truncation mutants to chromatin is significantly weaker than that of the intact protein, both in vitro and in living cells (Ueda et al. 2008).
Nucleosome interactions
All HMGNs bind to nucleosomes with very similar affinities (Ueda et al. 2008); however, their chromatin interactions are variant specific. Bimolecular fluorescence complementation assays in living cells (Cherukuri et al. 2008), chromatin immunoprecipiation analyses (Postnikov et al. 1997), and mobility shift assays of HMGN–CP complexes that have been supershifted with specific antibodies (Postnikov et al. 1997) reveal that, both in vitro and in vivo, 2 molecules of HMGN bind to CPs. These nucleosome–CP complexes contain either 2 molecules of HMGN1 or 2 molecules of HMGN2; mixed complexes containing 1 HMGN1 and 1 HMGN2 molecule on the same nucleosomes are not observed under physiological conditions (Postnikov et al. 1995; Cherukuri et al. 2008). The formation of homodimeric complexes raises the possibility that each HMGN variant produces unique structural modification in CPs. Site-directed cross-linking revealed that the proteins occupy distinct positions on the nucleosomes (Trieschmann et al. 1998; Ueda et al. 2008); however, the exact path of these proteins on the nucleosomal surface is not yet known.
Analyses of HMGN binding to chromatin in vivo by FRAP mobility and by visualization of their intranuclear organization, and in vitro by mobility shift assays suggest 2 different binding modes (Cherukuri et al. 2008; Ueda et al. 2008). The major mode is high-affinity binding to CPs, which involves specific interactions between the conserved NBD and the nucleosome CP, which leads to the formation of CP–HMGN homodimer complexes on chromatin. Mutation of any of the 4 amino acids in the R-S-RL motif described above abolishes this specific NBD-dependent nucleosome binding, resulting in FRAP recovery rates that are 10 times faster than those of the wild-type protein (Ueda et al. 2008). The minor mode is low-affinity binding to DNA, which probably does not involve homodimerization of the HMGN proteins. This type of binding is found in mitotic cells, where a high fraction of the HMGNs are phosphorylated (Cherukuri et al. 2008; Prymakowska-Bosak et al. 2001). Interestingly, when the specific binding of HMGN to nucleosomes is abolished, the protein mislocalizes to the nucleolus, an organelle from which the wild-type protein is excluded (Prymakowska-Bosak et al. 2001; Ueda et al. 2008). These findings suggest that the formation of homodimeric HMGN–CP complexes requires specific high-affinity interaction of these proteins with their nucleosomal targets.
Factors affecting the mobility and chromatin binding of HMGNs in live cells
Several factors were shown to affect the binding of HMGNs to nucleosomes in live cells: the primary structure of the HMGNs themselves, post-translational modifications, the structure of the chromatin fiber, and competition with other proteins for chromatin-binding sites.
Based on sequence similarity and in vitro chromatin-binding analyses, 3 distinct conserved elements were described in the primary structure of HMGNs: the NBD, the bipartite nuclear localization signals flanking the NBD, and a less well defined regulatory domain, which is located in the C-terminal region of the protein (Fig. 3). The regulatory domain was previously called the chromatin-unfolding domain (Bustin 2001); it was renamed because recent findings indicate that this region is involved in chromatin interactions and in modulating the HMGN-related effects on histone post-translational modifications in an HMGN-variant-specific manner (Ueda et al. 2006, 2008).
A comprehensive analysis of the kinetic and chromatin-binding properties of a battery of point and deletion mutants of HMGN2 revealed that several regions of the protein, most of which are located in the NBD and toward the C-terminal half, affect its chromatin affinity (Ueda et al. 2008). The C-terminal region affects the affinity of the protein for chromatin, while the specificity for nucleosome binding resides in the NBD. Detailed analyses of HMGN2 point mutants revealed that several residues in the NBD affect its binding to CPs; however, the specific interaction between HMGN2 and CP is dependent only on residues R22S24R26L27. Mutation in any of these 4 residues, which are absolutely conserved in all HMGN variants, will abolish the specific interaction of the HMGN proteins with CPs (Ueda et al. 2008). Previous biochemical studies (Crippa et al. 1992) and more recent FRAP analysis of HMGN swap mutants indicated that the NBD is an interchangeable chromatin-binding module that can act as an independent functional domain (Ueda et al. 2008). Thus, the NBD domain can be considered as a chromatin-binding module that specifically recognizes structural features in CPs, independent of DNA sequence or histone tail modifications.
Post-translational modifications in either HMGNs or in chromatin were also shown to affect their interaction. HMGNs are phosphorylated (Thomson et al. 1999; Prymakowska-Bosak et al. 2001; Soloaga et al. 2003) and acetylated (Herrera et al. 1999; Bergel et al. 2000; Luhrs et al. 2002) at multiple sites (Zhang and Wang 2008). The levels of phosphorylation are low in interphase nuclei, but elevated during mitosis or during response to cellular stress. Although in vitro analyses suggest that the phosphorylation of several sites in HMGN inhibit its CP binding (Prymakowska-Bosak et al. 2001), in vivo FRAP and fluorescence loss in photobleaching experiments suggest that only phosphorylation inside the NBD has significant effects on this binding (Prymakowska-Bosak et al. 2001; Catez et al. 2004; Lim et al. 2004). The most significant effect on chromatin interactions occurs upon phosphorylation of the conserved Ser residue, which is 1 of the 4 amino acids that determine the nucleosome-binding specificity of the protein (Prymakowska-Bosak et al. 2001; Catez et al. 2004; Lim et al. 2004; Ueda et al. 2008). Both HMGN1 and 2 are also acetylated on multiple residues (Bergel et al. 2000), a modification that has been shown to affect the interaction with CPs in vitro (Herrera et al. 1999). Interestingly, treatment of cells with histone deacetylase inhibitors reduces the binding of HMGN to chromatin in vivo. This treatment is known to elevate the acetylation of the nucleosomal histones; however, it is also possible that it leads to modifications in HMGNs, which in turn may affect their binding to chromatin (Luhrs et al. 2002; Ramirez et al. 2008).
Detailed comparison of in vivo and in vitro interaction between HMGNs and chromatin suggests that the major factors that affect the binding of HMGN to chromatin are operative at the level of the single nucleosome (Ueda et al. 2008). Nevertheless, the structure of the chromatin fiber and the degree of nucleosome compaction in chromatin seems to play a significant role in the intranuclear organization and their chromatin-binding affinity. In heterochromatin, where the CPs are relatively crowded, the mobility of HMGNs is at least 2 times slower and the FRAP recovery is less complete than in euchromatin, where the chromatin fiber is less compacted (Catez et al. 2002; Phair et al. 2004). Heterochromatin and euchromatin are also distinguished by their pattern of histone post-translational modifications (Ruthenburg et al. 2007; Trojer and Reinberg 2007), which might also affect the mobility of HMGN. Since intranuclear mobility is related to chromatin-binding affinity, it may seem that binding of HMGNs to the CPs in heterochromatin is stronger than to the CPs in euchromatin. However, it is also possible that the tight packing of the CPs in heterochromatin minimizes the relative time necessary to find a binding site, i.e., the “go” stage in HMGN mobility. Thus, at the level of the single CP, the interaction of HMGN with euchromatin may be similar to its binding to heterochromatin. In contrast, the interaction of the HMGNs with the chromatin fiber in the condensed mitotic chromosome seem to be fundamentally different than their binding to euchromatin (Cherukuri et al. 2008), perhaps because, in mitotic cells, a significant fraction of the HMGNs is phosphorylated (Prymakowska-Bosak et al. 2001).
FRAP analyses of living cells microinjected with various proteins revealed that HMGN variants compete among themselves and with variants of the linker histone H1 for chromatin-binding sites (Catez et al. 2002, 2004). These experiments suggest that HMGNs interact with chromatin as part of a network of chromatin-binding proteins, and that their binding to CPs may be affected by changes occurring in any member of the network. Thus, changes in the absolute levels, or even in the chemical properties of a competing component, will affect the binding of HMGN to chromatin. As elaborated elsewhere (Bustin et al. 2005), the competitive interaction among chromatin-binding proteins may lead to functional redundancy among these proteins, and may have significant effects on the cellular phenotype.
Perspective
Studies on the intranuclear organization of HMG proteins in living cells provided a new view on their intranuclear organization and into the mode by which these nuclear proteins interact with chromatin. Traditionally, these interactions were viewed as static, in which a specific molecule would continuously associate with a specific binding site until a specific stimulus actively caused its dissociation. In the new view, these interactions are highly dynamic and stochastic. Similar to other nuclear proteins, HMG proteins constantly move throughout the nucleus, scanning the chromatin landscape for binding sites (Phair et al. 2004). Their interactions with chromatin are regulated by multiple factors, including chemical changes in the protein or in their chromatin targets, and by competition with other nuclear proteins for chromatin-binding sites.
HMG proteins bind to chromatin as part of a protein network in which they compete with histone H1 variants, and with variants of their own HMG family for nucleosome-binding sites (Hill and Reeves 1997; Zlatanova and van Holde 1998; Catez et al. 2004). Competition of HMGs with other nuclear proteins can also be envisioned; however, this has not yet been experimentally demonstrated. Conceivably, a change in the expression level or in the binding affinity of a competitor could affect the binding of H1 or other HMG variants and trigger compensatory adjustments that would establish a new steady state of chromatin interactions. For example, induction of cell migration alters H1 histone post-translational modifications and increases its binding to chromatin (Gerlitz et al. 2007), while the binding of HMGN2 to chromatin seems to weaken (G. Gerlitz and M. Bustin, unpublished data). Similar situations could occur during the cell cycle or differentiation. Indeed, the mobility of H1 in embryonic stem cells differs from that in fully differentiated cells (Meshorer et al. 2006).
The key determinants shown to affect the binding of HMGs to chromatin in living cells are essentially the same as those affecting the in vitro binding of purified HMGs to isolated chromatin or nucleosomes. These interactions are operative at the level of the single nucleosome; the higher-order chromatin structure does not seem to have major effects on the binding of HMG protein to chromatin. The functional motifs characteristic of the 3 HMGs families are crucial for chromatin binding of the protein both in vivo and in vitro. Thus, in living cells, disruption of the NBD in HMGNs (Ueda et al. 2008), of the AT hook in HMGAs (Harrer et al. 2004), and of the HMG-Box in HMGBs (Agresti et al. 2005) abolish the interaction of the proteins with their chromatin targets. Likewise, studies in both living cells and with purified components have demonstrated that post-translational modifications in HMG proteins or in their binding sites affect chromatin interactions.
A major remaining unanswered question is whether the binding of these proteins to chromatin is specific. So far, there is no evidence that the proteins bind to chromatin with any sequence specificity; in fact, most in vitro studies failed to detect any preferential binding of the protein to chromatin containing unique sequences. Yet, emerging evidence suggests that HMG variants may affect the expression of specific genes or specific cellular processes. Three possible mechanisms for the preferential interaction of the proteins with specific sites in chromatin can be envisioned. First, and most likely, HMG proteins may be targeted to specific regions as part of multiprotein complexes. Indeed, HMG proteins are known to form such complexes and can interact with specific regulatory factors (Reeves and Beckerbauer 2001; Lim et al. 2002; Bianchi and Agresti 2005). Second, an HMG and a regulatory factor interact only on the surface of chromatin, as was shown for HMGB1 and glucocorticoid receptors (Agresti et al. 2005). In this scenario, stochastic assembly of HMG and a regulatory factor results in mutual strengthening of the chromatin interaction of each individual component. Such an interaction may nucleate the assembly of additional regulatory factors in a fashion similar to the stepwise, dynamic, assembly of polymerase I transcription complexes on ribosomal genes (Gorski et al. 2008). The third possibility, which has not yet been experimentally demonstrated, is that the various HMG variants preferentially bind to nucleosomes bearing specific histone post-translational modifications.
Studies with genetically altered mice suggest that HMG proteins affect developmental processes and play a role in the etiology of certain diseases (Hock et al. 2007). Elucidation of the factors that regulate the binding of the HMGs to the chromatin could provide insights into the molecular mechanisms by which these ubiquitous and abundant nuclear proteins affect the cellular phenotype.
Acknowledgments
Supported by the Intramural Program, CCR, NCI, NIH, and by a grant from the DFG.
Footnotes
This paper is one of a selection of papers published in this Special Issue, entitled CSBMCB’s 51st Annual Meeting – Epigenetics and Chromatin Dynamics, and has undergone the Journal’s usual peer review process.
Contributor Information
Gabi Gerlitz, Protein Section, Laboratory of Metabolism, National Cancer Institute, US National Institute of Health, 37 Convent Drive, Bldg. 37, Bethesda, MD 20892, USA.
Robert Hock, Department of Cell and Developmental Biology, Biocenter, University of Wuerzburg, Am Hubland, D-97074, Germany.
Tetsuya Ueda, Protein Section, Laboratory of Metabolism, National Cancer Institute, US National Institute of Health, 37 Convent Drive, Bldg. 37, Bethesda, MD 20892, USA.
Michael Bustin, Protein Section, Laboratory of Metabolism, National Cancer Institute, US National Institute of Health, 37 Convent Drive, Bldg. 37, Bethesda, MD 20892, USA.
References
- Agresti A, Bianchi ME. HMGB proteins and gene expression. Curr Opin Genet Dev. 2003;13:170–178. doi: 10.1016/S0959-437X(03)00023-6. [DOI] [PubMed] [Google Scholar]
- Agresti A, Scaffidi P, Riva A, Caiolfa VR, Bianchi ME. GR and HMGB1 interact only within chromatin and influence each other’s residence time. Mol Cell. 2005;18:109–121. doi: 10.1016/j.molcel.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Aizawa S, Nishino H, Saito K, Kimura K, Shirakawa H, Yoshida M. Stimulation of transcription in cultured cells by high mobility group protein 1: essential role of the acidic carboxyl-terminal region. Biochemistry. 1994;33:14690–14695. doi: 10. 1021/bi00253a006. [DOI] [PubMed] [Google Scholar]
- Assenberg R, Webb M, Connolly E, Stott K, Watson M, Hobbs J, Thomas JO. A critical role in structure-specific DNA binding for the acetylatable lysine residues in HMGB1. Biochem J. 2008;411:553–561. doi: 10.1042/BJ20071613. [DOI] [PubMed] [Google Scholar]
- Banks GC, Li Y, Reeves R. Differential in vivo modifications of the HMGI(Y) nonhistone chromatin proteins modulate nucleosome and DNA interactions. Biochemistry. 2000;39:8333–8346. doi: 10.1021/bi000378+. [DOI] [PubMed] [Google Scholar]
- Bergel M, Herrera JE, Thatcher BJ, Prymakowska-Bosak M, Vassilev A, Nakatani Y, et al. Acetylation of novel sites in the nucleosomal binding domain of chromosomal protein HMG-14 by p300 alters its interaction with nucleosomes. J Biol Chem. 2000;275:11514–11520. doi: 10.1074/jbc.275.15.11514. [DOI] [PubMed] [Google Scholar]
- Bianchi ME, Agresti A. HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev. 2005;15:496–506. doi: 10.1016/j.gde.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Bianchi ME, Manfredi AA. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev. 2007;220:35–46. doi: 10.1111/j. 1600-065X.2007.00574.x. [DOI] [PubMed] [Google Scholar]
- Bonaldi T, Langst G, Strohner R, Becker PB, Bianchi ME. The DNA chaperone HMGB1 facilitates ACF/CHRAC-dependent nucleosome sliding. EMBO J. 2002;21:6865–6873. doi: 10.1093/emboj/cdf692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin M. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol Cell Biol. 1999;19:5237–5246. doi: 10.1128/mcb.19.8.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin M. Chromatin unfolding and activation by HMGN(*) chromosomal proteins. Trends Biochem Sci. 2001;26:431–437. doi: 10.1016/S0968-0004(01)01855-2. [DOI] [PubMed] [Google Scholar]
- Bustin M, Reeves R. High-mobility-group chromosomal proteins: architectural components that facilitate chromatin function. Prog Nucleic Acid Res Mol Biol. 1996;54:35–100. doi: 10.1016/S0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- Bustin M, Catez F, Lim JH. The dynamics of histone H1 function in chromatin. Mol Cell. 2005;17:617–620. doi: 10.1016/j.molcel.2005.02.019. [DOI] [PubMed] [Google Scholar]
- Cao X, Clavijo C, Li X, Lin HH, Chen Y, Shih HM, Ann DK. SUMOylation of HMGA2: selective destabilization of promyelocytic leukemia protein via proteasome. Mol Cancer Ther. 2008;7:923–934. doi: 10.1158/1535-7163.MCT-07-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catez F, Brown DT, Misteli T, Bustin M. Competition between histone H1 and HMGN proteins for chromatin binding sites. EMBO Rep. 2002;3:760–766. doi: 10.1093/embo-reports/kvf156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catez F, Yang H, Tracey KJ, Reeves R, Misteli T, Bustin M. Network of dynamic interactions between histone H1 and high-mobility-group proteins in chromatin. Mol Cell Biol. 2004;24:4321–4328. doi: 10.1128/MCB.24.10.4321-4328. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catez F, Ueda T, Bustin M. Determinants of histone H1 mobility and chromatin binding in living cells. Nat Struct Mol Biol. 2006;13:305–310. doi: 10.1038/nsmb1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherukuri S, Hock R, Ueda T, Catez F, Rochman M, Bustin M. Cell cycle dependent binding of HMGN proteins to chromatin. Mol Biol Cell. 2008;19(5):1816–1824. doi: 10. 1091/mbc.E07-10-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill ME, Suzuki M. ‘SPKK’ motifs prefer to bind to DNA at A/T-rich sites. EMBO J. 1989;8:4189–4195. doi: 10.1002/j.1460-2075.1989.tb08604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus P, Schulze E, Wisniewski JR. Insect proteins homologous to mammalian high mobility group proteins I/Y (HMG I/Y). Characterization and binding to linear and four-way junction DNA. J Biol Chem. 1994;269:33042–33048. [PubMed] [Google Scholar]
- Crippa MP, Alfonso PJ, Bustin M. Nucleosome core binding region of chromosomal protein HMG-17 acts as an independent functional domain. J Mol Biol. 1992;228:442–449. doi: 10.1016/0022-2836(92)90833-6. [DOI] [PubMed] [Google Scholar]
- Cui T, Leng F. Specific recognition of AT-rich DNA sequences by the mammalian high mobility group protein AT-hook 2: a SELEX study. Biochemistry. 2007;46:13059–13066. doi: 10.1021/bi701269s. [DOI] [PubMed] [Google Scholar]
- Cui T, Wei S, Brew K, Leng F. Energetics of binding the mammalian high mobility group protein HMGA2 to poly(dA-dT)2 and poly(dA)-poly(dT) J Mol Biol. 2005;352:629–645. doi: 10.1016/j.jmb.2005.07.048. [DOI] [PubMed] [Google Scholar]
- Diana F, Sgarra R, Manfioletti G, Rustighi A, Poletto D, Sciortino MT, et al. A link between apoptosis and degree of phosphorylation of high mobility group A1a protein in leukemic cells. J Biol Chem. 2001;276:11354–11361. doi: 10.1074/jbc.M009521200. [DOI] [PubMed] [Google Scholar]
- Edberg DD, Bruce JE, Siems WF, Reeves R. In vivo posttranslational modifications of the high mobility group A1a proteins in breast cancer cells of differing metastatic potential. Biochemistry. 2004;43:11500–11515. doi: 10.1021/bi049833i. [DOI] [PubMed] [Google Scholar]
- Edberg DD, Adkins JN, Springer DL, Reeves R. Dynamic and differential in vivo modifications of the isoform HMGA1a and HMGA1b chromatin proteins. J Biol Chem. 2005;280:8961–8973. doi: 10.1074/jbc.M407348200. [DOI] [PubMed] [Google Scholar]
- Fan Y, Nikitina T, Zhao J, Fleury TJ, Bhattacharyya R, Bouhassira EE, et al. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell. 2005;123:1199–1212. doi: 10.1016/j.cell.2005. 10.028. [DOI] [PubMed] [Google Scholar]
- Frank O, Schwanbeck R, Wisniewski JR. Protein footprinting reveals specific binding modes of a high mobility group protein I to DNAs of different conformation. J Biol Chem. 1998;273:20015–20020. doi: 10.1074/jbc.273.32.20015. [DOI] [PubMed] [Google Scholar]
- Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nat Rev Cancer. 2007;7:899–910. doi: 10.1038/nrc2271. [DOI] [PubMed] [Google Scholar]
- Gerlitz G, Livnat I, Ziv C, Yarden O, Bustin M, Reiner O. Migration cues induce chromatin alterations. Traffic. 2007;8:1521–1529. doi: 10.1111/j.1600-0854.2007.00638.x. [DOI] [PubMed] [Google Scholar]
- Gorski SA, Snyder SK, John S, Grummt I, Misteli T. Modulation of RNA polymerase assembly dynamics in transcriptional regulation. Mol Cell. 2008;30:486–497. doi: 10.1016/j.molcel.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrer M, Luhrs H, Bustin M, Scheer U, Hock R. Dynamic interaction of HMGA1a proteins with chromatin. J Cell Sci. 2004;117:3459–3471. doi: 10.1242/jcs.01160. [DOI] [PubMed] [Google Scholar]
- Herrera JE, Sakaguchi K, Bergel M, Trieschmann L, Nakatani Y, Bustin M. Specific acetylation of chromosomal protein HMG-17 by PCAF alters its interaction with nucleosomes. Mol Cell Biol. 1999;19:3466–3473. doi: 10.1128/mcb.19.5.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Reeves R. Competition between HMG-I(Y), HMG-1 and histone H1 on four-way junction DNA. Nucleic Acids Res. 1997;25:3523–3531. doi: 10.1093/nar/25.17.3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Pedulla ML, Reeves R. Directional binding of HMG-I(Y) on four-way junction DNA and the molecular basis for competitive binding with HMG-1 and histone H1. Nucleic Acids Res. 1999;27:2135–2144. doi: 10.1093/nar/27.10.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock R, Furusawa T, Ueda T, Bustin M. HMG chromosomal proteins in development and disease. Trends Cell Biol. 2007;17:72–79. doi: 10.1016/j.tcb.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huth JR, Bewley CA, Nissen MS, Evans JN, Reeves R, Gronenborn AM, Clore GM. The solution structure of an HMG-I(Y)-DNA complex defines a new architectural minor groove binding motif. Nat Struct Biol. 1997;4:657–665. doi: 10.1038/nsb0897-657. [DOI] [PubMed] [Google Scholar]
- Isackson PJ, Bidney DL, Reeck GR, Neihart NK, Bustin M. High mobility group chromosomal proteins isolated from muclei and cytosol of cultured hepatoma cells are similar. Biochemistry. 1980;19:4466–4471. doi: 10.1021/bi00560a013. [DOI] [PubMed] [Google Scholar]
- Ito I, Fukazawa J, Yoshida M. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J Biol Chem. 2007;282:16336–16344. doi: 10.1074/jbc.M608467200. [DOI] [PubMed] [Google Scholar]
- Jiang X, Wang Y. Acetylation and phosphorylation of high-mobility group A1 proteins in PC-3 human tumor cells. Biochemistry. 2006;45:7194–7201. doi: 10.1021/bi060504v. [DOI] [PubMed] [Google Scholar]
- Kasinsky HE, Lewis JD, Dacks JB, Ausio J. Origin of H1 linker histones. FASEB J. 2001;15:34–42. doi: 10.1096/fj. 00-0237rev. [DOI] [PubMed] [Google Scholar]
- Kim TK, Maniatis T. The mechanism of transcriptional synergy of an in vitro assembled interferon-beta enhan-ceosome. Mol Cell. 1997;1:119–129. doi: 10.1016/S1097-2765(00) 80013-1. [DOI] [PubMed] [Google Scholar]
- King LM, Francomano CA. Characterization of a human gene encoding nucleosomal binding protein NSBP1. Genomics. 2001;71:163–173. doi: 10.1006/geno.2000.6443. [DOI] [PubMed] [Google Scholar]
- Lee KB, Thomas JO. The effect of the acidic tail on the DNA-binding properties of the HMG1,2 class of proteins: insights from tail switching and tail removal. J Mol Biol. 2000;304:135–149. doi: 10.1006/jmbi.2000.4206. [DOI] [PubMed] [Google Scholar]
- Lever MA, Th’ng JP, Sun X, Hendzel MJ. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature. 2000;408:873–876. doi: 10.1038/35048603. [DOI] [PubMed] [Google Scholar]
- Li O, Vasudevan D, Davey CA, Droge P. High-level expression of DNA architectural factor HMGA2 and its association with nucleosomes in human embryonic stem cells. Genesis. 2006;44:523–529. doi: 10.1002/dvg.20242. [DOI] [PubMed] [Google Scholar]
- Lim JH, Bustin M, Ogryzko VV, Postnikov YV. Metastable macromolecular complexes containing high mobility group nucleosome-binding chromosomal proteins in HeLa nuclei. J Biol Chem. 2002;277:20774–20782. doi: 10.1074/jbc. M200404200. [DOI] [PubMed] [Google Scholar]
- Lim JH, Catez F, Birger Y, West KL, Prymakowska-Bosak M, Postnikov YV, Bustin M. Chromosomal protein HMGN1 modulates histone H3 phosphorylation. Mol Cell. 2004;15:573–584. doi: 10.1016/j.molcel.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Lim JH, West KL, Rubinstein Y, Bergel M, Postnikov YV, Bustin M. Chromosomal protein HMGN1 enhances the acetylation of lysine 14 in histone H3. EMBO J. 2005;24:3038–3048. doi: 10.1038/sj.emboj.7600768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- Luhrs H, Hock R, Schauber J, Weihrauch M, Harrer M, Melcher R, et al. Modulation of HMG-N2 binding to chromatin by butyrate-induced acetylation in human colon adenocarcinoma cells. Int J Cancer. 2002;97:567–573. doi: 10.1002/ijc. 10098. [DOI] [PubMed] [Google Scholar]
- Maher JF, Nathans D. Multivalent DNA-binding properties of the HMG-1 proteins. Proc Natl Acad Sci USA. 1996;93:6716–6720. doi: 10.1073/pnas.93.13.6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merika M, Thanos D. Enhanceosomes. Curr Opin Genet Dev. 2001;11:205–208. doi: 10.1016/S0959-437X(00)00180-5. [DOI] [PubMed] [Google Scholar]
- Meshorer E, Yellajoshula D, George E, Scambler PJ, Brown DT, Misteli T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev Cell. 2006;10:105–116. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda TB, Webb KJ, Edberg DD, Reeves R, Clarke S. Protein arginine methyltransferase 6 specifically methylates the nonhistone chromatin protein HMGA1a. Biochem Biophys Res Commun. 2005;336:831–835. doi: 10.1016/j.bbrc.2005.08.179. [DOI] [PubMed] [Google Scholar]
- Munshi N, Merika M, Yie J, Senger K, Chen G, Thanos D. Acetylation of HMG I(Y) by CBP turns off IFN beta expression by disrupting the enhanceosome. Mol Cell. 1998;2:457–467. doi: 10.1016/S1097-2765(00)80145-8. [DOI] [PubMed] [Google Scholar]
- Munshi N, Agalioti T, Lomvardas S, Merika M, Chen G, Thanos D. Coordination of a transcriptional switch by HMGI(Y) acetylation. Science. 2001;293:1133–1136. doi: 10.1126/science.293.5532.1133. [DOI] [PubMed] [Google Scholar]
- Nightingale K, Dimitrov S, Reeves R, Wolffe AP. Evidence for a shared structural role for HMG1 and linker histones B4 and H1 in organizing chromatin. EMBO J. 1996;15:548–561. [PMC free article] [PubMed] [Google Scholar]
- Nissen MS, Reeves R. Changes in superhelicity are introduced into closed circular DNA by binding of high mobility group protein I/Y. J Biol Chem. 1995;270:4355–4360. doi: 10.1074/jbc.270.9.4355. [DOI] [PubMed] [Google Scholar]
- Nissen MS, Langan TA, Reeves R. Phosphorylation by cdc2 kinase modulates DNA binding activity of high mobility group I nonhistone chromatin protein. J Biol Chem. 1991;266:19945–19952. [PubMed] [Google Scholar]
- Pallier C, Scaffidi P, Chopineau-Proust S, Agresti A, Nordmann P, Bianchi ME, Marechal V. Association of chromatin proteins high mobility group box (HMGB) 1 and HMGB2 with mitotic chromosomes. Mol Biol Cell. 2003;14:3414–3426. doi: 10.1091/mbc.E02-09-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phair RD, Scaffidi P, Elbi C, Vecerova J, Dey A, Ozato K, et al. Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol Cell Biol. 2004;24:6393–6402. doi: 10.1128/MCB.24.14.6393-6402.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postnikov YV, Trieschmann L, Rickers A, Bustin M. Homodimers of chromosomal proteins HMG-14 and HMG-17 in nucleosome cores. J Mol Biol. 1995;252:423–432. doi: 10.1006/jmbi.1995.0508. [DOI] [PubMed] [Google Scholar]
- Postnikov YV, Herrera JE, Hock R, Scheer U, Bustin M. Clusters of nucleosomes containing chromosomal protein HMG-17 in chromatin. J Mol Biol. 1997;274:454–465. doi: 10. 1006/jmbi.1997.1391. [DOI] [PubMed] [Google Scholar]
- Prymakowska-Bosak M, Misteli T, Herrera JE, Shirakawa H, Birger Y, Garfield S, Bustin M. Mitotic phosphorylation prevents the binding of HMGN proteins to chromatin. Mol Cell Biol. 2001;21:5169–5178. doi: 10.1128/MCB. 21.15.5169-5178.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radic MZ, Saghbini M, Elton TS, Reeves R, Hamkalo BA. Hoechst 33258, distamycin A, and high mobility group protein I (HMG-I) compete for binding to mouse satellite DNA. Chromosoma. 1992;101:602–608. doi: 10.1007/BF00360537. [DOI] [PubMed] [Google Scholar]
- Ramirez T, Brocher J, Stopper H, Hock R. Sodium arsenite modulates histone acetylation, histone deacetylase activity and HMGN protein dynamics in human cells. Chromosoma. 2008;117:147–157. doi: 10.1007/s00412-007-0133-5. [DOI] [PubMed] [Google Scholar]
- Reeves R, Beckerbauer L. HMGI/Y proteins: flexible regulators of transcription and chromatin structure. Biochim Biophys Acta. 2001;1519:13–29. doi: 10.1016/s0167-4781(01)00215-9. [DOI] [PubMed] [Google Scholar]
- Reeves R, Nissen MS. The A.T-DNA-binding domain of mammalian high mobility group I chromosomal proteins. A novel peptide motif for recognizing DNA structure. J Biol Chem. 1990;265:8573–8582. [PubMed] [Google Scholar]
- Reeves R, Nissen MS. Interaction of high mobility group-I (Y) nonhistone proteins with nucleosome core particles. J Biol Chem. 1993;268:21137–21146. [PubMed] [Google Scholar]
- Reeves R, Wolffe AP. Substrate structure influences binding of the non-histone protein HMG-I(Y) to free nucleosomal DNA. Biochemistry. 1996;35:5063–5074. doi: 10.1021/bi952424p. [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Schwanbeck R, Gerharz M, Drung A, Rogalla P, Piekielko A, Blank C, et al. Point mutations within AT-hook domains of the HMGI homologue HMGIYL1 affect binding to gene promoter but not to four-way junction DNA. Biochemistry. 2000;39:14419–14425. doi: 10.1021/bi0011274. [DOI] [PubMed] [Google Scholar]
- Sgarra R, Diana F, Bellarosa C, Dekleva V, Rustighi A, Toller M, et al. During apoptosis of tumor cells HMGA1a protein undergoes methylation: identification of the modification site by mass spectrometry. Biochemistry. 2003a;42:3575–3585. doi: 10.1021/bi027338l. [DOI] [PubMed] [Google Scholar]
- Sgarra R, Diana F, Rustighi A, Manfioletti G, Giancotti V. Increase of HMGA1a protein methylation is a distinctive characteristic of leukaemic cells induced to undergo apoptosis. Cell Death Differ. 2003b;10:386–389. doi: 10.1038/sj.cdd.4401184. [DOI] [PubMed] [Google Scholar]
- Sgarra R, Rustighi A, Tessari MA, Di Bernardo J, Altamura S, Fusco A, et al. Nuclear phosphoproteins HMGA and their relationship with chromatin structure and cancer. FEBS Lett. 2004;574:1–8. doi: 10.1016/j.febslet.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Sgarra R, Lee J, Tessari MA, Altamura S, Spolaore B, Giancotti V, et al. The AT-hook of the chromatin architectural transcription factor high mobility group A1a is arginine-methylated by protein arginine methyltransferase 6. J Biol Chem. 2006;281:3764–3772. doi: 10.1074/jbc.M510231200. [DOI] [PubMed] [Google Scholar]
- Shirakawa H, Tanigawa T, Sugiyama S, Kobayashi M, Terashima T, Yoshida K, et al. Nuclear accumulation of HMG2 protein is mediated by basic regions interspaced with a long DNA-binding sequence, and retention within the nucleus requires the acidic carboxyl terminus. Biochemistry. 1997;36:5992–5999. doi: 10.1021/bi962487n. [DOI] [PubMed] [Google Scholar]
- Shirakawa H, Landsman D, Postnikov YV, Bustin M. NBP-45, a novel nucleosomal binding protein with a tissue-specific and developmentally regulated expression. J Biol Chem. 2000;275:6368–6374. doi: 10.1074/jbc.275.9.6368. [DOI] [PubMed] [Google Scholar]
- Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, et al. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003;22:2788–2797. doi: 10.1093/emboj/cdg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon MJ, Strauss F, Varshavsky A. A mammalian high mobility group protein recognizes any stretch of six A.T base pairs in duplex DNA. Proc Natl Acad Sci USA. 1986;83:1276–1280. doi: 10.1073/pnas.83.5.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos D, Maniatis T. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell. 1995;83:1091–1100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- Thomae AW, Pich D, Brocher J, Spindler MP, Berens C, Hock R, et al. Interaction between HMGA1a and the origin recognition complex creates site-specific replication origins. Proc Natl Acad Sci USA. 2008;105:1692–1697. doi: 10.1073/pnas. 0707260105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JO, Travers AA. HMG1 and 2, and related ‘architectural’ DNA-binding proteins. Trends Biochem Sci. 2001;26:167–174. doi: 10.1016/S0968-0004(01)01801-1. [DOI] [PubMed] [Google Scholar]
- Thomson S, Clayton AL, Hazzalin CA, Rose S, Barratt MJ, Mahadevan LC. The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J. 1999;18:4779–4793. doi: 10.1093/emboj/18.17.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trieschmann L, Martin B, Bustin M. The chromatin unfolding domain of chromosomal protein HMG-14 targets the N-terminal tail of histone H3 in nucleosomes. Proc Natl Acad Sci USA. 1998;95:5468–5473. doi: 10.1073/pnas.95.10.5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojer P, Reinberg D. Facultative heterochromatin: is there a distinctive molecular signature? Mol. Cell. 2007;28:1–13. doi: 10.1016/j.molcel.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Ueda T, Chou H, Kawase T, Shirakawa H, Yoshida M. Acidic C-tail of HMGB1 is required for its target binding to nucleosome linker DNA and transcription stimulation. Biochemistry. 2004;43:9901–9908. doi: 10.1021/bi035975l. [DOI] [PubMed] [Google Scholar]
- Ueda T, Postnikov YV, Bustin M. Distinct domains in high mobility group N variants modulate specific chromatin modifications. J Biol Chem. 2006;281:10182–10187. doi: 10.1074/jbc.M600821200. [DOI] [PubMed] [Google Scholar]
- Ueda T, Catez F, Gerlitz G, Bustin M. Delineation of the protein module that anchors HMGN proteins to nucleosomes in the chromatin of living cells. Mol Cell Biol. 2008;28:2872–2883. doi: 10.1128/MCB.02181-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson M, Stott K, Thomas JO. Mapping intramolecular interactions between domains in HMGB1 using a tail-truncation approach. J Mol Biol. 2007;374:1286–1297. doi: 10. 1016/j.jmb.2007.09.075. [DOI] [PubMed] [Google Scholar]
- Yie J, Liang S, Merika M, Thanos D. Intra- and intermolecular cooperative binding of high-mobility-group protein I(Y) to the beta-interferon promoter. Mol Cell Biol. 1997;17:3649–3662. doi: 10.1128/mcb.17.7.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka K, Saito K, Tanabe T, Yamamoto A, Ando Y, Nakamura Y, et al. Differences in DNA recognition and conformational change activity between boxes A and B in HMG2 protein. Biochemistry. 1999;38:589–595. doi: 10.1021/bi981834l. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Wang Y. Homeodomain-interacting protein kinase-2 (HIPK2) phosphorylates HMGA1a at Ser-35, Thr-52, and Thr-77 and modulates its DNA binding affinity. J Proteome Res. 2007;6:4711–4719. doi: 10.1021/pr700571d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Wang Y. High mobility group proteins and their post-translational modifications. Biochim Biophys Acta. 2008;1784(9):1159–1166. doi: 10.1016/j.bbapap.2008.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zhang K, Zou Y, Perna A, Wang Y. A quantitative study on the in vitro and in vivo acetylation of high mobility group A1 proteins. J Am Soc Mass Spectrom. 2007;18:1569–1578. doi: 10.1016/j.jasms.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatanova J, van Holde K. Linker histones versus HMG1/2: a struggle for dominance? Bioessays. 1998;20:584–588. doi: 10.1002/(SICI)1521-1878(199807)20:7<584::AID-BIES10>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Zou Y, Wang Y. Mass spectrometric analysis of high-mobility group proteins and their post-translational modifications in normal and cancerous human breast tissues. J Proteome Res. 2007;6:2304–2314. doi: 10.1021/pr070072q. [DOI] [PubMed] [Google Scholar]
- Zou Y, Webb K, Perna AD, Zhang Q, Clarke S, Wang Y. A mass spectrometric study on the in vitro methylation of HMGA1a and HMGA1b proteins by PRMTs: methylation specificity, the effect of binding to AT-rich duplex DNA, and the effect of C-terminal phosphorylation. Biochemistry. 2007;46:7896–7906. doi: 10.1021/bi6024897. [DOI] [PubMed] [Google Scholar]