

Abstract
Block copolymer micelles (BCMs) were prepared from amphiphilic diblock copolymers of poly(n-butyl acrylate) and poly(acrylic acid) partially modified with 2-hydroxyethyl acrylate. Radical polymerization of acrylamide in the presence of micellar crosslinkers gave rise to elastomeric hydrogels (BCM-PAAm) whose mechanical properties can be tuned by varying the BCM composition. Transmission electron microscopy (TEM) imaging revealed stretch-induced, reversible micelle deformation in BCM-PAAm gels. A model hydrophobic drug, pyrene, loaded into the micelle core prior to the formation of BCM-PAAm gels, was dynamically released in response to externally applied mechanical forces. The BCM-crosslinked hydrogels with combined strength and force-modulated drug release are attractive candidates for the repair and regeneration of mechanically-active tissues.
Keywords: Hydrogels, Block Copolymer Micelles, Microscopic Crosslinker, Mechano-Responsive, Drug Delivery
Hydrogel materials have attracted a great deal of attention from biomedical fields due to their high water content, tissue-like viscoelasticity1-6 and stimuli-responsive properties.7-15 Because most tissues in the body are subjected to various types of mechanical forces, and cells within these tissues have sophisticated mechano-transduction machinery, mechanical force can be considered as a universal stimulus in biological entities.16-18 We are interested in developing hydrogel materials with built-in mechano-sensing mechanisms for use as tissue engineering scaffolds or drug release devices. To this end, self-assembled block copolymer micelles (BCMs)19 with reactive handles were employed as the microscopic crosslinkers in place of the traditional multifunctional molecular crosslinkers20 for the construction of covalently crosslinked networks. Block copolymer micelles are dynamic entities whose stability is described by the association and dissociation equilibrium constants.21, 22 We envision that covalent entrapment of self-assembled BCMs in a hydrogel network will effectively restrict the micelle dissociation, at the same time, providing mechano-responsible elements to a hydrogel network. In our design, BCMs were assembled from amphiphilic diblock copolymers of poly(n-butyl acrylate) (PnBA) and poly(acrylic acid) (PAA) partially modified with acrylate. PAA was chosen as the hydrophilic block because of its susceptibility to chemical modification while PnBA was selected as the hydrophobic block to impart conformational flexibility to the assembled BCMs at physiological conditions owing to its low glass transition temperature (−49 to −55 °C).23 Poly(acrylamide) (PAAm)-based hydrogels were prepared using acrylamide as a water-soluble monomer and acrylated BCMs as the microscopic crosslinkers (Scheme 1). An additional advantage of micellar crosslinkers over traditional molecular crosslinkers is their ability to sequester hydrophobic molecules in the micelle cores.24, 25 Chemically crosslinked hydrogels integrated with drug-loaded BCMs are likely to exhibit controlled drug release profiles that are modulated by the external forces. To our knowledge, this type of responsive hydrogels has not yet been reported.
Scheme 1.

Synthesis of mechano-responsive hydrogels. (A) Self-assembly of (PAA-g-HEA)-b-PnBA into reactive BCMs; (B) Hydrogel formation by radical polymerization of AAm in the presence of crosslinkable BCMs.
To prepare BCM-crosslinked PAAm gels, acrylated block copolymers were first synthesized and characterized (Scheme 1A). Precursor diblock copolymers of poly(tert-butyl acrylate)-b-poly(n-butyl acrylate) (PtBA-b-PnBA) containing the same number of tBA repeats and three different PnBA lengths were synthesized by atom transfer radical polymerization (ATRP). Subsequent hydrolysis of the tert-butyl groups revealed the hydrophilic PAA blocks, through which acrylate groups were installed via the reaction with hydroxyethyl acrylate (HEA). Detailed description of polymer synthesis and characterization can be found in the supporting information (Scheme S1 and Figure S1-S5). Approximately 20 mol% of PAA was acrylated, irrespective of the copolymer molecular weight. As a result, three acrylated block copolymers were obtained as (PAA100-g-HEA20)-b-PnBA16, (PAA100-g-HEA20)-b-PnBA40 and PAA100-g-HEA20)-b-PnBA125. Partial esterification of the PAA block with HEA did not affect the micellization ability of the copolymers.26 Here, micelles assembled from (PAA100-g-HEA20)-b-PnBA16, (PAA100-g-HEA20)-b-PnBA40 and PAA100-g-HEA20)-b-PnBA125 are referred to as BCM16, BCM40 and BCM125, respectively.
Transmission electron microscopy (TEM) images of uranyl acetate-stained BCM samples demonstrated a spherical morphology and a relatively uniform particle size for all three micellar structures (Figure S6A-C). The average diameters of the micelles, based on 100 counts of particles from the respective TEM images, were estimated to be 23 ± 3, 28 ± 3, 45 ± 5 nm for BCM 16, BCM 40 and BCM 125, respectively. Dynamic light scattering (DLS, Figure S6D) analysis showed an average particle size of 28 ± 4 nm for BCM16, 42 ± 5 nm for BCM40 and 80 ± 3 nm for BCM125. Particle size estimated by TEM was smaller than the corresponding hydrodynamic size determined by DLS, probably due to the collapse of block copolymer chains in micelles of TEM cast-film samples. Such effect is manifested more dramatically in BCM40 and BCM125. Block copolymers with a longer PnBA block also had a lower critical micelle concentration (Figure S7), in good agreement with previous reports.25, 27
BCM-crosslinked PAAm (BCM-PAAm) gels were prepared by free radical polymerization of AAm in the presenece of various crosslinkable BCMs using a redox initiator pair (Scheme 1B, see Supporting Information for detailed description of hydrogel synthesis). Representative stress versus strain curves (Figure 1A) for all BCM-PAAm gels displayed a sigmoid shape characteristic of elastomeric materials with low modulus and a large deformation before fracture. Young’s modulus decreased progressively from 36.9 ± 4.9 kPa to 33.9 ± 4.5 kPa and 2.2 ± 0.8 kPa for PAAm gels crosslinked by BCM16, 40 and 125, respectively (Figure 1B). This trend is consistent with the swelling ratio results (Table S1). Hydrogels crosslinked by BCM16 and BCM40 are stiffer than those crosslinked by BCM125. PAAm gels crosslinked by BCM16 exhibited the highest tensile strength with an average stress at break of 152.5 ± 7.8 kPa, whereas PAAm crosslinked by BCM125 can be extended up to 542 ± 26% (Figure 1C, D). Interestingly, BCM-crosslinked gels reported here had a significantly lower sol fraction (Table S1) as compared to traditional covalent hydrogels,28 indicating that the reactive BCMs are more efficient crosslinkers. Among the three types BCM-PAAm gels investigated, BCM16-PAAm gels has the highest modulus and strength; they can completely recover to their original shape after repetitive loading/unloading up to 350% strain without failure.26 Therefore, we elect to conduct in-depth characterization of BCM16-PAAm gels to assess force-induced structural alterations in BCM-crosslinked hydrogels.
Figure 1.

Mechanical properties of BCM16-PAAm gel, BCM40-PAAm gel and BCM125-PAAm gel measured by dynamic mechanical analyzer (DMA). (A) stress-strain curves until failure; (B) Young’s modulus as a functional of hydrogel composition; (C) Maximum stress as a functional of hydrogel composition; (D) Maximum strain as a functional of hydrogel composition. *: statistically different from each other, p≤0.05.
Hydrogel samples were dehydrated at ambient temperature under nitrogen atmosphere at a fixed strain (0%, 60% or 200%) for at least 24 h before being microtomed to obtain ~75 nm thin sections for TEM imaging. Micelles appear as bright spherical particles in the gel matrix due to the negative staining introduced by uranyl acetate. The relaxed BCM16-PAAm gels contain spherical particles with an average diameter of 31 ± 4 nm (Figure 2A), in good agreement with TEM measurements for BCM alone (Figure S6A). Therefore, the radical crosslinking process did not compromise the structural integrity of the micelles. When BCM16-PAAm gels were stretched to 60% strain, the spherical micelles, which were covalently connected to the PAAm network, became ellipsoidal (Figure 2B and S8). With an average dimension of 35 ± 5 nm along the long axis and 28 ± 3 nm in the short axis, the deformed particles had an aspect ratio of 1.26 ± 0.13. TEM characterization of control gels devoid of BCMs showed a feature-less background stain at 0 and 60% strain (Figure S9A, B). At a strain of 200%, the micelle particles appeared more elongated, having an average dimension of 51 ± 7 nm × 34 ± 5 nm and an aspect ratio of 1.53 ± 0.23 (Figure 2C). The force-induced structural alteration is reversible, as evidenced by microscopic analyses of BCM16-PAAm gels before, during and after stretching to 200% (Figure 3). Upon removal of the external force, micelles in the gels effectively returned to their original spherical shape, with an average diameter of 32 ± 6 nm.
Figure 2.
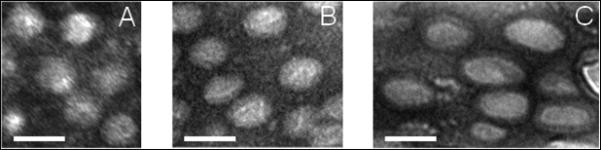
Cross-sectional TEM images of BCM16-PAAm gels stretched to 0% (A), 60% (B) and 200% strain (C). Similar numbers of BCMs are shown in each image. Samples were stained by uranyl acetate. Scale bar: 50 nm.
Figure 3.
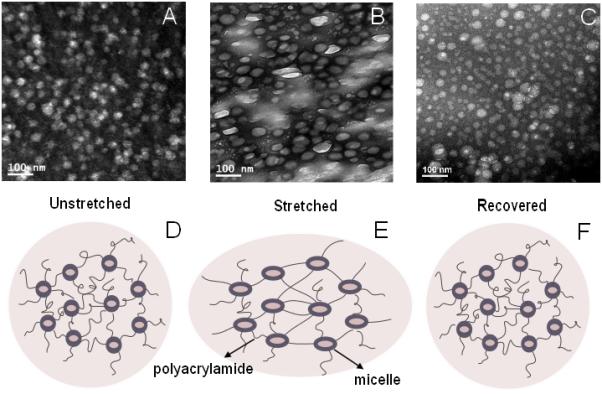
Cross-sectional TEM images show the microstructure of BCM16-PAAm gels imaged under various conditions: (A) relaxed, (B) stretched to 200%, (C) relaxed after stretching to 200%. Samples were stained by uranyl acetate. The cartoon below each TEM image (D, E, F) shows the proposed gel microstructures at the corresponding condition.
Our TEM results suggest that the macroscopic deformation was transmitted to the micellar building blocks via the covalent linkages between the PAAm chains and the BCM shells. The flexibility of the PnBA core provides an efficient energy dissipation mechanism through the elongation of the micelles along the stretch direction. Of note, individual micelles deformed to a lesser extent than the macroscopic gels. Such difference can be attributed to the presence of different mechano-responsive elements in the BCM-PAAm gels. In the crosslinked gels, the alignment of gel network was achieved by the relaxation and rotation of random-coil PAAm chains along the stretching direction, and the local deformation of block copolymer assemblies, occurred through intra-micellar chain relaxation and possible dissociation of the hydrophobic interactions among the PnBA chains in the hydrophobic core, as well as the weakening of the van der Waals interactions within the PAA corona. Consequently, affine deformation is not observed here. Micelles appeared larger when the BCM-PAAm gels were stretched to 200% than those under the strains of 60% and 0%. At a sufficiently high strain, a larger portion of the PnBA chains adopted an extended-chain conformation (rather than in a collapsed state) and the hydrophobic interaction among PnBA chains, as well as the van der Waals interactions in the PAA block, was significantly weakened. Void space could also be created through the separation of PnBA chains. These alterations collectively led to the increase in micelle dimension at 200% strain.
It is important to note that our BCM-PAAm gels are distinctly different from hydrogels derived from physically-associated micelles from AB diblock and ABA triblock copolymer.29, 30 These physical gels have poor mechanical properties and are not mechano-responsive owing to generally weak intermolecular interactions that contribute to network formation.30 Although we and others31, 32 have reported the force-induced deformation of micron-sized hydrogel particles crosslinked to a covalent network, to the best of our knowledge, this is the first report that confirmed, direct manipulation of nanosized micellar objects via the external mechanical forces. The discovery of mechano-responsive properties of BCM-crosslinked hydrogels not only improves our understanding of block copolymer assembly but also expands the range of applications of BCMs in biomedical fields. Indeed, our work was inspired by the diverse structural motifs in biological systems that change conformation over a range of mechanical forces.33 Force-induced structural changes, in turn, produce changes at the biochemical level that effectively direct cellular behaviors.16, 34
Here, we investigated whether force-induced micelle deformation could give rise to a systematic variation of chemical signals. Owing to its low water solubility (6 × 10−7 M), well-defined fluorescence property and sensitivity to its microenvironment, pyrene is expected to serve as “molecular beacon” signaling local mechanical deformations.35, 36 In our study, pyrene was first loaded into the pre-assembled BCM16 micelles. The amount of pyrene added to the micelle solution was well below its saturation concentration in water and pyrene-loaded micelles were thoroughly washed to minimize pyrene molecules that could be potentially associated to the micelle surface physically. Pyrene encapsulation efficiency was found to be 98.9%, confirming the ability of the PnBA cores to sequester hydrophobic molecules through hydrophobic association. Pyrene-loaded BCMs maintain their ability to crosslink PAAm and the resultant pyrene containing gels exhibit the same mechanical properties as those without the encapsulated pyrene (data not shown).
Figure 4 shows the release behavior of pyrene from BCM16-PAAm gels. A 5-min-on, 5-min-off stimulation regimen was applied to the samples using a custom-design, stepper-motor-driven stretcher (Figure S10) for a total of one hour at a strain of 0, 30 or 60% at a rate of 4 mm/s and 12 cycles per minute. In the absence of the mechanical stretch, pyrene was released from the gels at a rate of approximately 0.05 ± 0.01 ng/min (black curve, Figure 4B). A slightly faster release rate (0.08 ± 0.02 ng/min) was observed within the first minute, possibly due to the presence of trace amount of free pyrene on the micelle surface that was rapidly released. Pyrene release rate was significantly modulated when the BCM-PAAm gels were mechanically stretched. Within the first 5 min of dynamic stretching up to 30% strain (red curve, Figure 4B), pyrene was released at a rate of 0.19 ± 0.02 ng/min, approximately 2.5 times faster than the rate observed from the static controls. When the strain was increased to 60%, pyrene release rate was accelerated accordingly at an average rate of 0.54 ng/min (blue curve, Figure 4B), 10 times faster than the static controls. During the first 5-min-off period, the release rate for the stretched and the static samples resumed to a similar baseline level of approximately 0.02 ng/min. During the second 5-min-on period (10 min to 15 min in Figure 4B), pyrene was released at a rate of 0.19 ± 0.02 ng/min and 0.34 ± 0.01 ng/min from gels subjected to 30% and 60% strain, respectively. The release rate remained unchanged for the static controls. In the following 5-min-off period, the pyrene release rate returned to 0.04 ± 0.01 ng/min for all gels. During the subsequent cycles, similar release patterns were observed, with an accelerated release during stretch (5 min-on) and diminished release during the rest period (5 min off). While the undulating pattern of release rate was maintained throughout the course of the release study, the relative difference in magnitude among the three samples tested gradually narrowed. By the sixth on-off cyclic, pyrene was released at a rate of 0.12 ± 0.01 ng/min from gels subjected to 60% strain, still 2 fold faster than the static controls.
Figure 4.

Effects of periodic stretching forces (A) on the release rate (B) and cumulative release of pyrene (C) from BCM16-PAAm gels.
Figure 4C shows the cumulative release profiles over the course of 60 min. Only 10% of pyrene initially loaded was released in an hour, with a relatively constant rate of 1.6% per min. In the presence of the selected stretching regimen, the amount of pyrene release during each stretch period at 60% strain was approximately two times higher than that at 30 % strain, and 2.5 times higher than the static controls. Cumulatively, 15% and 25 % of pyrene was released after one hour as a result of 30% external mechanical stretch and 60% mechanical stretch, respectively.
Collectively, our results suggest that the release behavior of pyrene encapsulated in BCM16-PAAm gels can be tuned by external mechanical forces. The hydrophobic pyrene molecules, encapsulated in the hydrophobic micelle cores through the hydrophobic association, could diffuse through the hydrogel matrix with a diffusion coefficient dependent on the concentration gradient. The diffusion mechanism dominated the pyrene release in the absence of stretch. It is possible that trace amount of pyrene was physically associated at the micelles at the hydrophilic-hydrophobic interface rather than being entrapped in the hydrophobic core. These molecules will be released from the gel relatively faster due to the shorter diffusion path. This contributed to the faster release rate observed at the beginning of the static measurement. In the presence of the external mechanical stimulation, block copolymer micelles, which are covalently linked to the hydrogel matrix, deformed along the stretch direction. Simultaneously, water molecules can penetrate into the hydrophobic core,20 thereby rendering the local microenvironment less hydrophobic. Consequently, the hydrophobic association between pyrene and the micelle core became weaker and pyrene release rate increased accordingly. In our system, the overall pyrene release rate is mediated by the combined effects of the free diffusion of pyrene molecules driven by the concentration gradient, the π-π stacking of pyrene molecules 37 in the PnBA core and the mechanical stretch imposed to the gel network. As more and more pyrene is released from BCM-PAAm gels, the concentration gradient is decreased, making it more difficult for the remaining pyrene to be released. Consequently, a higher strain may be necessary to release the remaining pyrene entrapped in the BCMs. Throughout this study, the hydrogel stretching rate was maintained constant. The strain rate-dependent drug release from BCM-crosslinked hydrogels is the subject of our on-going investigation.
In summary, a new type of hydrogel with hierarchical structure and unique mechano-responsiveness was described in this study. Mono-disperse spherical nanoparticles were firstly assembled from amphiphilic block copolymers consisting of a hydrophobic, rubbery block (PnBA) and a hydrophilic, stealth block (PAA) partially decorated with reactive acrylates. Elastomeric hydrogels were constructed via radical polymerization of acrylamide using functional BCMs as the microscopic crosslinkers. The mechanical properties of the resultant BCM-PAAm gels can be readily tuned by varying the block copolymer composition. Cross-sectional TEM analysis showed that macroscopic deformation exerted on the gels was effectively transmitted to the immobilized micelles, causing a significant micelle deformation without compromising the mechanical integrity of the hydrogels. The BCMs return to their original shape after the force is removed. Force-induced micelle deformation was further utilized for the step-wise release of pyrene that was loaded into the hydrophobic core of BCMs prior to gelation, under the control of external stretching. If pyrene is replaced by biologically active molecules, mechanical forces can be readily converted into biochemical cues to guide cells through various developmental stages, facilitating the repair and regeneration of mechanically active tissues.
Supplementary Material
Acknowledgements
This work was supported by National Science Foundation (Career DMR-0643226 to XJ; DMR-0906815 to DJP) and National Institutes of Health (NIDCD: 3R01DC008965-03S2 to XJ). We thank the Keck Electron Microscopy Lab and Dr. Chaoying Ni and Mr. Frank Kriss for TEM assistance. We also appreciate Dr. Steve Bai for his help with NMR analyses.
Notes and References
- 1.Calvert P. Adv. Mater. 2009;21:743–756. [Google Scholar]
- 2.Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, Peppas NA. Adv. Mater. 2009;21:3307–3329. doi: 10.1002/adma.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jagur-Grodzinski J. Polym. Adv. Technol. 2010;21:27–47. [Google Scholar]
- 4.Hoare TR, Kohane DS. Polymer. 2008;49:1993–2007. [Google Scholar]
- 5.Kopecek J. Biomaterials. 2007;28:5185–5192. doi: 10.1016/j.biomaterials.2007.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wichterle O, Lim D. Nature. 1960;185:117–118. [Google Scholar]
- 7.Wu D-Q, Sun Y-X, Xu X-D, Cheng S-X, Zhang X-Z, Zhuo R-X. Biomacromolecules. 2008;9:1155–1162. doi: 10.1021/bm7010328. [DOI] [PubMed] [Google Scholar]
- 8.Chen GH, Hoffman AS. Nature. 1995;373:49–52. doi: 10.1038/373049a0. [DOI] [PubMed] [Google Scholar]
- 9.Onoda A, Arai N, Shimazu N, Yamamoto H, Yamamura T. J. Am. Chem. Soc. 2005;127:16535–16540. doi: 10.1021/ja052477m. [DOI] [PubMed] [Google Scholar]
- 10.Kokufata E, Zhang YQ, Tanaka T. Nature. 1991;351:302–304. [Google Scholar]
- 11.Miyata T, Asami N, Uragami T. Nature. 1999;399:766–769. doi: 10.1038/21619. [DOI] [PubMed] [Google Scholar]
- 12.Murakami Y, Maeda M. Biomacromolecules. 2005;6:2927–2929. doi: 10.1021/bm0504330. [DOI] [PubMed] [Google Scholar]
- 13.Shimoboji T, Larenas E, Fowler T, Kulkarni S, Hoffman AS, Stayton PS. Proc. Natl. Acad. Sci. U. S. A. 2002;99:16592–16596. doi: 10.1073/pnas.262427799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka T, Nishio I, Sun ST, Uenonishio S. Science. 1982;218:467–469. doi: 10.1126/science.218.4571.467. [DOI] [PubMed] [Google Scholar]
- 15.Xulu PM, Filipcsei G, Zrinyi M. Macromolecules. 2000;33:1716–1719. [Google Scholar]
- 16.Discher DE, Janmey P, Wang Y.-l. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 17.Lee KY, Peters MC, Anderson KW, Mooney DJ. Nature. 2000;408:998–1000. doi: 10.1038/35050141. [DOI] [PubMed] [Google Scholar]
- 18.Li L, Teller S, Clifton RJ, Jia X, Kiick KL. Biomacromolecules. 2011;12:2302–2310. doi: 10.1021/bm200373p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwon G, Naito M, Yokoyama M, Okano T, Sakurai Y, Kataoka K. J. Controlled Release. 1997;48:195–201. [Google Scholar]
- 20.Li ITS, Walker GC. Proc. Natl. Acad. Sci. U. S. A. 2011;108:16527–16532. doi: 10.1073/pnas.1105450108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gohy J-F. In: Adv. Polym. Sci. Abetz V, editor. vol. 190. Springer; Berlin / Heidelberg: 2005. pp. 65–136. [Google Scholar]
- 22.Riess G. Prog. Polym. Sci. 2003;28:1107–1170. [Google Scholar]
- 23.Colombani O, Ruppel M, Schubert F, Zettl H, Pergushov DV, Muller AHE. Macromolecules. 2007;40:4338–4350. [Google Scholar]
- 24.Natalya R. Prog. Polym. Sci. 2007;32:962–990. [Google Scholar]
- 25.Aliabadi HM, Mahmud A, Sharifabadi AD, Lavasanifar A. J. Controlled Release. 2005;104:301–311. doi: 10.1016/j.jconrel.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 26.Xiao LX, Liu C, Zhu JH, Pochan DJ, Jia XQ. Soft Matter. 2010;6:5293–5297. doi: 10.1039/C0SM00511H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shuai X, Ai H, Nasongkla N, Kim S, Gao J. J. Controlled Release. 2004;98:415–426. doi: 10.1016/j.jconrel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 28.Fisher JP, Timmer MD, Holland TA, Dean D, Engel PS, Mikos AG. Biomacromolecules. 2003;4:1327–1334. doi: 10.1021/bm030028d. [DOI] [PubMed] [Google Scholar]
- 29.Vogt AP, Sumerlin BS. Soft Matter. 2009;5:2347–2351. [Google Scholar]
- 30.Hunt JN, Feldman KE, Lynd NA, Deek J, Campos LM, Spruell JM, Hernandez BM, Kramer EJ, Hawker CJ. Adv. Mater. 2011;23:2327–2331. doi: 10.1002/adma.201004230. [DOI] [PubMed] [Google Scholar]
- 31.Jha AK, Hule RA, Jiao T, Teller SS, Clifton RJ, Duncan RL, Pochan DJ, Jia XQ. Macromolecules. 2009;42:537–546. doi: 10.1021/ma8019442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu J, Hiwatashi K, Kurokawa T, Liang SM, Wu ZL, Gong JP. Macromolecules. 2011;44:7775–7781. [Google Scholar]
- 33.Vogel V, Sheetz M. Nat. Rev. Mol. Cell Biol. 2006;7:265–275. doi: 10.1038/nrm1890. [DOI] [PubMed] [Google Scholar]
- 34.Ingber DE. FASEB J. 2006;20:811–827. doi: 10.1096/fj.05-5424rev. [DOI] [PubMed] [Google Scholar]
- 35.Allen C, Yu Y, Maysinger D, Eisenberg A. Bioconjugate Chem. 1998;9:564–572. doi: 10.1021/bc9702157. [DOI] [PubMed] [Google Scholar]
- 36.Keyes-Baig C, Duhamel J, Fung S-Y, Bezaire J, Chen P. J. Am. Chem. Soc. 2004;126:7522–7532. doi: 10.1021/ja0381297. [DOI] [PubMed] [Google Scholar]
- 37.Kalyanasundaram K, Thomas JK. J. Am. Chem. Soc. 1977;99:2039–2044. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.