

Abstract
The generation of transgenic mice by DNA microinjection is a powerful tool to investigate the molecular regulation of gene expression, development, and disease. The power of this technology is that foreign DNA can be introduced into every cell of a developing organism and the phenotypic impact of this genetic modification can be investigated in a system under the constraints of normal development and physiology. The generation of transgenic mice requires the preparation of the transgene DNA construction, collection of one-cell fertilized mouse embryos, injection of the transgene into mouse embryos, and transfer of the surviving embryos. Mice born from such manipulations are then screened for the presence of the transgene. The execution of these procedures requires a highly efficient system otherwise the cost of the generation of these mice can be cost prohibitive. However, the production of these animals can serve as an invaluable research resource.
Keywords: Transgenic, mouse, DNA microinjection
Introduction
One of the major quantum leaps in technology that has rapidly advanced biological research is the ability to modify the mammalian genome by either the introduction of foreign DNA sequences or by modification of endogenous DNA sequences (Brinster and Palmiter, 1984; Capecchi, 1989). The ability to introduce these modifications into the genome of mammalian species has allowed mechanistic investigation into how specific genes regulate or deregulate developmental and physiological processes. This technology is a result not only of advances in molecular biology, but also of research dedicated to understanding how altered gene expression impacts development, physiology, and diseases. These advances have allowed genomic modification technologies to evolve to a point where temporal control of gene expression and ablation can be achieved (Lewandoski, 2001). Continued progress in this field is expected to provide increasingly refined means to control gene expression; nonetheless, fundamental understanding of genome-level manipulation is required to fully and efficiently utilize novel advances. The goal of the following protocol, therefore, is to provide the researcher with the fundamental embryo manipulation skills required for the generation of transgenic mice by DNA microinjection into the one-cell mouse embryo.
One of the original breakthrough technologies in the generation of genetically engineered mouse models was successful introduction of foreign DNA into the pronucleus of the one-cell mouse embryo. The mice generated by this approach were termed “transgenic mice” and although the initial mice generated consisted of simple fusion DNA constructs containing relatively small (< 20 kilobase pairs) fragments of DNA, the technique quickly evolved to allow generation of transgenic mice using Bacterial Artificial Chromosomes (BAC) clones containing hundreds of base pairs of DNA.
Generation of transgenic mice is possible because introduced DNA integrates into the germ line. The foreign DNA can then direct transcription in a predictable manner, and the genetic manipulation is transmitted to future generations (due to integration into the germ line), thus establishing a new and novel strain of the organism. First reported decades ago by Gordon and coworkers, the fundamental principles remain largely true to this day (Gordon and Ruddle, 1981). The procedure consists of preparation of the transgene, embryo collection, microinjection, transfer of the embryos, and identification of the transgenic offspring; these steps have been described more recently by Nagy and coworkers in great detail.(Nagy et al., 2003).
Although there have been many technical improvements in this procedure (i.e. with respect to equipment and reagents), this protocol is geared towards the less-experienced investigator, and focuses on fundamentals by describing a basic, yet complete system for the generation of transgenic mice. This manuscript may be useful irrespective of whether one wishes to outsource the generation of transgenic mouse strains or to accomplish it in-house, since the underlying principles for creating genetically engineered mice have remained constant throughout the years, and thorough comprehension of the process is required prior to execution in-house or via a third party.
An overview of the entire procedure for generating genetically engineered mice is shown in Figure 1. Briefly, the protocols in this manuscript describe the following procedures:
Figure 1.

The overall scheme for the generation of genetically engineered mice.
Basic Protocol 1: Preparation of Transgene DNA for microinjection.
Basic Protocol 2: Superovulation of embryo donor mice and embryo collection.
Basic Protocol 3: DNA microinjection into one-cell mouse embryos.
Basic Protocol 4: Embryo transfer to the oviducts of synchronized pseudopregnant females.
Basic Protocol 5: Identification of transgenic founder mice.
Basic Protocol 1: Preparation of Transgene DNA
Prior to the preparation of the transgene DNA fragment for microinjection, there are several considerations in transgene design to be considered to ensure maximal success in expression of the transgene. Most transgene DNA constructions are fusion genes with tissue specific promoters driving specific genes of interest. In designing this construction, there are several rules that should be followed to ensure success; please refer to the Commentary for additional information.
The protocol below outlines the basic methods required to produce transgenic DNA for microinjection into embryos.
Materials
Restriction enzyme and appropriate reaction buffer
TAE Buffer
Agarose
Ethidium bromide
Gel tray and box
GENE CLEAN II kit
Tris
EDTA
UV/Vis Spectrophotometer
TE buffer (10 mM Tris pH 7.5, 0.25 mM EDTA)
NucleoBond® BAC 100 kit
P1 (50:10TE): 50mM Tris-HCl, 10mM EDTA, pH 8.0. Store at 4oC
P2: 200mM NaOH, 1% SDS. RT
P3: 2.8M K-Acetate, pH 5.5. Store at 4oC
7.5M K-Acetate: 184g K-Acetate in 250 ml sterile water. Filter sterilize or autoclave
0.1 mM EDTA
30 mM spermine
70 mM spermidine
100 mM NaCl
1000x Polyamine Stock
30 mM Spermine (Sigma, tetrahydrochloride, #S-1141)
70 mM Spermidine (Sigma, trihydrocholoride, #S-2501)
Dissolve the spermine and spermidine together in autoclaved distilled water, filter sterilize (0.2 micron filters), and store at -20oC. Since the polyamines are very hygroscopic, it is suggested that small quantities (1 gram) should be ordered and then all of it should be prepared at once.
1M Tris-HCl, pH 7.5 (autoclaved)
0.5 M EDTA, pH 8.0 (autoclaved)
5 M NaCl (autoclaved)
Preparation of Transgene DNA for microinjection
The transgene is usually separated from the vector sequences by agarose gel electrophoresis using the following protocol.
Cut 100 ug of your plasmid in 300 ul of reaction buffer. The plasmid must be cut with the appropriate restriction endonuclease(s) to clearly separate your transgene from all vector sequences.
Run digestion out on a 1.0% TAE agarose gel (Fisher Scientific, Fairlawn, NJ) with ethidium bromide (1 ul EtBr per 150 ul gel) and excise the appropriate band.
- Weigh band and use GENE CLEAN II (MP Biomedicals, Solon, OH) protocol with the following modifications:
- Use only 40 ul of glass milk to bind fragment.
- Elute fragment from gel with 50 ul of 10 mM Tris pH 7.4, 0.25 mM EDTA.
Dilute 5ul of isolated fragment into a total volume of 250 ul and take the OD at 260 and 280. Following this protocol, the concentration of the transgene should be at least 100 ng/ul. The 260:280 ratio has to be at least 1.7.
The DNA is then diluted to 2 ng/ul in microinjection TE buffer (See Reagents and Solutions). The DNA is then microinjected into the male pronucleus of one-cell mouse embryos. This buffer and concentration has been shown to be optimal for incorporation of the transgene into the murine genome. (Brinster et al., 1985)
Preparation of BAC DNA for injection
If large fragments of genomic DNA averaging 100-300 kb need to be injected, then Bacterial Artificial Chromosome (BAC) clones would be used.(Zhao, 2001) BAC clones allow faithful tissue specific expression of heterologous genes in transgenic mice. (Gong et al., 2003; Heintz, 2000) The protocol for BAC DNA isolation according to Han et al.(Han et al., 2009) utilizes NucleoBond® BAC 100 kit (Clontech Laboratories, Inc., Mountain View, CA) has modifications in accordance with the following references: Human Genome Sequencing Center [BCM] [steps 6-23], Welcome Trust Centre for Human Genetics and Institute of Molecular Medicine, Oxford [HGIMM] [steps 24-27], University of Michigan - Transgenic Animal Model Core [U. Mich.] [alkaline lysis, steps 6-15 & step 31], The Nest Group [Nest Grp] [step 33].
6. Inoculate 800 ml (2x400 ml) of LB medium containing relevant antibiotics with 0.5 ml of an overnight culture. It is very important to start from a single freshly streaked colony. Shake at 37oC overnight (full 16-18 h. No longer than 18h).
7. Pellet the cells at 2,000 g (4,000 rpm for a JA10 rotor) for 15 min.
8. Discard supernatant, making sure to drain fully.
9. Resuspend each pellet completely with 30 ml/400 ml starting culture of chilled buffer P1 (no RNase).
10. Add 100,000U Ready-Lyse (Epicentre Biotechnologies, Madison, WI) to each 400 ml starting volume, incubate at RT for 15min.
11. Carefully add 30ml of buffer P2 (by washing the side of the bottle with P2) and incubate at RT for 5min. DO NOT MIX OR VORTEX. If cell lysis is not well achieved, roll the tubes on bench top very gently.
12. Again, carefully add 30 ml of chilled buffer P3 (by washing the side of the bottle with P3) and incubate on ice for 15 min, DO NOT MIX OR VORTEX.
13. Spin at 15,000 g (9000 rpm for a JA10 rotor) for 30 min at 4oC.
14. Gently pour the supernatant into a clean bottle and repeat the spin for an additional 15 min at 15,000 g (9000 rpm for a JA10 rotor).
15. Transfer the supernatant through the Clontech filters to a third bottle and add 45 ml (half of the added volume of P1+P2+P3), mix gently, but thoroughly.
16. Spin at 6,000 g (7,500 rpm for a JA10 rotor) for 15 min at 4oC.
17. Decant the supernatant and drain the pellet completely.
18. Dissolve the pellet with 9 ml standard TE and transfer to 30 ml high-speed centrifugation tubes.
19. Add 4.5ml of 7.5M K-Acetate (autoclaved), mix very gently and place at -80oC for 30 min.
20. Completely thaw into a RT to 30oC water bath and centrifuge at 5,000 g (6500 rpm for a JA10 rotor) for 12 min.
21. Pour the supernatant into another 30 ml centrifugation tube and repeat the previous centrifugation step.
22. Mix with 27 ml of 100% ethanol and spin at 5,000 g (6500 rpm for a JA10 rotor) for 12 min.
23. Resuspend the pellet with 1 ml of buffer P1 (with RNase), incubate at 37oC for 30min.
24. Add 100ul of 10% SDS and mix. Add proteinase K to a final concentration of 0.2mg/ml. Digest at 50-60oC for 3h or overnight.
25. Extract the DNA solution with Buffered phenol (pH8). DO NOT VORTEX.
26. Extract the DNA solution with Chloroform, successively. DO NOT VORTEX.
27. Extract the DNA solution with Chloroform, successively. DO NOT VORTEX.
28. Add 7.5 M Ammonium Acetate for a final concentration of 2.5M (0.5vol). Precipitate DNA with 0.5vol isopropanol at room temperature, mix thoroughly.
29. Centrifuge in 1.5 ml eppendorf tubes at 12,000 rpm for 5 min.
30. Wash pellet with 1 ml 70% ETOH, add 300ul of 50:10 TE buffer to the pellet (to improve DNA stability), incubate at 37oC for 30 min and leave at RT overnight or for at least 3 hr.
31. Add 12ml of Clontech buffer N2 (Equilibration buffer) and proceed through pre-equilibrated columns as prescribed by the manufacturer. Resuspend in TE 50:10. Do not let the DNA dry too long (translucent, not white), incubate at 37oC for 30 min and let resuspend overnight.
32. Linearization: Digest with terminase (or other appropriate enzyme), for at least 3h to overnight.
33. Phenol:Chloroform DNA purification. Let the pellet dry for 15 min and dissolve in 20 ul of newly filtered BAC microinjection buffer for 30 min at 37oC.
DNA Quantification
34. Quantification: (a) OD reading: Microinjection DNA must meet the following parameters: 260/280 of 1.70 to 2.0, (<1.7 indicates protein contamination, >2.0 indicates phenol and/or Chloroform contamination. In both cases, you need to ethanol precipitate DNA one more time). (b) Digest with frequent cutter, like BamHI, and run on a 0.8% agarose gel, you should see a ladder pattern with sharp bands and no smear. (c) Run 1 ul of linearized DNA on a pulse field gel. You should see a sharp band of expected size, no smear nor genomic DNA contamination.
35. DNA storage: Store resuspended DNA at 4oC until microinjection day.
36. On the morning of microinjection, thaw DNA, do OD reading again, dilute into 1ng/ul in BAC microinjection buffer and keep on ice until injected. Higher concentration might be toxic.
Basic protocol 2: Embryo Collection
Normally, hybrid and outbred stains of mice respond well to hormonal induction of ovulation. However, use of hybrid animals severely limits the experiments that can be conducted once the mice are generated. Currently, two inbred stains are most commonly used to generate transgenic mice. These are the FvB/N and the C57BL/6 lines of mice. The former produces a large number of embryos with a higher efficiency of generating transgenic mice, primarily due to a slightly larger pronucleus and a more translucent cytoplasm.(Auerbach et al., 2003) However, the latter strain is the most popular since these mice are of a genetic background which can be bred to mice generated by ES cell technology (129Sv X C57Bl/6 or C57Bl6). Once the strain is selected, embryos can be collected by utilizing a superovulatory regimen of gonadotropins as described below.
Materials
4 week old female mice
Breeder male mice
PMS
hCG
PBS
TB syringes
Microdissection tools
Spherical depression slides (Electron Microscopy Science, Hatfield, PA)
Hyaluronidase
M2
KSOM
Superovulation and embryo collection
- Day 1, PMS (VWR International, Inc. Bridgeport, NJ Cat# 80056-608) (5 IU) is given at 10 AM four days before the day of microinjection.
- (The age of superovulation is critical since older mice respond less to hormonal induction of ovulation. The best response to superovulation is achieved using 3 week old females)
Day 3, hCG (purchase from local pharmacy, pregnyl hCG or substitute) (5 IU) is given 47 hours after PMS at 9 am, one day before the day of microinjection.
Place the females with intact males immediately after administering hCG.
Check for vaginal plugs at 7:00 - 8:00 a.m. on Day 4.
Female mice are then euthanized and oviducts excised.
The oviducts are placed in M2 (Millipore, Billerica, MA Cat# MR-015-D) media supplemented with 0.1 % hyaluronidase (Sigma-Aldrich, St. Louis, MO Cat# H3506-1G).
The ampulla of the oviduct where the embryos collect is gently torn with a 30 gauge needle or watchman forceps and the embryos are expelled into the M2 with hyaluronidase. (Figure 2.)
- Once the cumulus cells are removed by the hyaluronidase from the embryos, the embryos are transferred to M2.
- Do not let the embryos sit in the hyaluronidase too long, 2-3 minutes is generally enough time for the cumulus cells to be removed. Embryos left in hyaluronidase too long become sticky and are hard to inject.
Fertilized embryos are identified by the presence of pronucleus and are separated from unfertilized embryos.
The fertilized embryos are again washed in M2 media and prepared for microinjection.
Figure 2.
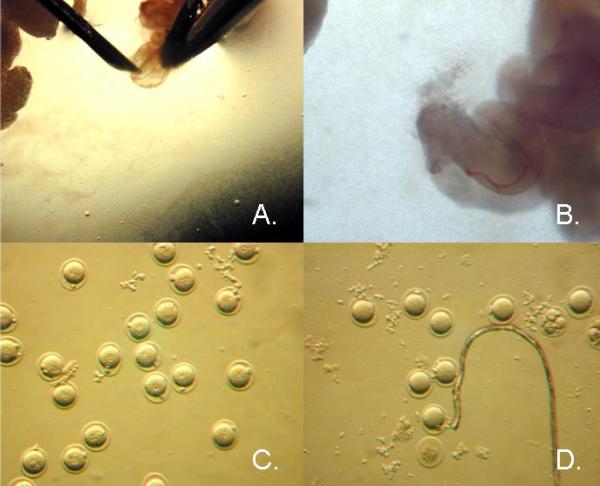
Embryos Collection. (A.) Ampulla of the mouse oviduct being gently torn to release embryos. (B.) Embryos being released from ampulla. (C.) Fertile one-cell embryos. (D.) Unfertilized ova and fragmented cells.
Basic Protocol 3: Microinjection
The embryos are microinjected with the aid of an inverted microscope equipped with differential contrast optics, usually Nomarski or Hoffman objective lenses, and micromanipulators. The embryos are injected with the aid of 2 needles. The Holding needle anchors the embryo in place with suction supplied by a micrometer syringe and the Injection needle which has a continuous flow of DNA being forced through its opening to allow the DNA to be introduced into the pronucleus. The DNA flow is given by positive pressure driven supplied by a micrometer syringe and allows the introduction of 2 pl of DNA at a concentration of 2 ng/ul. (Figure 3.)
Figure 3.

Inverted microscope with Hoffman objective lenses and micromanipulators. Holding and injection needles are in place and lowered into injection dish containing embryos.
Materials
Inverted microscope, such as Nikon, Zeiss
Narishige or Eppendorf Micromanipulators Manipulators – Coarse and Hydrolic
Injectors
Micrometer syringes for injectors
Teflon tubing
Paraffin oil
Borosilicate disposable micropipettes with filament
Sutter Micropipette Puller – P1000 or P-97
Disposable micropipettes
Sensaur Microforge
M2
Cytochalasin B
DOW Corning 200 fluid
KSOM
Petri dishes with tight lids M16
Preparation of needle
Injection Needle
The Injection Needles used are fashioned from Sutter Instrument Company glass disposable micropipettes. (Borosilicate with filament, catalog# BF100-78-10).
The pipettes are pulled on a Sutter micropipette puller. The setting will vary for different micropipette pullers. Refer to the Sutter Instrument P-1000 & P-97 Pipette Cookbook 2011 - http://www.sutter.com/contact/faqs/pipette_cookbook.pdf.
The needles are then filled with DNA. The needles chosen have a microfilament on the inside of the pipette. The DNA is filled into the needle by placing the base of the needle in an Eppendorf tube with 20 ul of transgene in injection buffer. Capillary action allows the DNA to migrate to the tip of the needle. The needle usually takes 5 to 10 minutes to allow a sufficient amount of DNA to migrate to the tip.
Once completed, the Injection needle is fitted to Teflon tubing, which is connected to a micrometer syringe filled with air. The needle is then mounted on the micromanipulator. Pressure is then increased in the needle by turning the micrometer syringe one-quarter turn.
Even though the injecting needle is not sealed, the opening is so minute that the DNA flow is very slow. The needle must be opened further. This is accomplished by viewing the needle under 400x magnification and using the micromanipulators to brush the injecting needle against the polished holding needle surface. This will open the injecting needle slightly. (See Figure 4.)
Figure 4.
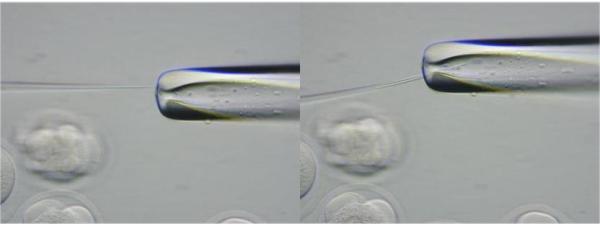
Enlarging opening of injection needle by brushing needle against the polished holding needle.
Holding Needle
6. The Holding needles used are fashioned from 10 ul glass disposable micropipettes from VWR.
7. The pipettes are pulled on a Sutter micropipette puller. The setting will vary for different micropipette pullers. Refer to the Sutter Instrument P-1000 & P-97 Pipette Cookbook 2011 - http://www.sutter.com/contact/faqs/pipette_cookbook.pdf.
8. The Holding needle is cut and polished using a Sensaur microforge.
9. Cutting the Holding Needle: This is accomplished by placing the needle against the microforge filament glass ball and heating the ball until the needle lightly fuses to the ball. The heat to the filament is then stopped. As the ball cools, it pulls away from the needle breaking the needle at the place where it was fused to the ball. The tip of the holding needle should be the size of the embryo (c 80 um). The approximate place to break the holding needle can only be learned with practice.
10. Polishing the Holding needle: After breaking the holding needle, its edges are polished by placing the needle tip near but not touching the glass ball of the microforge and heating it for about 3 seconds. The heat from the ball melts the edges of the holding needle. The edges should be, as flat as, possible, but not rounded. The inside of the holding needle should be small enough only to give proper suction.
11. Bending the Holding Needle: In some cases the needles must be bent to allow the needles to lie flat on the injection dish. The angle of bending is about 50. By holding the needle over the heated glass ball in the microforge, the proper angle can be made. The heat from the ball weakens the needle and causes it to bend around the ball. The needles are heated until the proper angle is achieved. This is done without closing the needle.
12. Once completed the holding needle is fitted to Teflon tubing, which is connected to a micrometer syringe prefilled with paraffin oil. The needle is then mounted onto the micromanipulator. Pushing paraffin oil through with the use of a Stoelting micrometer syringe fills the needles. The holding needle will fill easily.
As an alternative to pulling your own needles, needles can be purchased from Eppendorf or MicroJek.
DNA Pronuclear Injection
13. The embryos are placed in a 35 ul microdot of M2 media in the center of a petri dish (Fisher Scientific, Pittsburgh, PA Cat# 08-757-105) supplemented with Cytochalasin B.
Brinster and co-workers have reported that Cytochalasin B increases the survival of microinjected embryos by two fold (Brinster et al., 1980).
14. Make an injection chamber made using the lid of a Falcon 1006 petri dish. A 35 ul microdot of M2/CB is flattened in the center of the petri dish and covered with DOW CORNING® 200 FLUID 50CST (Ellsworth Adhesives, Germantown, WI).
15. Viewing the procedure under 40x, the injection needle is lowered into the microdot containing the embryos.
16. Under 40x the embryo is picked up with suction by the holding needle.
17. The injecting needle is positioned next to the embryo. (Figure 5)
Figure 5.
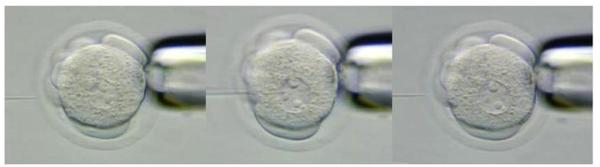
DNA microinjection into the male pronucleus of the one-cell mouse embryo.
18. Magnification is increased to 400x.
19. The microscope is focused until the larger, male pronucleus is visible; its nuclear membrane must not be confused with its nucleolar membrane.
20. Once the pronuclear membrane is in focus, the injecting needle is lowered until it is in the same focal plane as the nuclear membrane.
21. The injecting needle is then moved into the pronucleus and DNA injected while observing pronuclear swelling. (Figure 5.)
22. The amount of DNA you can inject into the pronucleus is about 2 pl. This is an approximate 50% increase in the diameter of the pronucleus and a doubling of its volume. Only practice will give you the ability to see how much you can swell the pronucleus without killing the embryo. If the pronucleus does not swell, you did not achieve a successful injection. (Figure 6.)
Figure 6.
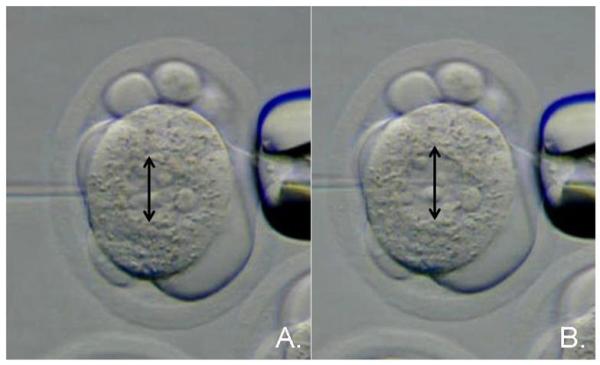
DNA microinjection into the male pronucleus of the one-cell mouse embryo. (A.) Injection needle moving into the male pronucleus of the one-cell embryo. (B.) Expansion of the male pronucleus upon delivery of DNA under hydrostatic pressure. The black arrows show that the pronucleus diameter increases in size by approximately 50%.
23. The injected embryo is separated from the non-injected embryos by moving the stage and releasing the embryo. Continue this same process until all embryos are injected.
24. After injection, the embryos are washed twice in M2 medium in order to remove the Cytochalasin B. The embryos, which survived microinjection, as judged by the appearance of normal morphology, are immediately transferred to pseudopregnant ICR females 0.5 dpc.
Note: If there are not enough recipients on the day of injection, the embryos may be cultured at 37°C, 5% CO2 KSOM (Millipore, Billerica, MA Cat# MR-106-D)
Lentiviral Injection – Subzonal Injection
25. Collect embryos as if for DNA microinjection.
26. Place embryos in a 50 ul microdot of M16 (Millipore, Billerica, MA Cat# MR-010P-5F) in the center of a petri dish (Fisher Scientific, Pittsburgh, PA Cat# 08-757-105) and cover with DOW CORNING® 200 FLUID 50CST (Ellsworth Adhesives, Germantown, WI).
27. Thaw an aliquot of the Lentiviral DNA (109 viral particles/ml) and centrifuge briefly at high speed (5 sec) to eliminate debris.
28. Assemble the micromanipulators as for DNA injection.
29. Place the dish of embryos onto microscope stage. Follow the same protocol as for DNA Microinjection for lowering the holding and injection needles into place.
30. Hold the embryos in place with the holding needle. Gently, push the injection needle through the zona pellucida and inject the perivitelline space without harming the embryo for 5-10 seconds.
31. Culture the embryos for 48 hours in M16 under oil at 37oC, 5% CO2.
32. Transfer surviving embryos to pseudopregnant ICR females 0.5 dpc.
Basic Protocol 4: Embryo Transfer
The success of generating genetically engineered mice is the ability to transfer the embryos that have undergone the specific manipulation to female mice and have these mice successfully gestate those embryos. The mouse is a favorable model for embryo transfer because the mouse can be induced to a stage that mimics pregnancy called pseudopregnancy.(Everett, 1969) (Vunder, 1970) If a female mouse is mated with a sterile male, the cervical stimulation by the act of coitus will trigger a neuroendocrine reflex that will allow the ovary to develop as if it were pregnant for 10 to 11 days. Therefore, the uterus will develop and be receptive for implantation 5 days after mating. If synchronized embryos are transferred to the reproductive tract prior to the receptive phase, implantation will occur and pregnancy will be maintained until term. If no embryos are transferred, the corpus luteum will regress and the female will begin to cycle. This process allows manipulated embryos to be transferred to mouse reproductive tracts without the presence of normal embryos from the recipient mother. Thus, the mouse can easily be made ready for embryo transfer without endocrine manipulation. Female mice are simply placed with vasectomized male mice and checked every morning for the presence of a post-coital vaginal plug. This plug is produced from the coagulation of the secretions of the male mouse accessory sex glands and indicates that coitus has occurred. Mice that have mated with the male mice, identified by the presence of a post-coital vaginal plug can be used for embryo transfer and will support embryo implantation on day 4.5 of pregnancy. (Day 0.5 is the morning of the plug).
Materials
ICR females >24 g
B6D2F1 vasectomized breeder males
Avertin
Hair clippers
Betadine
70% ETOH
Microdissection tools
Dissecting microscope
Embryo transfer pipettes
Mouth pipet and tubing
Wound clips and applicator
Preparation of recipients by natural mating
The most difficult process of transgenic mouse production is the embryo transfer. Successful embryo transfer depends not only on the viability of the embryo, but also on the ability of the foster mother to support a full term pregnancy.
1. ICR females (>24 g) are placed with vasectomized B6D2F1 males on the day prior to embryo collection.
By using males with hybrid vigor, the cost of the vasectomized male colony can be kept reasonable.
2. Females are checked for the presence of a vaginal plug at 7:00-8:00 a.m. on the day of injection.
Only females with visible plugs serve as recipients.
Embryo transfer
3. Anesthesia: Avertin (1.25% solution) is given at 0.2 ml/10 grams body weight IP of animal. The animal will reach surgical anesthesia in 3-5 min and be under for 20-30 minutes.
4. The hair on the dorsolateral side of the female is clipped.
5. The female is laid on her side and her dorsolateral side is washed with betadine and then 70% ETOH.
6. A small incision between the rib cage and the knee is made. Grasping the periovarian fat pad with a forceps exposes the ovary/oviduct.
7. Using iris scissors, an incision is made in the ovarian bursa to expose the fimbria of the oviduct.
8. The transfer pipette, as made for embryo collection, is filled with medium, a small air bubble and the embryos. (The volume containing the embryos must be minimal). The air bubble serves as a marker to show where the embryos have been transferred.
9. Under 4x magnification, the pipette is inserted through the fimbria into the oviduct and embryos expelled until you can see the air bubble in the oviduct.
10. The oviduct is then placed back into the peritoneal cavity.
11. The wound is closed with wound clips.
12. The mouse is placed on a heating pad (40oC) until it recovers from anesthesia.
13. Transfers are done to one side only, with 15 - 20 embryos transferred per mouse.
Support Protocol: Embryo culture
There are a number of embryo culture media that have been developed over the years. These media were designed to allow the efficient in vitro development of mouse embryos from the one-cell to blastocyst stage. The original mouse media were bicarbonate buffered salt solutions supplemented with glucose, lactate pyruvate and bovine serum albumin. The initial culture media were M16(Whittingham, 1971) and BMOC3 (Brinsters minimum ova culture media)(Brinster, 1968). Over the years, the culture media have improved and media such as CZB(Chatot et al., 1990) and KSOM(Erbach et al., 1994) are superior media to culture mouse embryos. However, all media rely on bicarbonate to buffer the pH. Therefore, all culture media must be equilibrated with carbon dioxide to be at the appropriate pH to support the embryo. Equilibration can be achieved by placing the media in the CO2 incubator over night to allow for equilibration. In order to manipulate the mouse embryo outside of a CO2 incubator, the embryos must be manipulated in a Hepes buffered media. The media of choice for such manipulation is M2 media.(Quinn et al., 1982)
Materials
35 mm Culture dishes
DOW Corning 200 fluid
M2
Appropriate culturing media, such as KSOM
Embryos are cultured in microdrops of medium covered with Silicon Oil (Dow Corning 200 Fluid) in 60 × 15 mm culture dishes.
4 microdrops of approximately 50ul of medium are spotted onto a 60 × 15 mm culture dish.
10 ml of Silicon (Dow Corning 200 Fluid) Oil are then poured on top of the drops. Embryos can be placed in the drops and cultured in an atmosphere of 5% C02 in air at 37oC.
Basic Protocol 5: Identification of Transgenic Mice
Once the mice born from the microinjection of DNA have been weaned, they are ready to be genotyped for the presence of the transgene. DNA is routinely isolated from tail biopsies and screened by either Southern Blot Analysis (Southern, 1975) or polymerase chain reaction, PCR. PCR screening of transgenic mice is by far the most rapid and easiest way to identify mice. However, limited information can be deemed by PCR screening of transgenic mice. Southern blot analysis, although slower and more laborious than PCR, is extremely useful to gain some initial information regarding the genetics of the founder transgenic mice, F0s. Below is a working protocol for DNA extraction from mouse tails for Southern blot analysis or PCR analysis of transgenes.
Materials
Avertin
Eartag or ear punch
TNES
Proteinase K
1.5 ml tubes
Vortex
NaCl
95% ETOH
Pasteur pipettes
70% ETOH
TE buffer
DNA extraction from mouse tails
Anesthetize mouse with Avertin
Number the mouse by eartag or by ear punch.
Cut 5 to 10 mm of tail tissue and put into a 1.5 ml microtube, placing on ice.
Prepare amount of TNES and Proteinase K, PK, (600ul of TNES + 10ul PK) for each tail needed for all tails cut and mix in a 15 or 50 ml tube. e.g. For 20 tails: 20 tails x 600 ul TNES = 12,000ul of TNES and 20 tails x 10 ul PK = 200ul of PK.
Vortex briefly (1 –2 seconds) to mix buffer.
Add 610 ul of TNES/PK buffer to each tail sample and incubate at 550C for 5 hours to overnight.
Add 200.4 ul NaCl and immediately shake vigorously (by hand) for 10 to 15 seconds.
Spin for 10 minutes at 14,000 x g.
Transfer supernatant to a new 1.5 ml microtube and add 800 ul of cold 95% ETOH.
Invert tubes individually several times to precipitate DNA (white thread).
Spool DNA onto forged Pasteur pipette tip.
Place spooled DNA and tip into cold 70% ETOH to wash.
Place spooled DNA and tip into another 1.5 ml tube.
Dry spooled DNA for 1 hour.
Resuspend pellet in 150 ul TE buffer.
Leave sample O/N at room temp or stored at 4oC.
Reagents and Solutions
Avertin Stock Solution
Dissolve 25 g Tribromoethannol (Sigma-Aldrich, St. Louis, MO Cat# 2,2,2-tribromoethanol - T48402-25G) in 15.5 ml Amylene hydrate (Fisher Scientific, Pittsburgh, PA Cat# A730-1). NOTE: This is very noxious. It is best to use an amber bottle (glass only) with a magnet for stirring. Let this dissolve over night in a chemical hood. Store in refrigerator.
Avertin Working Solution
Dilute 1.0 ml of Avertin Stock Solution in 79.0 ml PBS. Store in refrigerator.
Cytochalasin B
A stock of 1 mg/ml Cytochalasin B (Sigma-Aldrich, St. Louis, MO Cat# C6762) in DMSO is kept in 5 ul aliquots at -20. Prior to use 2.5 ul Cytochalasin B is added to 497.5 ul media.
TAE
Add the following to 900ml distilled H2O
242g Tris base
57.1ml Glacial Acetic Acid
18.6 g EDTA
Adjust volume to 1L with additional distilled H2O
Microinjection TE buffer
10mM Tris pH 7.5
0.25 mM EDTA)
TNES: 50 mM Tris, 0.4M NaCl, 1 mM EDTA, 0.5% SDS
100 ml stock solution:
TNES: 1M Tris, pH 7.85 ml
5M NaCl 8 ml
0.5M EDTA 0.2 ml
10% SDS 5 ml
dHOH 82 ml
Commentary
Background Information
When preparing transgenic DNA, there are several rules that must be followed to maximize success in generating transgenic mice. First, the promoter used should be one that has been proven to drive a transgene in mice at a level that gives a phenotypic response. Since transgenes integrate randomly in the genome and their expression is most often influenced by the site of integration, it is important that the promoter used has all the elements needed for appropriate tissue specific expression. Second, the gene of interest should be the genomic version of the gene containing the endogenous intervening sequences. This is necessary because transgenes generated from cDNA of the genes do not express as frequently and at the level of the genomic versions of the genes. Since most genes are too large to include all the endogenous introns, most investigators choose to use minigenes containing some of the endogenous introns or use heterologous introns. Third, the constructions must contain a proven polyadenylation signal. Usually, the poly adenylation sequences from the growth hormone gene will suffice. Finally, the construction must be able to be freed from the prokaryotic plasmid sequences. This latter point is critical. Prokaryotic sequences will dampen transgene expression.(Clark et al., 1997) The incorporation of prokaryotic sequences inhibits the expression of the transgene in the murine genome.
Critical Parameters and Troubleshooting
Basic Protocol 1
General consideration for BAC purification & Handling:
Maximize precautions using BAC DNA. Do not vortex. Do not pipette up/down. Do not repeatedly freeze thaw. Always use wide bore tips if repeated manipulations of the DNA are necessary. Store BAC DNA at 4°C (several weeks) or freeze aliquots at -80°C.
Concentration measurements by UV spectrophotometer are not very reliable since you can have absorbing contaminants, as well as, significant amounts of E.Coli DNA. Therefore, pulse field gel electrophoresis of Not I digested BAC DNAs is the method of choice to evaluate the integrity, quality and quantity of BAC DNA
Basic Protocol 2
In the process of generating transgenic mice, one must consider the genetic background in which to generate transgenic mice. In choice of mouse strain, there are several considerations. First, scientifically, which strain is more amenable to investigating the hypothesis being tested? For example, C57BL/6 mice may be best suited for metabolic studies while BALB/c and FvB mice may be more suited for the investigation of mammary gland biology. The second consideration is the practical one. Can enough embryos be collected to generate transgenic mice with economic feasibility? Since the frequency of transgenic mice from microinjection is 1 to 2 % per embryo injected, it is critical to choose a strain of mice which can be induced to produce a large number of embryos by hormonal induction (Brinster et al., 1985).
Basic Protocol 3
Maximizing success: The means of maximizing the production of transgenic mice is dependent upon not only the amount of DNA delivered to the male pronucleus, but also the expansion of the pronucleus. It is speculated that the hydrostatic pressure of the microinjection of the DNA causes breaks in the chromatin. The DNA repair process then integrates the transgene into the chromatin. Therefore, it is important to deliver not only a specified amount of DNA, but also the appropriate volume of injection medium. The 2 pl of 2 ng/ul of DNA is the suitable compromise. However, there is a fine line between the injection of the maximum volume of DNA and the volume tolerated by the embryo for survival. The usual survival after microinjection is between 70 to 80%. Higher survival rates than this in our hands is associated with a low frequency of incorporation. The efficiency of the generation of transgenic mice is measured by the number of transgenic mice generated per number of embryos injected and is 1 to 2 % (Brinster et al., 1985).
Basic Protocol 4
Successful embryo transfer requires the synchronous transfer of embryos to the reproductive tract of recipient mice (Review (Yoshinaga, 1988)). Embryos must be transferred to the uterus prior to the stage of “uterine receptivity otherwise the uterus will become refractory to implantation and the embryos will die. Therefore, when embryos are transferred to the uterus in an asynchronous fashion and the embryo is at an earlier stage of postovulatory development than the reproductive tract, the reproductive tract will advance beyond the receptive phase and become refractory to implantation before the embryo can initiate implantation. This type of embryo transfer will be unsuccessful. However, if the embryos are transferred at a more advanced postovulatory stage than the reproductive tract, the embryo will be held in a state of suspended animation until the uterus becomes receptive and the embryo will implant (Psychoyos, 1966). This embryo transfer will be successful. A basic rule for embryo transfer is: “the embryo will wait for the uterus to catch up but the uterus waits for no one.”
Basic Protocol 5
Southern blot analysis can be used to determine if the integrated transgene is intact, as well as, the number of copies of the transgene that have been integrated into the murine genome. This latter information is important should the transgene have integrated at more than one site in the murine genome or if the transgene expression is proportional to the number of integrated transgenes, the latter being a rare phenomenon.
Anticipated Results
When the procedures set forth in this manuscript are carefully followed, the investigator should have ample transgenic mice to use in their research. When at least 200 embryos are injected per construction, the efficiency of the generation of transgenic mice as measured by the number of transgenic mice generated per number of embryos injected is 1 to 2 % (Brinster and Palmiter, 1984). This should result in at least 2 to 4 independent transgenic mice per construction.
Time Considerations
The generation of transgenic mice is a long and arduous process that is made less cumbersome with practice and attention to detail. However, once the procedure of generating transgenic mice set forth in this manuscript is mastered and special attention is given to the details, the resulting mouse models are invaluable. Generally, it takes the investigator less than 1 month to design a construct, complete the plasmid prep, and isolate the DNA for microinjection. Mice will then be born 21 days after microinjection and transgenic mice are usually genotyped after weaning at 21 days of age. Once the transgenic mice are identified, expression analysis can then be conducted on offspring of the Fos. This requires waiting until the founder animals are of breeding age at 8 weeks. Generation of offspring will then take a minimum waiting of an additional 42 days. Therefore, from microinjection to identification of transgenic mice expressing the transgene will require a minimum of 5 to 6 months from the initial microinjection.
Literature Cited
- Auerbach AB, Norinsky R, Ho W, Losos K, Guo Q, Chatterjee S, Joyner AL. Strain-dependent differences in the efficiency of transgenic mouse production. Transgenic Res. 2003;12:59–69. doi: 10.1023/a:1022166921766. [DOI] [PubMed] [Google Scholar]
- Brinster RL. In vitro culture of mammalian embryos. J Anim Sci. 1968;27(Suppl 1):1–14. [PubMed] [Google Scholar]
- Brinster RL, Chen HY, Trumbauer ME, Avarbock MR. Translation of globin messenger RNA by the mouse ovum. Nature. 1980;283:499–501. doi: 10.1038/283499a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Chen HY, Trumbauer ME, Yagle MK, Palmiter RD. Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proc Natl Acad Sci U S A. 1985;82:4438–4442. doi: 10.1073/pnas.82.13.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Palmiter RD. Introduction of genes into the germ line of animals. Harvey Lect. 1984;80:1–38. [PMC free article] [PubMed] [Google Scholar]
- Capecchi MR. Altering the genome by homologous recombination. Science. 1989;244:1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- Chatot CL, Lewis JL, Torres I, Ziomek CA. Development of 1-cell embryos from different strains of mice in CZB medium. Biol Reprod. 1990;42:432–440. doi: 10.1095/biolreprod42.3.432. [DOI] [PubMed] [Google Scholar]
- Clark AJ, Harold G, Yull FE. Mammalian cDNA and prokaryotic reporter sequences silence adjacent transgenes in transgenic mice. Nucleic Acids Res. 1997;25:1009–1014. doi: 10.1093/nar/25.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbach GT, Lawitts JA, Papaioannou VE, Biggers JD. Differential growth of the mouse preimplantation embryo in chemically defined media. Biol Reprod. 1994;50:1027–1033. doi: 10.1095/biolreprod50.5.1027. [DOI] [PubMed] [Google Scholar]
- Everett JW. Neuroendocrine aspects of mammalian reproduction. Annu Rev Physiol. 1969;31:383–416. doi: 10.1146/annurev.ph.31.030169.002123. [DOI] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Gordon JW, Ruddle FH. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science. 1981;214:1244–1246. doi: 10.1126/science.6272397. [DOI] [PubMed] [Google Scholar]
- Han SJ, O’Malley BW, DeMayo FJ. An estrogen receptor alpha activity indicator model in mice. Genesis. 2009;47:815–824. doi: 10.1002/dvg.20572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz N. Analysis of mammalian central nervous system gene expression and function using bacterial artificial chromosome-mediated transgenesis. Hum Mol Genet. 2000;9:937–943. doi: 10.1093/hmg/9.6.937. [DOI] [PubMed] [Google Scholar]
- Lewandoski M. Conditional control of gene expression in the mouse. Nat Rev Genet. 2001;2:743–755. doi: 10.1038/35093537. [DOI] [PubMed] [Google Scholar]
- Nagy A, Gertsenstein M, Vintersten K, Behringer R. Manipulating the Mouse Embryo: A laboratory Manual. 3rd ed Cold Spring Harbor Press; Cold Spring Harbor, N.Y: 2003. [Google Scholar]
- Psychoyos A. [Study of the relations of the ovum and ondometrium during delay of nidation or the first phases of the nidation process in the female rat] C R Acad Sci Hebd Seances Acad Sci D. 1966;263:1755–1758. [PubMed] [Google Scholar]
- Quinn P, Barros C, Whittingham DG. Preservation of hamster oocytes to assay the fertilizing capacity of human spermatozoa. J Reprod Fertil. 1982;66:161–168. doi: 10.1530/jrf.0.0660161. [DOI] [PubMed] [Google Scholar]
- Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975;98:503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- Vunder PA. Hormonal regulation of the process of ovum implantation] Usp Sovrem Biol. 1970;70:106–119. [PubMed] [Google Scholar]
- Whittingham DG. Culture of mouse ova. J Reprod Fertil Suppl. 1971;14:7–21. [PubMed] [Google Scholar]
- Yoshinaga K. Uterine receptivity for blastocyst implantation. Ann N Y Acad Sci. 1988;541:424–431. doi: 10.1111/j.1749-6632.1988.tb22279.x. [DOI] [PubMed] [Google Scholar]
- Zhao S. A comprehensive BAC resource. Nucleic Acids Res. 2001;29:141–143. doi: 10.1093/nar/29.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]