

Abstract
Chronic myeloproliferative diseases without the Philadelphia chromosome marker (Ph-), although first described 60 years ago, only became the subject of interest after the turn of the millennium. In 2001, the World Health Organization (WHO) defined the classification of this group of diseases and in 2008 they were renamed myeloproliferative neoplasms based on morphological, cytogenetic and molecular features. In 2005, the identification of a recurrent molecular abnormality characterized by a gain of function with a mutation in the gene encoding Janus kinase 2 (JAK2) paved the way for greater knowledge of the pathophysiology of myeloproliferative neoplasms. The JAK2 mutation is found in 90-98% of polycythemia vera and in about 50% essential thrombocytosis and primary myelofibrosis. In addition to the JAK2 mutation, other mutations involving TET2 (ten-eleven translocation), LNK (a membrane-bound adaptor protein); IDH1/2 (isocitrate dehydrogenase 1/2 enzyme); ASXL1 (additional sex combs-like 1) genes were found in myeloproliferative neoplasms thus showing the importance of identifying molecular genetic alterations to confirm diagnosis, guide treatment and improve our understanding of the biology of these diseases. Currently, polycythemia vera, essential thrombocytosis, myelofibrosis, chronic neutrophilic leukemia, chronic eosinophilic leukemia and mastocytosis are included in this group of myeloproliferative neoplasms, but are considered different situations with individualized diagnostic methods and treatment. This review updates pathogenic aspects, molecular genetic alterations, the fundamental criteria for diagnosis and the best approach for each of these entities.
Keywords: Myeloproliferative disorders, Thrombocytosis, Polycythemia vera, Primary myelofibrosis
Introduction
In 1951, William Dameshek was the first author to report on the clinical and laboratory similarities of the four main hyperproliferative myeloid entities: chronic myeloid leukemia (CML), polycythemia vera (PV), essential thrombocytosis (ET), and primary myelofibrosis (PMF).(1) In 1975, the Group for the Study of Polycythemia Vera (PVSG) defined the diagnostic criteria and classification of Philadelphia (Ph) chromosome negative chronic myeloproliferative disorders. These parameters were redefined by Pearson in 1996.(2)
In 2001, the World Health Organization (WHO) proposed a new classification of these disorders, adding laboratory findings, imaging and evaluation of the bone marrow as diagnostic criteria.(3)
In 2008, through advances in cytogenetics and molecular biology, myeloproliferative disorders were renamed by the WHO as myeloproliferative neoplasms (MPN). In this classification, new groups were added taking into consideration relevant aspects such as clinical behavior, morphologic features, cytogenetics, and molecular changes.(4) Table 1 lists the diseases encompassed in the MPN group.
Table 1.
The 2008 World Health Organization Classification of Ph chromosome-negative myeloproliferative neoplasms 4
| Polycythemia vera |
| Essential thrombocytosis |
| Primary myelofibrosis |
| Chronic eosinophilic leukemia |
| Chronic neutrophil leukemia |
| Mastocytosis |
| Unclassifiable myeloproliferative neoplasm |
| Neoplasms associated with eosinophilia and PDGFR abnormalities |
The WHO 2008 classification of MPN included bone marrow (BM) histology as a diagnostic criterion as follows:(5)
- cellularity, maturation status, and topography of hematopoietic elements;
- morphology, arrangement and topography of megakaryocytes;
- evaluation of reticulin fibers, collagen deposits and hemosiderin contents;
- evaluation of anomalous infiltrating elements and atypically located blast aggregates;
- cell histogenesis by immunohistochemistry, allied with morphological and topographic evaluation;
- evaluation of CD34 and or CD117 expression by immunohistochemistry;
- presence of selected chromosome aberrations, by fluorescent in situ chromogenic hybridization.
Polycythemia vera
In the WHO 2008 classification of PV, a minor criterion refers to the BM findings: "hypercellularity with trilineage growth, prominent erythroid, granulocytic and megakaryocytic proliferation". Neither iron-laden macrophages nor reticulin fibers are conspicuous (increased 1+ or 2+ in less than 20% of the cases). Megakaryocytic alterations are important to distinguish PV from secondary polycythemia. In the myelofibrotic stage, which occurs in around 30% of the cases 10-12 years after diagnosis, it may be very difficult, if not impossible, to distinguish this from primary myelofibrosis.(6) [ Table 2; Figure 1A & 1B
Table 2.
Systematic histological bone marrow findings, which aid in the discrimination of chronic myeloproliferative neoplasms
| Histological findings | Polycythemia vera | Essential thrombocythemia | Primary myelofibrosis | |||
| Cellular phase | Fibrotic stage | |||||
| Increased cellularity | ++ | +++++ | ++ | |||
| Granulopoiesis | Increased | +++++ | ++++ | 0 | ||
| Left shift | ++++ | +++++ | +++ | ++ | ||
| Erythropoiesis | Increased | +++++ | + | + | 0 | |
| Left shift | +++++ | + | ++ | + | ||
| Megakaryopoiesis | Increased | ++++ | +++++ | ++++ | +++ | |
| Size | Small | +++ | 0 | +++ | +++ | |
| Medium | +++ | ++ | ++ | ++ | ||
| Large | +++ | +++ | ++++ | ++ | ||
| Giant | ++ | +++ | ++ | + | ||
| Topography | Endosteal translocation | ++ | ++ | +++ | +++ | |
| Cluster size | Small (≥ 3) | ++ | ++ | ++++ | ++++ | |
| Large (>7) | + | 0 | +++ | +++ | ||
| Cluster type | Dense | + | 0 | +++ | ++++ | |
| Loose | +++ | +++ | ++++ | ++ | ||
| Nuclei shape | Hypolobulation | ++ | + | ++++ | ++++ | |
| Hyperlobulation | ++++ | ++++ | ++++ | +++++ | ||
| Maturation defects | 0 | 0 | ++++ | +++++ | ||
| Naked nuclei | +++ | +++ | ++++ | +++++ | ||
| Fibers | Increased reticulin | ++ | 0 | ++++ | +++++ | |
| Increased collagen | 0 | 0 | 0 | +++++ | ||
| Osteosclerosis | 0 | 0 | 0 | |||
| Iron deposits | 0 | +++ | ++ | + | ||
| Lymphoid nodules present | ++ | 0 | ++ | + | ||
Semi-quantification: 0 usually absent; + rare; ++ slight; +++ moderate; ++++ manifest; ++++ overt
Adapted from: Thiele J, Kvasnicka HM. The 2008 WHO diagnostic criteria for polycythemia vera, essential thrombocythemia and primary myelofibrosis
Figure 1.
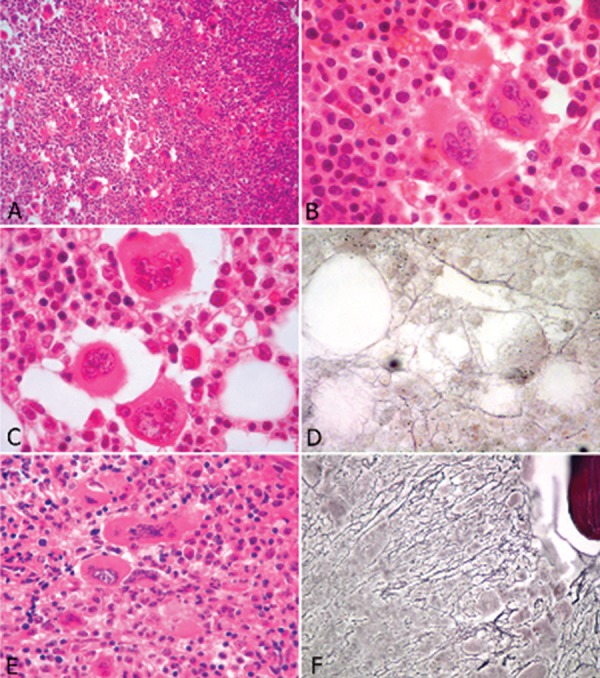
Histological features of chronic myeloproliferative neoplasms
A & B: Polycythemia Vera - Hypercellular bone marrow with increased numbers of erythroblasts and pleomorphic megakaryocytes (A:hematoxylin and eosin - magnification 200 x). Megakaryocytes show hyperlobated nuclei and are arranged in loose clusters (B: hematoxylin and eosin - magnification 1000 x)
C & D: Essential thrombocythemia - No significant alterations of the myeloid and erythroid series. Megakaryocytes are significantly increased in number, show hyperlobated nuclei and are isolated or arranged in loose clusters (C: hematoxylin and eosin - magnification 1000 x). Reticulin fibers are present in a loose network with some intersections (D: Gomori argentic impregnation - 1000 x)
Essential thrombocythemia
One of the four major criteria that define ET in the WHO 2008 classification is: "bone marrow biopsy showing proliferation mainly of the megakaryocytic lineage with increased numbers of enlarged, mature megakaryocytes. No significant increase or left-shift of neutrophil granulopoiesis or erythropoiesis". The nuclei of megakaryocytes are hyperlobulated or intensely folded, dispersed or loosely clustered. There is no or only a slight increase in reticulin fibers. In follow-up biopsies, it was shown that increases of reticulin fibers is indicative of progression to myelofibrosis.(7,8) [Table 2; Figure 1C& 1D]
Primary myelofibrosis
PMF is characterized in the WHO 2008 classification (major criterion) as: "presence of megakaryocyte proliferation and atypia, usually accompanied by reticulin and/or collagen fibrosis, or in the absence of significant reticulin fibrosis, the megakaryocytic changes must be accompanied by an increased bone marrow cellularity characterized by granulocytic proliferation and often decreased erythropoiesis (i.e., prefibrotic cellular-phase disease). There may be a moderate increase in reticulin fibers, but no collagen is seen. Megakaryocyte alterations are rather discriminating: anomalous topography, with prominent morphological abnormalities: pleomorphism, aberration of the nuclear cytoplasmic ratio due to large, bulbous and hyperchromatic cloud-shaped nuclei, disorganization of nuclear lobulation, naked megakaryocytes and maturation arrest. Early fibrosis leads to sinus dilatation as a result of stromal retraction.(9) [ Table 2; Figure 1E & 1F]
Cytogenetics
The identification of molecular genetic changes in chronic MPN is important to confirm the diagnosis, direct treatment, and improve our understanding of the biology of a disease.
The karyotype provides a comprehensive assessment of chromosomal abnormalities. It should be indicated whenever there is suspicion of MPN with leukocytosis, eosinophilia, monocytosis and/or thrombocytosis.(10) The identification of the Ph chromosome [t(9;22)(q34,q11)] or its variants confirm diagnosis of CML.(10) However, the investigation should be extended in the presence of other chromosomal abnormalities (Table 3).(1) The fluorescence in situ hybridization (FISH) and molecular cytogenetic methods are specific, sensitive and fast. These techniques have been recommended for situations in which there is no metaphases for analysis or in the absence of Ph karyotype.(11) In some cases, the study of molecular mutations can be made by polymerase chain reaction (PCR) or reverse transcriptase PCR (RT-PCR). At the diagnosis of PV, 10 to 30% of karyotypes are changed, but with the progression of the disease, the rate of cytogenetic abnormalities affects over 80% of cases, particularly in the fibrotic phase or at transformation to acute leukemia after polycythemia.(12) Approximately 50 to 60% of patients with PMF present altered karyotypes. Cases of ET show a smaller percentage of abnormalities at diagnosis ( Table 3).(11,12) But differentiation with myelodysplastic syndrome (MDS) should be made with the identification of del(5q) and t(3, 3)/inv(3).
Table 3.
Most frequent chromosomal alterations in myeloproliferative neoplasms
| Disease | Karyotype | FISH | Molecular changes |
| Chronic myeloid leukemia | t(9;22)(q34;qll) | BCR-ABL1 | BCR-ABL1 |
| Polycythemiavera | +8,+9, 20q-, 1q+, 1q-, 13q- | Centromere 8, 9, 20q-, 13q- | JAK2V617F>90% |
| Frequently homozygous | |||
| Primarymyelofibrosis | 8,+1q, +9,+21, 13q-,20q- | Centromere 8, 9, 20q-, 13q- | JAK2V617F < 50% |
| MPLW515K/L < 10% | |||
| Essentialthrombocytosis | 13q-, +8,+9 | Centromere 8, 9 | JAK2V617F < 50% |
| MPLW515K/L < 10% |
Molecular aspects
The first clues to the understanding of molecular basis of MPN were only revealed 50 years after Dameshek's insights. In 2005, four groups, using different methodologies, identified the first recurrent molecular abnormality in patients with MPN - a gain-of-function mutation of the gene encoding Janus kinase 2 (JAK2). The presence of the JAK2V617F mutation distinguishes PV from secondary polycythemia.(13) The JAK2V617F mutation has been found in 90-98% of PV patients, in about 50% of those with ET or PMF, in some patients with myelodysplastic/myeloproliferative neoplasms and in rare cases of acute myeloid leukemia (AML). The JAK2V617F mutation is a somatic guanine-to-thymine substitution at nucleotide 1849 in exon 14 that results in a valine-to-phenylalanine substitution in codon 617 (V617F).(14,15) JAK2 is a cytoplasmic tyrosine kinase that plays a central role in signal transduction from multiple hematopoietic growth factor receptors. It is associated with a growth factor-independent phenotype, leading to abnormal hematopoiesis.(15) Additionally, rare cases of MF and ET may have a mutation in MPLW515K/L.
In MPN, the presence of JAK2V617F has been associated with higher hemoglobin level, higher leukocyte count and lower platelet count.(16) In PV, a higher JAK2V617F allele burden has been associated with pruritus and transformation into myelofibrosis, but it does not seem to affect the risk of thrombosis, leukemic transformation or survival.(16) In patients with ET, JAK2V617F does not appear to be related with survival or leukemic transformation, but its association with thrombotic events is controversial. In PMF, a lower mutant allele burden has been associated with a lower survival rate.(17) The intriguing fact that a few PV patients and almost half of those with ET and PMF do not have the JAK2V617F mutation has prompted interest in studying other candidate genes that could be involved in the pathogenetic mechanisms of JAK2V617F-negative MPN.
These studies led to the discovery of JAK2 exon 12 mutations in ~3% of JAK2V617F-negative PV patients; thrombopoietin receptor gene (MPL - Myeloproliferative leukemia virus oncogene) mutations in ~8.5% of ET and 10% of PMF patients and TET2 (TET oncogene family member 2) gene mutations in ~ 16% of PV, 5% of ET and ~17% PMF patients.(18)
More recently, other mutations involving LNK (a membrane-bound adaptor protein), IDH1/2 (isocitrate dehydrogenase 1/2 enzyme), ASXL1 (additional sex combs-like 1), CBL (casitas B-lineage lymphoma proto-oncogene), IKZF1 (IKAROS family zinc finger 1) and EZH2 (an histone methyltransferase) genes were identified in MPN at lower frequencies.(18,19)
Essential thrombocytosis
Definition
ET is a clonal disorder. It is characterized by megakaryocytic proliferation with consequent increased peripheral platelets. Its course is chronic and may progress to myelofibrosis or acute leukemia.(20)
Epidemiology
The incidence varies between 0.6 to 2.5 cases per 100,000 persons/year. It is a disorder of middle age and is rare in childhood (one case per one million children).(21) It is predominant in females (2:1). There is possibly family involvement.
Clinical features
Vasomotor symptoms predominate: headache, dizziness, tinnitus, syncope, tingling, visual changes, acrocyanosis or erythromelalgia, translated as burning sensation in the feet and hands associated with redness and warmth. Between 25 to 35% of patients are asymptomatic and discover thrombocytosis accidentally. Mild splenomegaly is found in up to 40%. When platelets exceed 1000 x 109/L, there is increased risk of spontaneous bleeding in the gastrointestinal tract.(22) Bleeding and thrombotic events, the last represented by deep vein thrombosis, pulmonary embolism or Budd-Chiari syndrome, are responsible for morbidity and mortality. It is important to remember that angina, myocardial infarction, transient ischemic attack (TIA), stroke, pulmonary embolism or priapism can be the first manifestation of thrombocytosis.
Pathophysiology
For decades, ET was attributed to the proliferative advantage of megakaryocytic factors related to thrombopoietin (TPO) and its cellular receptor (c-Mpl). The mutations involved in MPN start early in the stem cell. Megakaryocytic precursors are hypersensitive to TPO and the action of cytokines: TGF beta, Interleukins 3 (IL-3), IL-6 and IL-11, which interact with the bone marrow microenvironment, promoting the growth of megakaryocytes.(23) In 35 to 70% of cases, acquired somatic mutation occur in the Janus kinase gene 2(JAK2V617F), responsible for phosphorylation and signaling activators of transcription (STAT). Three novel somatic mutations in the juxtamembrane region at codon 515 MPL (W515L, W515K, S505N) explain defects in signaling pathways of molecules STAT3, STAT5, mitogen activator protein (MAPK) and phosphatidylinositol-3kinase ATK (PI3K/AKT), giving a gain in function and proliferative advantage to megakaryocytes.(23)
Diagnostic criteria
WHO 2008 and British Committee for Standards in Hematology 2010(24)
Requires A1-A4
A1) platelet count = 450 x109/L sustained over more than six months without apparent cause;
A2) bone marrow biopsy: megakaryocytic hyperproliferation, in which megakaryocytes are increased in number and size with mature hyperlobulated and grouped.
A3) Absence of WHO criteria for CML, PV (normal red cell mass), PMF (no fibrosis), del 5q, MDS
A4) MPL and JAK2 mutations positive; if negative, no evidence for reactive thrombocytosis
British Committee for Standards in Hematology (BCSH) 2010
Requires A1-A3 or A3 + A3-A5
A.1) platelet count = 450 x109/L sustained over more than six months without apparent cause
A.2) JAK2 mutations and MPL positive
A.3) Absence of WHO criteria for CML, PV (normal red cell mass), PMF (no fibrosis), del 5q, MDS
A.4) no evidence of reactive thrombocytosis and normal iron stores
A.5) bone marrow biopsy: megakaryocytic proliferation with megakaryocytes increased in number and size, hyperlobulated with abundant cytoplasm.
Differential diagnosis
ET must be distinguished from other MPN and the processes that lead to transient reactive thrombocytosis: acute hemorrhage, recovery of thrombocytopenia, infections, inflammatory diseases, tissue damage, iron deficiency, hemolytic anemia, post splenectomy, cryoglobulinemia, medications (steroids, adrenaline, trans-retinoic acid), neoplasms, myelodysplasia Del 5q.
Risk stratification
Risk stratification considering the possibility of thrombosis. Several retrospective and prospective studies have shown two major factors for vascular complications: 1) age over 60 years and 2) previous thrombotic events. The platelet count above 1500 x 109/L, is associated with acquired von Willebrand disease, linked to risk of bleeding.(20,23)
Patients with ET are divided into risk groups:
High risk
- Age = 60years
and/or
- Episodes of bleeding or prior thromboembolic phenomena and/or
- Platelet count = 1500 109/L
Intermediate risk
- Age = 40 and < 60 years
- Presence of cardiovascular risk factors (smoking, hypertension, dyslipidemia, diabetes)
- Platelet count less than 1500 x 109/L
This group is still discussed and is not universally accepted.
Low risk
- Age < 40 years
- No previous thromboembolic phenomena
- No risk factors
- Platelet count < 1500 x 109/L
Over the past two years, meta-analyses have included the JAK2 and/or
Leukocytosis = 10 x 109/L as higher risk for thrombosis in ET(25)
Management
Conducts aim to prevent major bleeding and thrombosis, without increasing the risk of transformation to myelofibrosis or AML.(26)
Low Risk: the recommendation in asymptomatic patients is observation "watch and wait" (evidence - grade B). Some studies have suggested that aspirin prevents thrombotic complications and decreases microvascular disorders such as erythromelalgia, acrocyanosis, headache, dizziness, visual disorders and paresthesia of the extremities (palms and soles).(25) However, there is lack of evidence that it has effective prophylactic effects.
Intermediate risk: the therapy should be individualized. Platelets = 600 x 109/L in the presence of cardiovascular factors: Aspirin 100 mg/day and/or thrombocythemia reducing agents (evidence - grade C).(27) There is no consensus on what the best approach should be to patients younger than 60 years of age, platelet counts of less than 1000 x 109/L but greater than 600 x 109/L, no history of vasomotor and/or thrombohemorrhagic phenomena, but in the presence of cardiovascular factors.
High Risk: Patients over 60 years, platelets 600 x 109/L in the presence of cardiovascular risk factors are candidates for therapy. The first line is Hydroxyurea (HU) at an initial dose of 15 mg/kg/day in order to reduce the platelet count to less than 450 x 109/L.(28) Patients with indication for cytoreduction, but who fail HU therapy due to intolerance or resistance, should migrate to second-line therapy, where anagrelide and interferon alpha are utilized.(29)
Hematopoietic stem cell transplantation (HSCT) is not part of the conventional therapies for ET. However, allogeneic HSCT can be recommended in patients aged < 60 years old, with transformation to myelofibrosis or acute leukemia.(30)
JAK2 inhibitors are being tested in ET, but their benefits are unclear. It is not known if they will impact on progression or survival.(31)
Primary myelofibrosis
Clinical aspects
PMF has a heterogeneous clinical presentation. Around 30% of the patients are asymptomatic and present splenomegaly on physical exam or have a routine blood analysis that shows anemia, leukocytosis and/or thrombocytosis. In the initial phase, PMF may resemble ET because the initial finding may be only thrombocytosis. Patients also may present constitutional symptoms such as dyspnea, fatigue, night sweats, weight loss, fever, and bleeding. Some patients present renal stones and gouty arthritis due to hyperuricemia. Splenomegaly is present in 90% of the cases.(32)
Epidemiology and etiology: the disease occurs in 0.5-1.5 cases per 100,000 people per year, most commonly in the sixth or seventh decades of life and affecting both genders. Exposure to benzene or ionizing radiation is present in some cases and rare familial cases have been documented.(32)
The initial mutation responsible for PMF is not known. However, the majority of patients present JAK2V617F and a minority present MPL, LNK, CBL, TET2, ASXL1, IDH, IKZF1 or EZH2 mutations.(18) The JAK2V617F mutation is present in approximately 50-65% of the cases. Besides the clonal myeloproliferation present in PMF, a secondary inflammatory state occurs, characterized by bone marrow stromal changes and abnormal cytokine expression. Plasma levels of proinflammatory cytokines are elevated in PMF and might be pathogenetically linked to disease-associated constitutional symptoms and cachexia and also related to worse overall (OS) and leukemia-free survival (LFS).(33)
Diagnosis and classification
PMF is classified as a clonal BCR-ABL negative MPN by the WHO classification.(32) Diagnosis is based on findings of physical exam, peripheral blood, bone marrow morphology, cytogenetics and molecular markers and exclusion of other diseases ( Table 4 ). The typical peripheral blood smear shows leukoerythroblastosis and anisopoikilocytosis with teardrop-shaped red cells. The disease may be diagnosed in a prefibrotic stage (early stage) or more frequently, in an over fibrotic stage.(34)
Table 4.
WHO Diagnostic criteria for primary myelofibrosis (requires all 3 major criteria and more than 2 minor criteria)(32)
| I. Major criteria | |
| a. Megakaryocyte proliferation, including small-to-large megakaryocytes, with aberrant nuclear/cytoplasmic ratio and hyperchromatic and irregularly folded nuclei and dense clustering accompanied by either reticulin and/or fcollagen ibrosis or , in the absence of reticulin fibrosis (i.e. prefibrotic), megakaryocyte changes must be accompanied by increased marrow cellularity, granulocytic proliferation. | |
| b. Not meeting WHO criteria for chronic myelogenous leukemia, polycythemia vera, myelodysplastic syndromes or other myeloid neoplasms | |
| c. Demonstration of JAK2V617F or other clonal marker or, in the absence of a clonal marker, no evidence of reactive marrow fibrosis (infection, auto immune disorder, hairy cell leukemia, lymphoid neoplasm, metastatic malignancies and toxic myelopathies) | |
| II. Minor criteria | |
| a. Leukoerythroblastosis | |
| b. Increased serum lactate dehydrogenase | |
| c. Anemia | |
| d. Palpable splenomegaly | |
Differential diagnosis
Includes bone marrow fibrosis associated with non-neoplastic or other neoplastic conditions, such as metastatic cancer, lymphoid neoplasm and other myeloid malignancies, especially CML, MDS, chronic myelomonocytic leukemia (CMML), or AML.
Risk stratification
Accurate risk stratification of patients in terms of OS and LFS is necessary for treatment decisions regarding the timing for allogeneic transplantation, palliative treatment or participation in clinical trials.
The International Working Group for Myelofibrosis Research and Treatment (IWG-MRT developed the International Prognostic Scoring System (IPSS)(35) best used for assessing survival at diagnosis. Subsequently, the same group developed the dynamic IPSS (DIPSS)(36) model, which can be used for estimating survival at any point in the disease course. DIPSS also predicts the risk of progression to AML in PMF. More recently, the DIPSS plus model included red cell transfusion need,(37) unfavorable karyotype (+8, -7/7q, i(17q), -5/-12p-, inv(3), 11q23) or complexes ≥ 3)(38) and thrombocytopenia.
Treatment and role of bone marrow transplantation
Currently there is no curative treatment for PMF with the exception of bone marrow transplantation. However, this approach is restricted to a few patients, considering the morbidity and mortality of the procedure in this disease.
1) Treatment in low or intermediate 1 risk disease
For these patients with a long life expectancy (6-15 years) BMT is not justified. If the patient is symptomatic, conventional drugs such as hydrea or interferon should be used to control splenomegaly, leukocytosis and thrombocytosis.(33)
2) Treatment in high or intermediate 2 risk disease
These patients can be treated with conventional drug therapy, splenectomy, radiotherapy, allogeneic stem cell transplantation (aSCT) or experimental drug therapy. The main objective is to control anemia, symptomatic splenomegaly, constitutional symptoms and disease complications from extra medullary hematopoiesis (EMH).
Erythropoiesis stimulating agents (ESA) can be used in patients with hemoglobin (Hb) < 10 g/dL, not transfusion dependent, with erythropoietin levels < 125 U/L. Patients that fail this approach or who are not candidates for ESA can be treated with steroids, androgens, danazol, thalidomide (50 mg/d) with or without prednisone (0.25 mg/d) or lenalidomide (10 mg/d) with or without prednisone. Response rates and durations for each one of these treatment modalities are similar.(33) Aspirin should be given to patients with a platelet count of = 500 x 109/L due to the risk of thrombosis during thalidomide or lenalidomide use.
Splenectomy can be indicated for drug-refractory splenomegaly (more than 10 cm from the left costal margin) in symptomatic and transfusion dependent patients.(39) However, splenectomy has a perioperative mortality rate of 5-10% and 25% of morbidity with abdominal vein thrombosis, operative site bleeding and infections. The median survival after splenectomy is around two years. The hepatomegaly that occurs in ~20% of patients after splenectomy can be treated with hydroxyurea, cladribine or JAK inhibitors. Low-dose radiotherapy (100 cGy in 5-10 fractions) can induce a transient (3-6 months) reduction in spleen size however, this is often associated with severe and protracted pancytopenia.(40)
Role of bone marrow transplantation
BMT is the only curative option for PMF, but it is associated with high morbidity and mortality. Taking all the risks into consideration, BMT should not be indicated for low or intermediate-1 risk disease.(41)
New drugs in clinical trials
The discovery of the activating mutation JAK2 (V617F), present in more than 70% of patients with MPNs led to the development of new biochemically selective JAK2 inhibitors. JAK2 inhibitors are not specific for the JAK2V617F protein. They inhibit the JAK2-signal transducer and STAT pathway and can be used for any patient with MPN independent of JAK2 mutational status. In PMF these agents have decreased pathologic splenomegaly and improved disease-associated symptoms.
However, the ability of these agents to significantly impact disease-associated cytopenias, JAK2 allele burden or bone marrow histologic features remains unclear. Longer follow-ups are needed to evaluate the long-term safety profile.(42,43)
Polycythemia vera
PV is a pathologic condition presenting an increase of red blood cells as a result of the autonomous and clonal proliferation of bone marrow stem cells. Described for the first time by Vaquez(44) in 1892 and singled out as a clinical entity by Osler in 1903,(45) it was considered a slow but fatal disease up to a few years ago. However, in the last decade, changes in the physiopathologic criteria and the identification of the JAK2 protein mutation (JAK2V617F) led the way to diagnostic advances, better risk classification and the development of new therapies.
Clinical findings
Signs and symptoms of PV are caused by a higher erythrocytic load, leading to increases in blood viscosity and as a consequence to the decrease of its flow. It is seen in middle age individuals who characteristically present a cyanotic blush, more easily seen in the face, mainly lips, nose tip and ears. The same color alteration is also found in the distal portion of the limbs.
Increased blood pressure is frequently found in PV and this disease may be the cause of imbalance in the arterial hypertension. Common complaints are headaches, tinnitus, paresthesia and erythromelalgia which are related to the vaso-occlusive phenomena. Pruritus may occur after a hot or tepid baths and there can be some exacerbation of gout.(27) All of these symptoms tend to improve or disappear with treatment. The majority of patients present splenomegaly of varying degrees at diagnosis or during the evolution of the disease. They complain about feeling early satiety, increased abdominal volume, left hypochondriac pain or bowel constipation.
Epistaxis and ecchymosis are common. Ischemic manifestations of the central nervous system (CNS) are potentially debilitating and should be feared. Thrombosis may occur at any arterial or venous site, but thrombosis in the splanchnic territory are frequently related to MPNs which should always be investigated.(46,47)
Pathophysiology
The description of JAK2V617F, present in 95% of cases of PV, led to great advances in the understanding of its pathogenesis. This mutation is not exclusive to this entity; however, in PV there is a greater rate of homozygosis and a higher allele burden compared to ET and PMF(48,49). Other mutations were described but the one found in JAK2 exon 12 occurs in up to 2% of V617F negative patients.(49)
The natural evolution of PV is bone marrow fibrosis and extramedullary hematopoiesis while there is a low incidence of progression to myelodysplasia or acute leukemia.
Diagnosis
Currently, the diagnosis is based on the British guidelines(24) and on the World Health Organization (WHO) 2008 criteria(50) and requires the integration of clinical, laboratorial, and histology factors (Table 5 ). These are important points for proving the hematic changes by measurements of the erythrocytic mass and Hb, as well as demonstrating the presence of JAK2V617F or other mutations having a similar function.
Table 5.
WHO diagnostic criteria (50)
| 1. Major criteria | |
| 1. Hb > 18.5 g/dL for men and 16.5 g/dL for women or evidence of erythrocytic mass increase (over 25% of expected value) | |
| 2. Presence of JAK2V617F or other mutation functionally similar to the one of JAK2 exon 12. | |
| II. Minor criteria | |
| 1. Hypercellular bone marrow biopsy in the erythrocyte, granulocyte and megakaryocyte sectors (panmyelosis) | |
| 2. Serum EPO less than normal | |
| 3. In vitro formation of endogenous erythroid colonization | |
The bone marrow is hypercellular in all sectors, mainly in the erythrocytic and megakaryocytic lineages. Erythroid and myeloid precursors are morphologically normal whereas megakaryocytes may present in loose and hyperlobulated groups located close to the bone trabecula.
The requirement is the presence of both major criteria and at least one of the minor ones, or the first major criterion associated to two minor criteria.
Compliance to these parameters discards other pathologies presenting secondary polyglobinulemia, as pulmonary fibrosis, cyanotic cardiopathy or chronic obstructive pulmonary disease (COPD)
Risk stratification
Risk stratification is based on the probability of a thrombotic event. Several studies identified age over 60 years and earlier thrombosis as high risk predictors,(51) whereas their absence is classified as low risk. Cardiovascular risk factor and leukocytosis(52) seem to have some influence and may be considered as average risk.
Treatment
The goal of the therapy is to control the hematocrit level and decrease risk of morbidity. At the moment, it is still not possible to clearly define a safe hematocrit level(51) however, according to current guidelines,(53) the aim should be to keep it below 45%. All patients benefit from the use of low dose aspirin, as shown by the ECLAP study; low risk patients should be managed with phlebotomy.
Cytoreducing is indicated for high risk patients or for those who do not tolerate phlebotomy. Findings of progressive myeloproliferation with leak or thrombocytosis, symptomatic splenomegaly, pruritus or constitutional symptoms also stress the need of cytoreduction.
The first line drug is hydroxyurea (HU).(54) The initial dose varies from 500 to 1000 mg/d with later drug adjustment for each patient. In general, it is well tolerated, but may present some side effects such as cytopenia, gastrointestinal tract symptoms, mucocutaneous ulcers, alopecia and fever. The association with phlebotomy is possible when there is limitation to dose augmentation due to cytopenias.
Interferon alpha, which has not been approved for this indication by some health agencies yet, including the Food and Drug Administration of the USA (FDA) or the European Medicines Agency (EMA), has shown excellent response with both conventional and pegylated forms, the latter has greater tolerability. This drug is also indicated for pregnant patients as it does not present any risk to the fetus.
Although JAK2 inhibitors are being tested in PV, they are better indicated for the myelofibrosis phase for there is no evidence of changes in the evolution of the disease. There is also great interest in the histone deacetylase inhibitors (HDAC)(55) vorinostat and givinostat; however, further studies are required to attest their effectiveness and safety.
Chronic neutrophil leukemia
It is a rare disease characterized by neutrophilia in the peripheral blood and BM in the absence of Ph or BCR-ABL1 rearrangement.(56)
Clonal eosinophilia is defined according to the pathophysiological characteristics or associated molecular genetic changes as myeloid or lymphoid neoplasms with eosinophilia and abnormalities of PDGFR alpha, PDGFR beta or FGFR1.(57,58) They are characterized by myeloid or lymphoproliferation thus manifesting chronic eosinophilic leukemia (CEL), AML or lymphoma with persistent eosinophilia usually = 1500/µL, organ infiltration by eosinophils or mast cells, the presence of rearrangements involving PDGFRalfa, PDGFRbeta or FGFR1, absence of Ph or BCR-ABL1 rearrangements and < 20% blasts in BM(57,58). CEL is more common in men with a peak incidence around 40 years. About 10% of patients are asymptomatic. In others, symptoms such as fever, fatigue, cough, angioedema, muscle aches, rash and diarrhea are frequent, as well as anemia, thrombocytopenia, mucosal ulceration, endomyocardial fibrosis and splenomegaly.(59)
We must distinguish CEL clonal hematopoietic diseases in which eosinophilia is part of the neoplastic clone, such as CML, PV, PMF and ET, MDS and AML, especially myelomonocytic with inv(16) or the CBFβ/MYH11 rearrangement and maturation with t(8;21) or ETO/AML1 rearrangement (RUNX1/RUNXT1).(59) Clonal unspecified chronic eosinophilia is characterized by a persistently high number of eosinophils in peripheral blood (= 1500/µL), organ and endomyocardial fibrosis, presence of blasts in BM or peripheral blood (< 20%), absence of MPN or other evidence of MDS, the absence of Ph (BCR/ABL1), PDGFRα, PDGFRβ, FGFR1, inv(16), t(16,16) or t(5,12). Idiopathic hypereosinophilic syndrome is similar to the above but without the presence of blasts, without clonality and more than 6 months of eosinophilia.(56)
Mastocytosis
Mastocytosis is due to clonal proliferation of mast cells that accumulate in different organs such as the bone marrow, gastrointestinal tract, liver, spleen and lymph nodes. The disease is heterogeneous, varying from itchy skin lesions that can regress spontaneously to widespread and aggressive infiltration with general symptoms. It can occur at any age. It is classified according to the distribution: cutaneous form, indolent systemic disease associated with clonal hematologic damage, systemic aggressive mast cell leukemia, mast cell and mast cell extracutaneous sarcoma.(59) Most adults with mastocytosis present point mutations in c-KIT which consists in the substitution of valine for aspartic acid (ASP816VAL or D816V) in mononuclear cells of bone marrow.(59)
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interest
References
- 1.Tefferi A, Vainchenker W.Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J Clin Oncol. 2011; 29(5): 573-82 Comment in: J Clin Oncol. 2011;29(18):e564-5 [DOI] [PubMed] [Google Scholar]
- 2.Laszlo J.Myeloproliferative disorders (MPD): myelofibrosis, myeloscrerosis, extramedullary hematopoiesis, undifferentiated MPD, and hemorrhagic thrombocythemia. Semin Hematol. 1975; 12(4): 409-32 [PubMed] [Google Scholar]
- 3.Vardiman JW, Harris NL, Brunning R.The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002; 100(7): 2292-302 Comment in: Blood. 2003;101(7):2895-6 [DOI] [PubMed] [Google Scholar]
- 4.Tefferi A, Vardiman JW.Classification and diagnosis of myeloproliferative neoplasms: The 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008; 22(1): 14-22 Comment in: Leukemia. 2008;22(11):2118-9 [DOI] [PubMed] [Google Scholar]
- 5.Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009; 114(5): 937-51 Comment in: Blood. 2010;115(3):748-9; author reply 749-50 [DOI] [PubMed] [Google Scholar]
- 6.Thiele J, Kvasnicka HM, Facchetti F, Franco V, Van der Walt J, Orazi A.European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005; 90(8): 1128-32 [PubMed] [Google Scholar]
- 7.Thiele J, Kvasnicka HM, Diehl V.Standardization of bone marrow features - does it work in hematopathology for histological discrimination of different disease patterns? Histol Histopathol. 2005; 20(2): 633-44 [DOI] [PubMed] [Google Scholar]
- 8.Thiele J, Kvasnicka HM.The 2008 WHO diagnostic criteria for polycythemia vera, essential thrombocythemia and primary myelofibrosis. Curr Hematol Malig Rep. 2009; 4(1): 33-40 [DOI] [PubMed] [Google Scholar]
- 9.Ahmed A, Chang CC.Chronic idiopathic myelofibrosis: clinicopathologic features, pathogenesis and prognosis. Arch Pathol Lab Med. 2006; 130(8): 1133-43 [DOI] [PubMed] [Google Scholar]
- 10.Chauffaille ML.Neoplasias mieloproliferativas: revisão dos critérios diagnósticos e dos aspectos clínicos. Rev Bras Hematol Hemoter. 2010; 32(4): 308-16 [Google Scholar]
- 11.Maciel JF, de Lourdes Chauffaille M, Inaoka RJ, Colleoni GW, Yamamoto M.Essential Thrombocythemia after treatment of non-Hodgkin's Lymphoma. Leuk Res. 2007; 31(11): 1593-5 [DOI] [PubMed] [Google Scholar]
- 12.Gangat N, Tefferi A, Thanarajasingam G, Patnaik M, Schwager S, Ketterling R, et al. Cytogenetic abnormalities in essential thrombocythemia: prevalence and prognostic significance. Eur J Haematol. 2009; 83(1): 17-21 [DOI] [PubMed] [Google Scholar]
- 13.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR, Cancer Genome Project Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005; 365(9464): 1054-61 Erratum in: Lancet. 2005;366(9480):122 [DOI] [PubMed] [Google Scholar]
- 14.Levine RL, Pardanani A, Tefferi A, Gilliland DG.Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007; 7(9): 673-83 [DOI] [PubMed] [Google Scholar]
- 15.Barosi G, Bergamaschi G, Marchetti M, Vannucchi AM, Guglielmelli P, Antonioli E, Massa M, Rosti V, Campanelli R, Villani L, Viarengo G, Gattoni E, Gerli G, Specchia G, Tinelli C, Rambaldi A, Barbui T, Gruppo Italiano Malattie Ematologiche Maligne dell'Adulto (GIMEMA) Italian Registry of Myelofibrosis JAK2 V617F mutational status predicts progression to large splenomegaly and leukemic transformation in primary myelofibrosis. Blood. 2007; 110(12): 4030-6 [DOI] [PubMed] [Google Scholar]
- 16.Kantarjian H, Schiffer C, Jones D, Cortes J.Monitoring the response and course of chronic myeloid leukemia in the modern era of BCR-ABL tyrosine kinase inhibitors: practical advice on use and interpretation of monitoring methods. Blood. 2008; 111(4): 1774-80 [DOI] [PubMed] [Google Scholar]
- 17.Guglielmelli P, Barosi G, Specchia G, Rambaldi A, Lo Coco F, Antonioli E, et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood. 2009; 114(8): 1477-8 [DOI] [PubMed] [Google Scholar]
- 18.Tefferi A, Lasho TL, Huang J, Finke C, Hanson CA, Mesa RA, et al. Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia. 2008; 22(4): 756-61 [DOI] [PubMed] [Google Scholar]
- 19.Tefferi A.Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010; 24(6): 1128-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tefferi A, Thiele J, Orazi A, Kvasnicka HM, Barbui T, Hanson CA, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007; 110(4): 1092-7 Comment in: Blood. 2008;111(3):1741; author reply 1742 [DOI] [PubMed] [Google Scholar]
- 21.Johansson P.Epidemiology of the myeloproliferative disorders polycythemia vera and essential thrombocythemia. Semin Thromb Hematost 2006; 32(3): 171-3 [DOI] [PubMed] [Google Scholar]
- 22.Giuglielmelli P, Tefferi A.Advances in understanding and management of myeloproliferative neoplasms. CA Cancer J Clin. 2009; 59(3): 171-91 [DOI] [PubMed] [Google Scholar]
- 23.Girodon F, Bonicelli G, Schaffer C, Mounier M, Carillo S, Lafon I, et al. Significant increase in the apparent incidence of essential thrombocythemia related to new WHO diagnostic criteria: a population-based study. Haematologica. 2009; 94(6): 865-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harrison CN, Bareford D, Butt N, Campbel P, Conneally E, Drummond M, Erber W, Everington T, et al. Guideline for investigation and management of adults and children presenting with a thrombocytosis. Br J Haematol. 2010: 352-75 [DOI] [PubMed] [Google Scholar]
- 24.Lussana F, Caberlon S, Pagani C, Kamphuisen PW, Buller HR, Cattaneo M.Association of V617F Jak2 mutation with the risk of thrombosis among patients with essential thrombocythaemia or idiopathic myelofibrosis: a systematic review. Thromb Res. 2009; 124(4): 409-17 [DOI] [PubMed] [Google Scholar]
- 26.Biergegard G.Long-term management of thombocytosis in essential thrombocythaemia. Ann Hematol. 2009; 88(1): 1-10 [DOI] [PubMed] [Google Scholar]
- 27.Landolfi R, Gennaro L.Prevention of thrombosis in polycythemia vera and essential thrombocythemia. Haematologica. 2008; 93(3): 331-5 Comment on: Haematologica. 2008;93(3):372-80 [DOI] [PubMed] [Google Scholar]
- 28.Finazzi G, Ruggeri M, Rodeghiero F, Barbui T.Efficacy and safety of long-term use of hydroxyurea in young patients with essential thrombocythemia and a high risk of thrombosis. Blood. 2003; 101(9): 3749 Comment on: Blood. 2001;97(4):863-6. [DOI] [PubMed] [Google Scholar]
- 29.Beer P, Erber W, Campbell P, Green A.How I treat essential thrombocythemia. Blood. 2011; 117(5): 1472-82 Comment in: Blood. 2011;118(4):1179-80; author reply 1180-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kerbauy DM, Gooley TA, Sale GE, Flowers ME, Doney KC, Georges GE, et al. Hematopoietic cell transplantation as curative therapy for idiopathic myelofibrosis, advanced polycythemia vera, and essential thrombocythemia. Biol Blood Marrow Transplant. 2007; 13(3): 355-65 [DOI] [PubMed] [Google Scholar]
- 31.Spivak J.Narrative review: Thrombocytosis, polycythemia vera, and JAK2 mutations: The phenotypic mimicry of chronic myeloproliferation. Ann Intern Med. 2010; 152(5): 300-6 [DOI] [PubMed] [Google Scholar]
- 32.Swerdlow SH, International Agency for Research on Cancer. World Health Organization. Louis A.Duhring Fund.: WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed Lyon, France: International Agency for Research on Cancer; 2008 [Google Scholar]
- 33.Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011; 29(10): 1356-63 [DOI] [PubMed] [Google Scholar]
- 34.Barosi G, Mesa RA, Thiele J, Cervantes F, Campbell PJ, Verstovsek S, Dupriez B, Levine RL, Passamonti F, Gotlib J, Reilly JT, Vannucchi AM, Hanson CA, Solberg LA, Orazi A, Tefferi A, International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia. 2008; 22(2): 437-8 [DOI] [PubMed] [Google Scholar]
- 35.Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, Vannucchi AM, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009; 113(13): 2895-901 Comment in: Blood. 2010;115(3):745; author reply 745-6 [DOI] [PubMed] [Google Scholar]
- 36.Passamonti F, Cervantes F, Vannucchi AM, Morra E, Rumi E, Cazzola M, et al. Dynamic International Prognostic Scoring System (DIPSS) predicts progression to acute myeloid leukemia in primary myelofibrosis. Blood. 2010; 116(15): 2857-8 [DOI] [PubMed] [Google Scholar]
- 37.Tefferi A, Siragusa S, Hussein K, Schwager SM, Hanson CA, Pardanani A, et al. Transfusion-dependency at presentation and its acquisition in the first year of diagnosis are both equally detrimental for survival in primary myelofibrosis - prognostic relevance is independent of IPSS or karyotype. Am J Hematol. 2010; 85(1): 14-7 Comment in: Am J Hematol. 2010;85(1):4-5 [DOI] [PubMed] [Google Scholar]
- 38.Caramazza D, Begna KH, Gangat N, Vaidya R, Siragusa S, Van Dyke DL, et al. Refined cytogenetic-risk categorization for overall and leukemia-free survival in primary myelofibrosis: a single center study of 433 patients Leukemia. 2011; 25(1): 82-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mesa RA, Nagorney DS, Schwager S, Allred J, Tefferi A.Palliative goals, patient selection, and perioperative platelet management: outcomes and lessons from 3 decades of splenectomy for myelofibrosis with myeloid metaplasia at the Mayo Clinic. Cancer. 2006; 107(2): 361-70 [DOI] [PubMed] [Google Scholar]
- 40.Elliott MA, Tefferi A.Splenic irradiation in myelofibrosis with myeloid metaplasia: a review. Blood Rev. 1999; 13(3): 163-70 [DOI] [PubMed] [Google Scholar]
- 41.Ballen KK, Shrestha S, Sobocinski KA, Zhang MJ, Bashey A, Bolwell BJ, et al. Outcome of transplantation for myelofibrosis. Biol Blood Marrow Transplant. 2010; 16(3): 358-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010; 363(12): 1117-27 Comment in: N Engl J Med. 2010;363(12):1180-2; N Engl J Med.2010;363(25):2464; author reply 2464-5; discussion 2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, Stone RM, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011; 29(7): 789-96 J Clin Oncol. 2011; 29(7): 781-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaquez H.Sur une forme spéciale de cyanose s'accompgnant d'hyperglobulie excessive et persistant. Comptes rendus de La Société de Biologie, Paris, 1892; 44: 384-88 [Google Scholar]
- 45.Osler W.Chronic cyanosis with polycythemia and enlarged spleen: a new clinical entity. Am J Med Sci. 2008; 335(6): 411-7 Comment in: Am J Med Sci. 2008;335(6):418-9 [DOI] [PubMed] [Google Scholar]
- 46.Berlin NI.Diagnosis and classification of the polycythemias. Semin Hematol. 1975; 12(4): 339-5 [PubMed] [Google Scholar]
- 47.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005; 352(17): 1779-90 Comment in: N Engl J Med. 2005;353(13):1416-7; author reply 1416-7; N Engl J Med. 2005;352(17):1744-6 [DOI] [PubMed] [Google Scholar]
- 48.Passamonti F, Rumi E, Pietra D, Della Porta MG, Boveri E, Pascutto C, et al. Relation between JAK2(V617F) mutation status, granulocyte activation and constitutive mobilization of CD34+ cells into peripheral blood in myeloproliferative disorders. Blood. 2006; 107(9): 3676-82 [DOI] [PubMed] [Google Scholar]
- 49.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic eritrocytosis. N Engl J Med. 2007; 356(5): 459-68 Comment in: N Engl J Med. 2007;356(5):444-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McMullin M, Reilly JT, Campbell P, Bareford D, Green A, Harrison C.Amendment to the guideline for diagnosis and investigation of polycythaemia/erythrocytosis. Br J Haematol 2007; 138: 812-23 [DOI] [PubMed] [Google Scholar]
- 51.Crisà E, Venturino E, Passera R, Prina M, Schinco P, Borchiellini A, et al. A retrospective study on 226 polycythemia vera patients: impact of median hematocrit value on clinical outcomes and survival improvement with anti-thrombotic prophylaxis and non-alkylating drugs. Ann Hematol. 2010; 89(7): 691-9 [DOI] [PubMed] [Google Scholar]
- 52.Barbui T, Carobbio A, Rambaldi A, Finazzi G.Perspectives on thrombosis in essential thrombocythemia and polycythemia vera: is leukocytosis a causative factor? Blood. 2009; 114(4): 759-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barosi G, Birgegard G, Finazzi G, Griesshammer M, Harrison C, Hasselbalch HC, et al. Response criteria for essential thrombocythemia and polycythemia vera: result of a European LeukemiaNet consensus conference. Blood. 2009; 113(20): 4829-33 [DOI] [PubMed] [Google Scholar]
- 54.Barbui T, Barosi G, Birgegard G, Cervantes F, Finazzi G, Griesshammer M, Harrison C, Hasselbalch HC, Hehlmann R, Hoffman R, Kiladjian JJ, Kröger N, Mesa R, McMullin MF, Pardanani A, Passamonti F, Vannucchi AM, Reiter A, Silver RT, Verstovsek S, Tefferi A, European LeukemiaNet Philadelphia-Negative Classical Myeloproliferative Neoplass: Critical Concepts and Management Recommendations from European Leukemianet. J Clin Oncol. 2011; 29(6): 761-70 Comment in: J Clin Oncol. 2011;29(18):e564-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rambaldi A, Dellacasa CM, Salmoiraghi S, et al. A phase 2 A study of the histone-deacetylase inhibitor in patients with JAK2V617F positive myeloproliferative neoplasms. {abstract} Blood 2008; 112:100 (não localizada) [DOI] [PubMed] [Google Scholar]
- 56.Chauffaille, MLLFNeoplasias mieloproliferativas: revisão dos critérios diagnósticos e dos aspectos clínicos. Rev Bras Hematol. Hemoter. 2010; 32(4); 308-16 [Google Scholar]
- 57.Tefferi A, Patnaik MM, Pardanani A.Eosinophilia: secondary, clonal and idiopatic. Br J Haematol. 2006; 133(5): 468-92 [DOI] [PubMed] [Google Scholar]
- 58.Fletcher S, Bain B.Diagnosis and treatment of hypereosinophilic syndromes. Curr Opin Hematol. 2007; 14(1): 37-42 [DOI] [PubMed] [Google Scholar]
- 59.Metcalfe DD.Mast cells and mastocytosis. Blood. 2008; 112(4): 946-56 [DOI] [PMC free article] [PubMed] [Google Scholar]