

Abstract
In humans, congenital myopathy-linked tropomyosin mutations lead to skeletal muscle dysfunction, but the cellular and molecular mechanisms underlying such dysfunction remain obscure. Recent studies have suggested a unifying mechanism by which tropomyosin mutations partially inhibit thin filament activation and prevent proper formation and cycling of myosin cross-bridges, inducing force deficits at the fiber and whole-muscle levels. Here, we aimed to verify this mechanism using single membrane-permeabilized fibers from patients with three tropomyosin mutations (TPM2-null, TPM3-R167H and TPM2-E181K) and measuring a broad range of parameters. Interestingly, we identified two divergent, mutation-specific pathophysiological mechanisms. (i) The TPM2-null and TPM3-R167H mutations both decreased cooperative thin filament activation in combination with reductions in the myosin cross-bridge number and force production. The TPM3-R167H mutation also induced a concomitant reduction in thin filament length. (ii) In contrast, the TPM2-E181K mutation increased thin filament activation, cross-bridge binding and force generation. In the former mechanism, modulating thin filament activation by administering troponin activators (CK-1909178 and EMD 57033) to single membrane-permeabilized fibers carrying tropomyosin mutations rescued the thin filament activation defect associated with the pathophysiology. Therefore, administration of troponin activators may constitute a promising therapeutic approach in the future.
INTRODUCTION
Sarcomeres, the fundamental contractile units of skeletal muscle, are sophisticated macromolecular complexes composed of interdigitating thick filaments and thin filaments. Skeletal muscle thin filaments are copolymers of actin subunits, tropomyosin/troponin regulatory complexes, nebulin stabilizing molecules and CapZ and tropomodulin (Tmod) capping molecules. A key feature of skeletal muscle contraction and force generation is thin filament activation. According to the steric model of thin filament regulation (1), activation involves various thin filament states and protein movements initiated by Ca2+ binding, which provokes the opening of the hydrophobic pocket of troponin C and induces the movement of troponin I toward the hydrophobic pocket, relieving the inhibition of tropomyosin and actin. Tropomyosin can then move toward the inner domain of the thin filament, exposing sites on actin that allow weak binding of myosin cross-bridges (displacement from the B- to C-state, with both considered to be ‘off’ states). The weak-to-strong transition induces additional tropomyosin movement (to the M-state, known as the ‘on’ state), exposing more sites on the thin filament, permitting additional binding of myosin cross-bridges and, ultimately, leading to muscle shortening and steady-state force production.
Thin filament activation and skeletal muscle contraction are deregulated in nemaline myopathy (NM) (2–4). NM is one of the most common congenital myopathies, with an estimated incidence of 1:50 000 (2). The clinical spectrum of NM ranges from severe cases with antenatal or neonatal onset and early death to adult-onset cases with slow progression (5). The classical form of NM is defined by weakness with limb, facial, masticatory and respiratory muscle involvement (5). Notably, NM is characterized by mutations in the TPM2 and TPM3 genes, which encode β- and γ-tropomyosin, respectively (2,6). Recent studies have shown that certain β- and γ-tropomyosin mutants act as ‘poison’ proteins in skeletal muscle by partially inhibiting thin filament activation (3,7) and preventing proper formation and cycling of myosin cross-bridges (3,4). This triggers a deficit in force-generating capacity at the fiber level and contributes to generalized skeletal muscle weakness (3,4,7). Here, we aimed to verify such molecular and cellular events in skeletal muscle harboring three TPM2 and TPM3 mutations (TPM2-null, TPM3-R167H and TPM2-E181K). Hence, we used skinned human skeletal muscle fibers and subjected these fibers to a broad range of functional and structural assays. Our findings demonstrate that congenital myopathy-causing tropomyosin mutations induce contractile pathophysiology via two distinct mechanisms, which are specified in a mutation-specific manner. Specifically, the TPM2-null and TPM3-R167H mutations decreased cooperative thin filament activation with simultaneous reductions in myosin cross-bridge number and force production. In contrast, the TPM2-E181K mutation increased thin filament activation, cross-bridge binding and force generation.
In addition to well-regulated thin filament activation, correct thin filament length specification is essential for skeletal muscle function and may be perturbed in NM. The giant protein nebulin coextends with actin along ∼0.9–1.0 μm of the length of skeletal muscle thin filaments, while a nebulin-free, Tmod-capped pointed-end extension specifies muscle-specific thin filament lengths beyond the N-terminal M1M2M3 domain of nebulin in chicken, mouse, rabbit and human muscles (8–11). Indeed, it has been shown that NM-causing nebulin mutations, in addition to altering thin filament activation, can also destabilize thin filaments, leading to shorter thin filament length and a leftward shift and narrowing of the force-sarcomere length curve (12). To date, tropomyosin mutations have not been found to affect thin filament lengths in striated muscle sarcomeres, despite the well-known role for tropomyosin in stabilizing actin filaments in vitro (13,14). However, the nebulin-free actin filament comprising the pointed-end extension is associated with tropomyosin, indicating that tropomyosin may play a role in stabilizing this portion of the thin filament and specifying muscle-specific thin filament lengths independently of nebulin. Therefore, we used distributed deconvolution (DDecon), a high-resolution immunofluorescence and computational image analysis technique, to test whether the TPM2-null, TPM3-R167H and TPM2-E181K mutations might lead to thin filament shortening by destabilizing the thin filament pointed-end extension. While skeletal muscle fibers harboring the TPM2-null and TPM2-E181K mutations had normal thin filament lengths, TPM3-R167H muscle displayed abnormally short thin filaments, which adversely impacted myosin cross-bridge formation and force production at long sarcomere lengths. Hence, thin filament shortening represents a novel mechanism underlying the development of tropomyosin-based congenital myopathy.
To date, no efficient therapeutic intervention for NM has been discovered. As thin filament dysfunction appears to play a key role in NM (2–4), modulating thin filament activation may restore skeletal muscle contractility. Existing pharmacological drugs such as troponin activators may act accordingly, as they facilitate the response of the thin filament to Ca2+ binding in striated muscle (15,16) in response to various disease states (16,17). To confirm this, we utilized skinned human skeletal muscle fibers and tested the effects of troponin activators (CK-1909178 and EMD 57033) in the presence of two of the aforementioned NM-causing tropomyosin mutations (TPM2-null and TPM3-R167H). Importantly, we prove that most of the NM-related physiological alterations are reversed via the administration of troponin activators. Taken together, the studies described here highlight diverse pathophysiological mechanisms that lead to thin filament dysfunction in tropomyosin-based NM and introduce one novel therapeutic strategy for reversing the contractile symptoms of NM.
RESULTS
Divergent effects of tropomyosin mutations on skeletal muscle contractility
A host of studies have demonstrated that NM-associated mutations in genes encoding thin filament structural proteins—including tropomyosin—alter cross-bridge cycling kinetics and skeletal muscle contractility (3,4,12,18–20). Therefore, we examined the effects of the TPM2-null, TPM3-R167H and TPM2-E181K mutations on the contractile properties of isolated muscle fibers. For TPM3-R167H and TPM2-E181K, because of a predominance of fibers expressing the type I Myosin heavy-chain (MyHC) isoform (21), most of the analyses were confined to this type of fibers (Fig. 1). For TPM2-null, given the small numbers of hybrid (type I/IIa and type IIa/IIx) and pure type IIx fibers, comparisons were restricted to fibers expressing type I and type IIa MyHC (Fig. 1).
Figure 1.
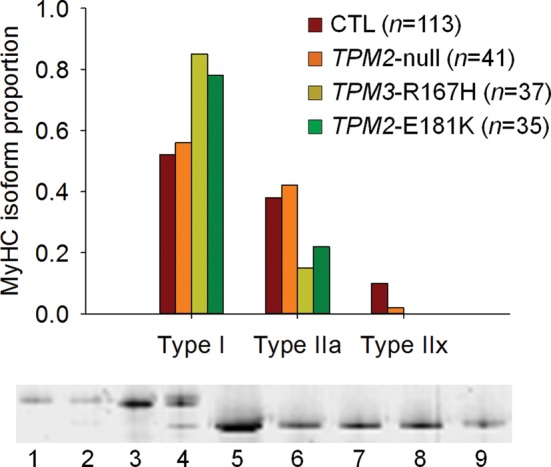
MyHC isoform composition. A predominance of type I fibers was observed for TPM3-R167H and TPM2-E181K, whereas TPM2-null had a similar MyHC isoform distribution as CTL. In the figure, examples of electrophoretic separations are displayed. Lanes 1–3 and 5–9 are single fibers, whereas lane 4 is a muscle homogenate showing the three different MyHC isoforms (I, IIa and IIx, from bottom to top).
At saturating [Ca2+] (pCa 4.50) and optimal sarcomere length (between 2.60 and 2.80 µm), steady-state isometric force production was not different among TPM2-null, TPM3-R167H, TPM2-E181K and control (CTL) fibers (Fig. 2). Nevertheless, at a higher sarcomere length (3.20 µm), force production was significantly lower in TPM3-R167H fibers when compared with TPM2-null, TPM2-E181K and CTL fibers (Fig. 3). To investigate the effects of these mutations on force production at non-saturating [Ca2+], relative force–pCa relationships (pCa between 6.30 and 4.50; sarcomere length between 2.6 and 2.8 µm) were constructed (Fig. 4A). These data were fitted using the following sigmoid Hill equation:
| (1) |
where F is force, −log10[Ca50] is the midpoint (pCa50 or Ca2+ sensitivity), and nH is the Hill coefficient. Our experimental force–pCa curves revealed that the Ca2+ sensitivity of force production was significantly lower in TPM2-null and TPM3-R167H fibers but higher in TPM2-E181K fibers when compared with CTL fibers (Table 1).
Figure 2.
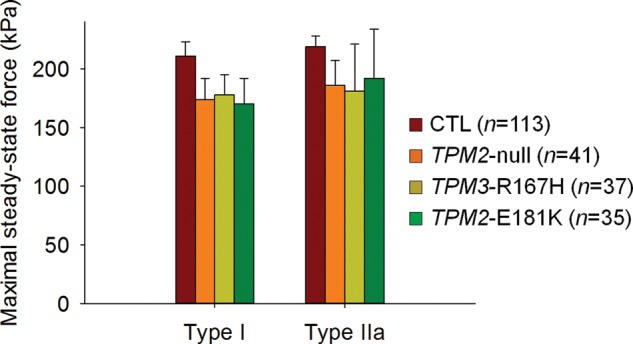
Maximal fiber force production. Maximal isometric steady-state force generation was not significantly modified by the various tropomyosin mutations at saturating [Ca2+] (pCa 4.50) and optimal sarcomere length (between 2.60 and 2.80 µm).
Figure 3.
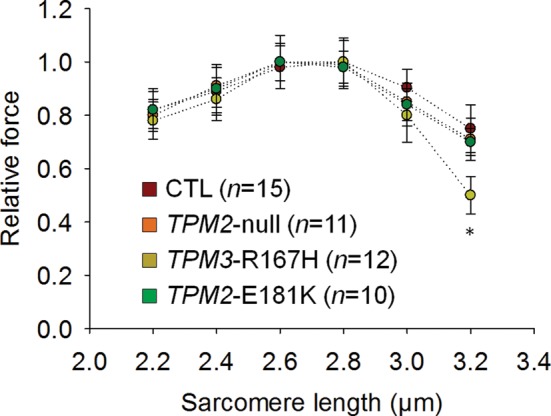
Force generation as a function of sarcomere length. The mean relative force–sarcomere length relationships of fibers expressing the type I MyHC isoform (pCa 4.50) clearly demonstrates a lower force generation at 3.20 µm for TPM3-R167H when compared with other groups. In the figure, asterisk indicates a significant difference when compared with CTL (P < 0.05).
Figure 4.
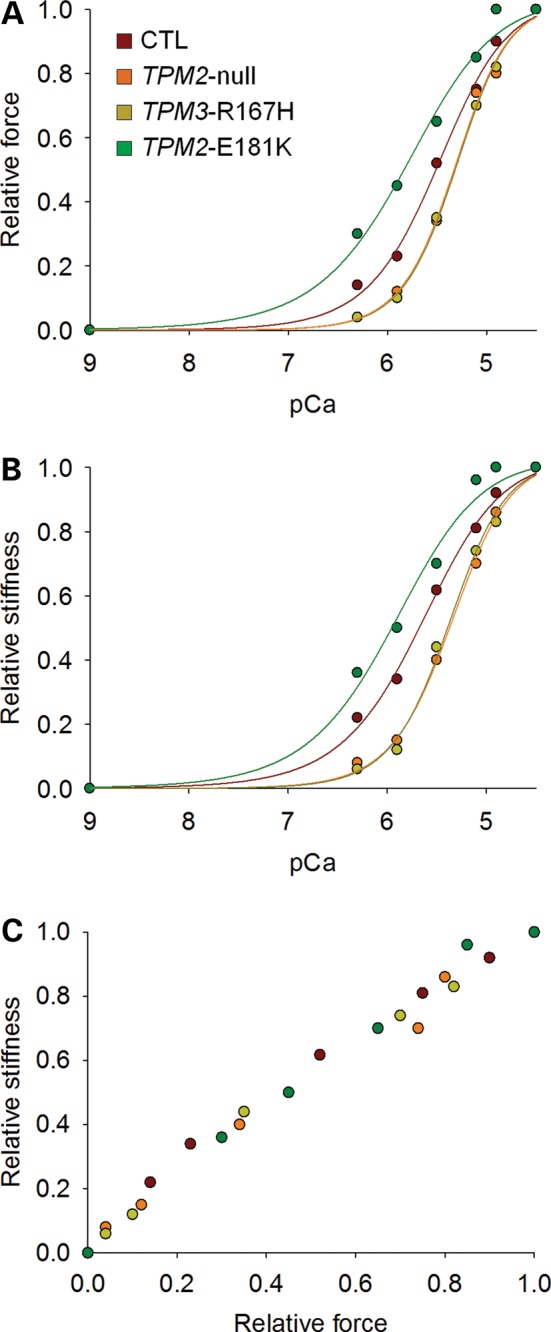
Fiber force production and stiffness at non-saturating [Ca2+]. The various typical curves for fibers expressing the type I MyHC isoform highlight the decrease (TPM3-R167H and TPM2-null) or increase (TPM2-E181K) in Ca2+ sensitivities of force and stiffness when compared with CTL.
Table 1.
Ca2+ sensitivity and Hill coefficient
| Fiber type | Genotype | Compound | pCa50 of force | nH of force | pCa50 of stiffness | nH of stiffness |
|---|---|---|---|---|---|---|
| I | CTL | — | 5.66 ± 0.06 (n = 68) | 3.73 ± 0.55 (n = 68) | 5.77 ± 0.13 (n = 68) | 3.54 ± 0.98 (n = 68) |
| 30 µm EMD | 6.00 ± 0.06# (n = 20) | 3.18 ± 0.97 (n = 20) | 5.89 ± 0.09 (n = 20) | 3.34 ± 1.29 (n = 20) | ||
| TPM2-null | — | 5.22 ± 0.08* (n = 18) | 2.86 ± 1.21 (n = 18) | 5.25 ± 0.10* (n = 18) | 2.77 ± 1.39 (n = 18) | |
| 30 µm EMD | 5.67 ± 0.07# (n = 10) | 3.47 ± 1.02 (n = 10) | 5.66 ± 0.11# (n = 10) | 3.28 ± 0.96 (n = 10) | ||
| TPM3-R167H | — | 5.11 ± 0.08* (n = 32) | 4.05 ± 0.74 (n = 32) | 5.16 ± 0.20* (n = 32) | 3.89 ± 0.58 (n = 32) | |
| 30 µm EMD | 5.55 ± 0.09# (n = 16) | 3.87 ± 0.58 (n = 16) | 5.49 ± 0.07# (n = 16) | 3.66 ± 0.71 (n = 16) | ||
| TPM2-E181K | — | 5.86 ± 0.04* (n = 29) | 3.84 ± 0.82 (n = 29) | 5.94 ± 0.02* (n = 29) | 3.58 ± 0.69 (n = 29) | |
| 30 µm EMD | x | x | x | x | ||
| IIa | CTL | — | 5.49 ± 0.06 (n = 45) | 4.20 ± 0.64 (n = 45) | 5.45 ± 0.13 (n = 45) | 3.82 ± 1.21 (n = 45) |
| 30 µm CK | 5.77 ± 0.10# (n = 18) | 3.51 ± 0.81 (n = 18) | 5.74 ± 0.08# (n = 18) | 3.68 ± 0.74 (n = 18) | ||
| TPM2-null | — | 5.12 ± 0.05* (n = 23) | 3.99 ± 0.87 (n = 23) | 5.17 ± 0.06* (n = 23) | 2.87 ± 0.66 (n = 23) | |
| 30 µm CK | 5.69 ± 0.15# (n = 12) | 3.78 ± 1.08 (n = 12) | 5.68 ± 0.15# (n = 12) | 3.02 ± 1.09 (n = 12) |
Values appear as the mean ± standard error. CK corresponds to CK-1909178, and EMD corresponds to EMD 57033. x, not tested.
*Significant difference when compared with CTL (P < 0.05) for the same fiber type, as determined by its MyHC isoform (I or IIa).
#Significant difference when compared with its without-compound value (P < 0.05) for the same group of fibers.
To obtain additional insights into the physiological mechanisms that produce these observed changes in contractility and evaluate the contribution of the number of strongly bound myosin cross-bridges in such impairment, stiffness measurements were performed, and relative stiffness-pCa curves were constructed (Fig. 4B). Fiber stiffness at non-saturating [Ca2+] (pCa between 6.30 and 4.50; sarcomere length between 2.6 and 2.8 µm) followed the same trend as force. Thus, the Ca2+ sensitivity of stiffness was significantly lower in TPM2-null and TPM3-R167H fibers but higher in TPM2-E181K fibers when compared with CTL fibers (Table 1). Note that the relative stiffness-relative force relationships did not reveal any differences among TPM2-null, TPM3-R167H, TPM2-E181K and CTL fibers (Fig. 4C).
In addition to measuring force and stiffness, we calculated the rate of force development (ktr) in order to assess whether the TPM2-null, TPM3-R167H and TPM2-E181K mutations affect the kinetics of force generation. At saturating [Ca2+] (pCa 4.50) and optimal sarcomere length (between 2.60 and 2.80 µm), ktr was significantly lower in TPM3-R167H and TPM2-E181K fibers when compared with CTL and TPM2-null fibers (Fig. 5A). At such [Ca2+], ktr reflects the myosin cross-bridge cycling turnover rate and, according to the two-state cross-bridge model, is proportional to fapp + gapp, where fapp is the rate constant for attachment and gapp is the rate constant for detachment (22,23). To evaluate the effects of the TPM2-null, TPM3-R167H and TPM2-E181K mutations at submaximal [Ca2+], relative ktr–pCa curves (pCa between 6.30 and 4.50; sarcomere length between 2.6 and 2.8 µm) were constructed (Fig. 5B). These relative ktr–pCa curves revealed elevated relative ktr values for TPM2-null and TPM3-R167H fibers at pCa 6.30 and 5.90 when compared with TPM2-E181K and CTL fibers (Fig. 5B). At these [Ca2+] values, ktr does not solely depend on cross-bridge kinetics; it also depends on the level of thin filament activation and cooperative mechanisms (24,25). To distinguish between these possibilities, N-ethylmaleimide (NEM)-S1, a strongly binding analog of myosin subfragment-1 that does not generate force but activates the thin filament and traps it in an open conformation, was employed (26). To avoid any binding competition between the compound and actual myosin cross-bridges that would deteriorate steady-state force, we used a suboptimal concentration of 3 µm NEM-S1. This concentration of NEM-S1 modified the relative ktr–pCa relationships of TPM2-null, TPM3-R167H, TPM2-E181K and CTL fibers in a similar manner (Fig. 5C). This demonstrates that perturbations at the level of thin filament activation and cooperative mechanisms are likely to explain the elevated relative ktr at pCa 6.30 and 5.90 in TPM2-null and TPM3-R167H fibers.
Figure 5.
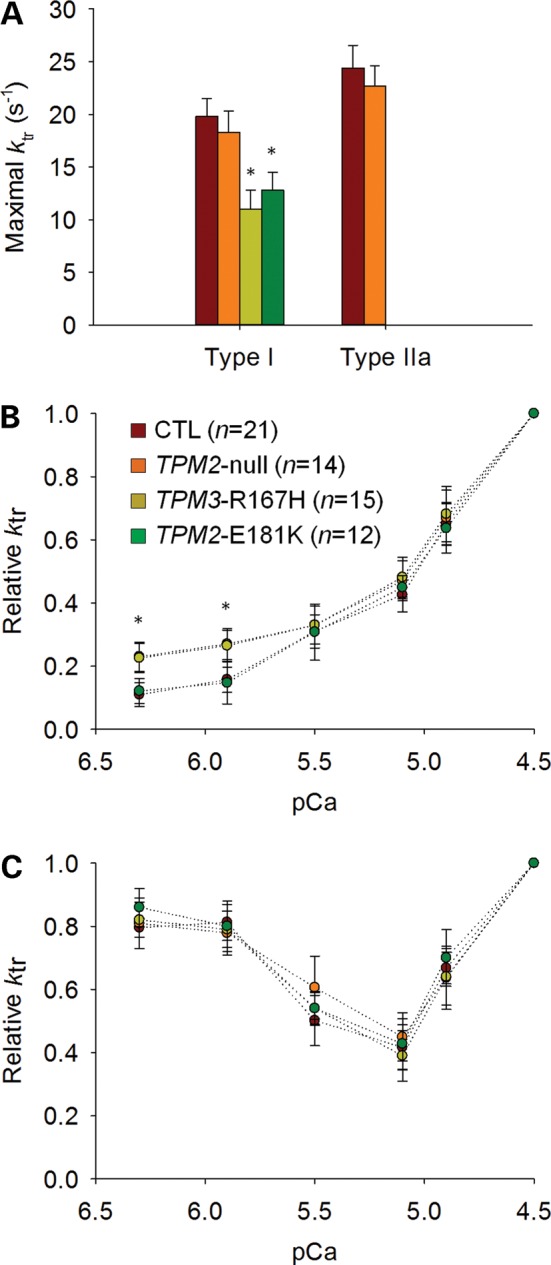
Rate of force development. In fibers expressing the type I MyHC isoform, at saturating [Ca2+] (pCa 4.50) (A), ktr was significantly lower for TPM3-R167H and TPM2-E181K when compared with TPM2-null and CTL while at submaximal [Ca2+] (pCa 6.30 and 5.90) (B), relative ktr was significantly higher for TPM3-R167H and TPM2-null when compared with TPM2-E181K and CTL. In the presence of 3 µm of NEM-S1 (C), the elevated relative ktr returned to normal values. In the figure, asterisk indicates a significant difference when compared with CTL (P < 0.05).
An important consideration in the aforementioned experiments is that the contractile modifications induced by the TPM2-null, TPM3-R167H and TPM2-E181K mutations may simply be secondary phenotypes caused by altered myofibrillar protein levels secondary to tropomyosin haploinsufficiency. However, this is unlikely, as measurements of protein levels using 12% SDS–PAGE revealed no changes in relative myosin, actin and tropomyosin content in TPM2-null, TPM3-R167H and TPM2-E181K fibers, with the exception of compensatory tropomyosin isoform up-regulation in TPM2-null fibers (Fig. 6). Therefore, altered myofibrillar protein content does not account for the contractile changes that occur in fiber harboring the TPM2-null, TPM3-R167H and TPM2-E181K mutations.
Figure 6.

Relative myosin, actin and tropomyosin content. The relative contents of myosin, actin and tropomyosin were not different between TPM2-null, TPM3-R167H, TPM2-E181K and CTL, except for a tropomyosin isoform switch in type I [C] and IIa [D] fibers of TPM2-null. In [C], a typical electrophoretic separation of actin, α-, β- and γ-tropomyosin is shown for a single fiber from CTL1 expressing the type I MyHC isoform.
Reduced thin filament length in TPM3-R167H fibers
Previous studies have demonstrated that, in a subset of NM patients (specifically, those harboring nebulin mutations), muscle weakness is partly due to thin filament instability and resultant thin filament depolymerization and shortening, which alter the shape of the force–sarcomere length relationship (12). To test whether the TPM2-null, TPM3-R167H and TPM2-E181K mutations also affect thin filament lengths, we used immunofluorescence staining and DDecon analysis to measure thin filament lengths based on Tmod4 localization with respect to the Z-line as well as the position of the N-terminal M1M2M3 domain of nebulin. In both CTL and mutant fiber bundles, the architecture of the myofibrillar lattice was well-preserved, as indicated by regular striated arrays of F-actin and α-actinin (Fig. 7A). In CTL fiber bundles, thin filament lengths ranged from 1.26 ± 0.05 to 1.38 ± 0.07 μm (Table 2), consistent with the inter-individual and inter-muscle heterogeneity of thin filament lengths reported previously (9). The TPM2-null and TPM2-E181K fiber bundles had thin filament lengths that were similar to CTL bundles, but the TPM3-E167H fiber bundle displayed a markedly shorter thin filament length of 1.03 ± 0.08 μm (Fig. 7B and C; Table 2). In contrast, the position of the nebulin M1M2M3 domain with respect to the Z-line was relatively uniform across the CTL and mutant fiber bundles, ranging from 0.90 ± 0.05 to 0.99 ± 0.05 μm (Table 2), consistent with previous measurements in human muscles (9). Therefore, the TPM3-E167H mutation did not affect the length of the nebulin-stabilized core region of the thin filament. Instead, the TPM3-E167H mutation reduced the length of the nebulin-free, Tmod-capped pointed-end extension (8–10). We conclude that the TPM3-E167H mutation prevents thin filament pointed-end extensions from achieving their appropriate in vivo lengths, either by inhibiting actin monomer addition or by enhancing actin depolymerization at thin filament pointed ends.
Figure 7.

Immunofluorescence microscopy of skeletal muscle thin filament components. (A) Longitudinal cryosections of skeletal muscle fiber bundles from CTL subjects and TPM2-null, TPM3-R167H and TPM2-E181K patients were phalloidin-stained for F-actin, immunostained for Tmod4 to visualize thin filament pointed ends and immunostained for α-actinin to visualize Z-lines. (B) Higher-magnification views of CTL and TPM3-R167H bundles qualitatively suggest that modest thin filament shortening in TPM3-R167H bundles, as evidenced by slightly wider F-actin-free gaps (H-zones) spanning Tmod4 doublets even when sarcomere length and as determined by the distance between successive α-actinin striations, is held constant. (C) Immunostaining for the nebulin M1M2M3 domain reveals no overt changes in the position of the nebulin M1M2M3 domain in TPM3-R167H fiber bundles. Both thin filament length and the position of the nebulin M1M2M3 domain were quantified using DDecon (Table 2). Bars, 3 μm. Yellow brackets indicate thin filament arrays between F-actin-free H-zones.
Table 2.
Distances of Tmod4 and the nebulin M1M2M3 domain from the Z-line, as determined by DDecon analysis of fluorescence images
| Subject | Tmod4 |
Nebulin M1M2M3 |
||||
|---|---|---|---|---|---|---|
| Mean ± SD (μm) | Min–max (μm) | n | Mean ± SD (μm) | Min–max (μm) | n | |
| CTL1 | 1.30 ± 0.05a | 1.19–1.37 | 79 | 0.93 ± 0.06a | 0.82–1.06 | 102 |
| CTL2 | 1.35 ± 0.04b | 1.16–1.41 | 104 | 0.93 ± 0.05a | 0.78–1.02 | 38 |
| CTL3 | 1.38 ± 0.07c | 1.28–1.51 | 133 | 0.99 ± 0.05b | 0.90–1.07 | 115 |
| CTL4 | 1.26 ± 0.05d | 1.20–1.35 | 87 | 0.90 ± 0.05c | 0.82–1.10 | 112 |
| TPM2-null | 1.30 ± 0.08a | 1.19–1.52 | 129 | 0.95 ± 0.03a | 0.89–1.00 | 116 |
| TPM3-R167H | 1.03 ± 0.08e | 0.90–1.18 | 96 | 0.92 ± 0.02a | 0.85–1.09 | 82 |
| TPM2-E181K | 1.33 ± 0.06b | 1.23–1.56 | 63 | 0.96 ± 0.07d | 0.87–1.07 | 51 |
Different superscripts within a single column indicate significantly different values, as determined by one-way ANOVA with post hoc Fisher's PLSD tests. n, number of myofibrils.
Administration of troponin activators reverses contractile dysfunction
Because abnormal thin filament activation appears to be a unifying mechanism underlying skeletal muscle weakness in NM (3,4,18–20,27), we speculated that modulating thin filament activation might rescue the aberrant contractile properties of muscle fibers harboring NM-causing tropomyosin mutations. To test this, we used troponin activators, which are known to enhance the thin filament response to Ca2+ binding in striated muscle (15,16) under a wide spectrum of disease states (16,17). Two pharmacological drugs were selected: EMD 57033, which is known to be efficient in cardiac cells and fibers expressing type I MyHC (15,28), and CK-1909178, which is active in fibers expressing type II MyHC isoforms (16). In the presence of 30 µm EMD 57033, in fibers expressing type I MyHC, the Ca2+ sensitivities of force production and stiffness were significantly increased in TPM2-null, TPM3-R167H and CTL fibers (Table 1). Similarly, with 30 µm CK-1909178, in fibers expressing type IIa MyHC, the Ca2+ sensitivities of force and stiffness were significantly enhanced in TPM2-null and CTL fibers (Table 1). Based on these observations, troponin activators appear to be capable of rescuing most of the pathological contractile phenotype of TPM2-null and TPM3-R167H muscle fibers.
DISCUSSION
Tropomyosin mutations associated with congenital myopathy alter skeletal muscle contractile function, but, unlike initially thought, the underlying mechanisms are variable and mutation-specific. The TPM3-R167H and TPM2-null mutations severely reduce the proportion of strong myosin cross-bridges bound to actin filaments at submaximal [Ca2+], and subsequently dramatically depress force production at non-saturating [Ca2+], as attested by the changes in the Ca2+ sensitivities of stiffness and force and by the preserved relative stiffness-relative force relationships (Fig. 4). In contrast, the TPM2-E181K mutation has opposite effects and enhances the sensitivities of force and stiffness to [Ca2+] (Fig. 4). Hence, this particular defect facilitates strong myosin cross-bridge binding and resultant force generation at non-saturating [Ca2+]. How do various congenital myopathy-related tropomyosin mutations result in distinct effects on thin filament activation, cross-bridge number and force production?
The single amino acid substitution in TPM3-R167H does not produce a major charge modification and is located in the outer domain of the protein strand, in the ‘f’ position of the heptad repeat. Therefore, it is unlikely that the defect disrupts the coiled-coil dimeric structure of tropomyosin by modifying the chain–chain interaction between the two strands. Similarly, considering that the nearest troponin complex-anchoring region is ∼23 residues away from the TPM3-R167H mutation (29), a change in tropomyosin's affinity for troponin T is unlikely. Nevertheless, the defect may locally disrupt binding to actin's amino acid residues, altering tropomyosin stability and flexibility (30–32). This would affect thin filament activation by modifying the equilibria governing the transitions among the B-, C- and M-states in favor of tropomyosin's ‘off’ states (1). More specifically, it would be expected to disrupt cooperative thin filament activation (25). Hence, fewer myosin cross-bridge binding sites are exposed on the thin filaments, and fewer myosin cross-bridges can be formed in the strong binding state, limiting force production at submaximal [Ca2+].
While NM-causing nebulin mutations have been shown to reduce thin filament length (12), this study is the first to demonstrate that a specific NM-causing tropomyosin mutation (TPM3-R167H) can also reduce thin filament length (from 1.26–1.38 μm in CTL, TPM2-null and TPM2-E181K myofibrils down to 1.03 μm in TPM3-R167H myofibrils), specifically by shortening the nebulin-free, Tmod-capped extension at the pointed end of the thin filament (Fig. 7). This length change is functionally relevant, as thin filament shortening depresses force development at long sarcomere lengths (9,12,33–35). Indeed, our experimental force-sarcomere length curves for CTL and TPM3-R167H fibers demonstrate depressed force generation at long sarcomere lengths in TPM3-R167H fibers (Fig. 3). Moreover, reduced thin filament length in TPM3-R167H myofibrils may account for observed changes in thin filament activation in TPM3-R167H fibers, as shorter thin filaments have proportionally fewer sites available for binding of myosin heads during actomyosin cross-bridge cycling. However, this proposed relationship between thin filament length and thin filament activation remains to be experimentally validated.
It should be noticed that, here, we used DDecon to measure thin filament lengths. DDecon is a quantitative, super-resolution microscopy technique that enables length measurements to be made with a precision of up to ∼10–20 nm (36) which is well beyond the limits of conventional line-scan analysis of confocal images. Thus, by extensively sampling myofibrils throughout the muscle biopsy specimens, we were able to apply DDecon to detect subtle thin filament length changes that were not detectable in another study of TPM3-R167H using conventional image analysis methods (4).
What might be the mechanism underlying the shorter thin filament lengths in the TPM3-R167H mutant but not in the other mutants? Tropomyosin isoforms are known to reduce actin subunit off rates from actin filament pointed ends, with their efficacies related to the relative affinity of each tropomyosin isoform for binding along the actin filament (13,14). Weaker binding of TPM3-R167H to actin (see above) would therefore be expected to be associated with an increased rate of actin subunit loss from filament ends, leading to shorter filaments. Future studies comparing the binding of the TPM3-R167H and the TPM2-E181K mutant proteins to actin filaments, and their effects on actin elongation and depolymerization rates, will help address this mechanism. Tropomyosins can also mechanically stabilize/stiffen actin filaments (37,38) and, via such a mechanism, are likely to be involved in protecting actin filaments from disassembly by myosin activity during force-generating ATPase cycles (39). Thus, altered or weaker binding of the TPM3-R167H mutant to actin could mechanically destabilize the filament, leading to filament breakage and/or depolymerization during muscle contraction. Notably, the length of the proximal, nebulin-associated portion of the thin filament remained unchanged in the TPM3-R167H mutant, which is consistent with nebulin's role in mechanically and chemically stabilizing a 0.9–1.0-μm-long core of the thin filament (8,11,40–42).
In the case of the TPM2-null mutation, the observed molecular and cellular dysfunction is undoubtedly related to the lack of β-tropomyosin that results in overexpression of the γ- and α-tropomyosin isoforms (43). Even though these three tropomyosin isoforms share a large number of residues (86–91%) (44), some differences exist throughout the tropomyosin molecule. The replacement of β- by γ-tropomyosin may induce local electrostatic changes, destabilizing cooperative thin filament activation. It may also increase the probability of tropomyosin existing in its ‘off’ states, lowering the number of recruited myosin cross-bridges and subsequent force generation at submaximal [Ca2+]. This is consistent with previous experiments in β- and γ-tropomyosin-overexpressing mouse hearts (45–47) and detergent-extracted fiber bundles, which exhibit increases and decreases, respectively, in the Ca2+ sensitivity of force (45–47). In the present study, the replacement of β- by α-tropomyosin may also be detrimental for thin filament activation. Indeed, biophysical studies have demonstrated that β- and α-tropomyosin exhibit differences in their preferred position on the thin filament, with α-tropomyosin located further in the outer domain of the actin filament and, thus, further from an activated position (48).
Like the TPM3-R167H mutation, the TPM2-E181K mutation is positioned in the ‘f’ position of tropomyosin heptad repeat; consequently, it may not directly modify tropomyosin's chain–chain interactions. However, the single amino acid replacement has a net charge change. Moreover, as this particular mutation is located close to one of the two troponin T-binding areas (29) and directly binds to actin filaments (31), it may perturb propagation of Ca2+ signals along the thin filament, disrupting the equilibria among the B-, C- and M-states in favor of tropomyosin's ‘on’ state (1). Thus, the TPM2-E181K mutation facilitates thin filament cooperative activation and myosin cross-bridge attachment to actin monomers, promoting force generation at submaximal [Ca2+]. Such facilitation has been previously reported for the hypertrophic cardiomyopathy-linked E180G α-tropomyosin mutation (49–51). The fact that the TPM2-E181K mutation enhances force production at the skinned single-fiber level is unlikely to explain skeletal muscle weakness in the patient but the hyper-contraction may well provoke the observed joint contractures, stiffness and arthrogryposis. Other phenomena prior to Ca2+ binding to the thin filament (i.e. from neural drive to excitation–contraction coupling and Ca2+ handling) may be involved in the development of muscle weakness. In contrast, contractile dysfunction due to the TPM2-null and TPM3-R167H mutations may very well contribute to muscle weakness. Indeed, skeletal muscles are often submaximally activated in vivo. Therefore, the ability to respond to submaximal [Ca2+] is of great importance. In skeletal muscle harboring the TPM3-R167H and TPM2-null mutations, a higher [Ca2+] is required to activate the muscle cells at the same level as CTLs (16,28). One may suggest that this leads to an increased sarcoplasmic reticulum Ca2+-ATPase (SERCA) activity at the single-fiber and whole-muscle levels, decreasing the overall efficiency of contraction (52). Further experiments supporting this particular point are needed.
Seeking potential therapeutic interventions to rescue the reduced strong myosin cross-bridge number at submaximal [Ca2+] and subsequent lack of force generation in tropomyosin-related NM is clinically relevant. Here, we successfully modified thin filament activation via the use of troponin activators that are known to resolve similar problems in cardiac muscle (15,53). EMD 57033 is notably active in fibers expressing the type I MyHC isoform, whereas CK-1909178 is efficient in cells carrying the type II MyHC isoform. According to NMR and solution studies, EMD 57033 binds to the cardiac/slow skeletal troponin C isoform, whereas CK-1909178 attaches to the fast skeletal isoform of the same protein (16,54,55). During contraction, both promote troponin I displacement toward troponin C and favor thin filament protein movement toward the M-state (open state). Here, despite tropomyosin mutations, when the compounds are applied (30 µm), we observed the complete restoration of strong myosin cross-bridge binding to actin filaments at submaximal [Ca2+], leading to normal Ca2+ sensitivity of force production in both TPM3-R167H and TPM2-null skeletal muscle fibers. Consequently, tropomyosin defects and the subsequent movement blockade toward the M-state can be fully repaired. Hence, targeting thin filament activation as therapeutic approach appears relevant for these mutations and may be warranted in the future. However, certain limitations exist. As stated above, EMD 57033 binds to the troponin C isoform present in cardiac muscle and may produce unwanted side effects on the heart. CK-1909178 may overcome this by binding to the fast skeletal isoform of the same protein. Nevertheless, in some patients with NM and other neuromuscular diseases, skeletal muscle fibers expressing the type I MyHC isoform are predominant, and type II fibers are scarce. Therefore, considerable work remains to be done regarding this class of compounds.
In conclusion, in this study, we addressed the functional effects of three tropomyosin mutations. Two of these mutations (TPM2-null and TPM3-R167H) reduce the number of strongly bound myosin cross-bridges at submaximal [Ca2+], inducing a dramatic decrease in the Ca2+ sensitivity of force, adversely affecting contractile function. The TPM3-R167H mutation also induces a concomitant and substantial reduction in thin filament length, which may partially contribute to the reduction in numbers of strongly bound myosin cross-bridges. In contrast, the TPM2-E181K mutation induces an entirely different mechanism of muscle dysfunction, as it facilitates cross-bridge binding and force production at non-saturating [Ca2+]. Modulating thin filament activation in TPM2-null and TPM3-R167H skeletal muscle via the use of troponin activators reverses most of these impairments, suggesting that the administration of troponin activators may constitute a promising therapeutic approach to minimize generalized skeletal muscle weakness in NM.
MATERIALS AND METHODS
Subjects
This study involved patients carrying three different tropomyosin mutations termed TPM2-null, TPM3-R167H and TPM2-E181K (Table 3). The clinical histories, muscle morphologies and genotype analyses of two of these patients have been described in detail elsewhere (21,43,56). Seven healthy subjects (Table 3) with no history of neuromuscular disease served as CTLs. Informed consents were obtained from the patients and CTL subjects enrolled in this study. A local ethics committee at Uppsala University approved the protocol, and the experiments were carried out according to the guidelines of the Declaration of Helsinki.
Table 3.
CTL and patient muscle biopsies
| Subject ID | Biopsy location | Gene mutation | Protein change | Sex | Age (years) | Clinical features |
|---|---|---|---|---|---|---|
| TPM2-null | Gastrocnemius | c.628C>T | p.Gln210Stop absence of β-tropomyosin | M | 9 | Severe hypotonia at birth and delayed acquisition of motor milestones. Mild joint contractures. No respiratory or swallowing difficulties (43) |
| TPM3-R167H | Deltoid | c.503G>A | p.Arg167His | M | 58 | No delay in motor milestones, poor physical performances since childhood. Nocturnal non-invasive ventilation started at 50-year old (21) |
| TPM2-E181K | Deltoid | c.541G>A | p.Glu181Lys | F | 46 | Torticolis and arthrogryposis at birth, severe joint contractures and spine stiffness, mild distal weakness (56) |
| CTL1 | Vastus lateralis | — | — | F | 26 | — |
| CTL2 | Vastus lateralis | — | — | M | 39 | — |
| CTL3 | Tibialis anterior | — | — | F | 52 | — |
| CTL4 | Tibialis anterior | — | — | M | 67 | — |
| CTL5 | Vastus lateralis | — | — | M | 35 | — |
| CTL6 | Vastus lateralis | — | — | M | 26 | — |
| CTL7 | Tibialis anterior | — | — | F | 49 | — |
Muscle biopsies and fiber treatment
For the patients, open muscle biopsies of the deltoid muscle or lateral gastrocnemius muscle were performed under anesthesia. For the CTLs, percutaneous conchotome muscle biopsy specimens were obtained from the tibialis anterior or vastus lateralis muscles, also under anesthesia. The biopsy specimens were placed in relaxing solution at 4°C. Bundles of ∼50 fibers were dissected free and then tied to glass capillary tubes at slightly stretched lengths using surgical silk. Fiber bundles were then treated with skinning solution (relaxing solution containing glycerol; 50:50 v/v) for 24 h at 4°C, after which they were transferred to −20°C. In addition, fiber bundles were treated with sucrose, a cryoprotectant, within 1–2 weeks for long-term storage (57). Fiber bundles were detached from the capillary tubes and snap-frozen in liquid nitrogen-chilled propane and stored at −160°C.
Solutions
Relaxing and activating solutions contained (in mm) 4 Mg-ATP, 1 free Mg2+, 20 imidazole, 7 EGTA, 14.5 creatine phosphate and KCl to adjust the ionic strength to 180 mm. The pH was adjusted to 7.0. The concentrations of free Ca2+ were 10−9.00 m (relaxing solution) and 10−6.30, 10−5.90, 10−5.50, 10−5.10, 10−4.90 and 10−4.50 m (activating solutions), expressed as pCa (−log10[Ca2+]). The apparent stability constants for Ca2+-EGTA were corrected for temperature (15°C) and ionic strength (180 mm). The computer program by Fabiato (58) was used to calculate the concentrations of each metal, ligand and metal–ligand complex. Troponin activators were CK-1909178 and EMD 57033, prepared as 10 mm stock solutions in DMSO and diluted in relaxing solution to obtain a final CK-1909178 or EMD 57033 concentration of 30 µm and DMSO concentration of 0.3% (v/v). CTL solutions were prepared with an equivalent volume of DMSO, which had no effect on fiber function.
Preparation and use of NEM-S1
Myosin S1 was purified from rabbit fast-twitch skeletal muscle and modified with NEM, as described previously (26). A stock solution of NEM-S1 (30 μm) was prepared by overnight dialysis against a solution of 20 mm imidazole, pH 7.0, and 1 mm DTT. A working solution of NEM-S1 (15 μm) was prepared immediately before use by mixing equal volumes of 2× stock of pCa 9.0 and NEM-S1 stock. The final concentration of NEM-S1 was adjusted by adding the appropriate amount of 1× pCa 9.00 solutions. Before activation, each fiber was first incubated for 15 min at 15°C in the solution of pCa 9.00 containing 3 μm NEM-S1. It was subsequently transferred to pre-activating solution for 1 min and then into activating solutions of varying pCa (i.e. pCa 6.30–4.50) without NEM-S1. After each activation, the fiber was returned to solution of pCa 9.00 containing NEM-S1. Thus, the fiber was incubated in NEM-S1-free solutions for no more than 2 min, during which time negligible amounts of NEM-S1 would be debound (26).
Fiber functional analyses
On the day of the experiment, fiber bundles were de-sucrosed and transferred to relaxing solution, and single fibers were dissected. A 1–2-mm-long fiber segment was placed between connectors leading to a force transducer (model 400A, Aurora Scientific) and a lever arm system (model 308B, Aurora Scientific) (59,60). The two ends of the fiber segment were tightly attached to the connectors, as described previously (59). Sarcomere length was controlled using a high-speed video analysis system (model 901A HVSL, Aurora Scientific). Prior to the mechanical experiments, fiber diameter and depth were measured through a microscope (320× magnification) using an image analysis system. Cross-sectional area (CSA) was calculated from the diameter and depth, assuming an elliptical circumference, and was corrected for the 20% swelling that is known to occur during skinning (59). Mechanical experiments were done at 15°C and included force measurements (normalized to CSA) at several sarcomere lengths (from 2.20 to 3.20 µm) and in various activating solutions with different [Ca2+] values (pCa's varying from 9.00 to 4.50).
In addition to force measurements, stiffness calculations were performed. Once steady-state isometric force was attained, small-amplitude sinusoidal length changes (ΔL, ±0.2% of fiber length) were applied to one end of the fiber at 500 Hz (61). The resultant force response (ΔF) was measured, and the means of 20 consecutive ΔL and ΔF readings were used to determine stiffness. The fiber stiffness was calculated as the difference between the stiffness in the activating solutions and the resting stiffness measured in the same segment in the relaxing solution. Fiber stiffness was calculated as follows (62):
| (2) |
As with the force measurements, stiffness was measured in solutions with various pCa's (from 9.00 to 4.50).
The rate of force development was also estimated. Once steady-state isometric force was reached at various pCa's (from 9.00 to 4.50), a slack by 20% of the original fiber length was introduced within 1–2 ms at one end of the fiber, resulting in a rapid reduction of force to near zero. This was followed by a brief period of unloaded shortening (20 ms), after which the preparation was quickly restretched to its original length and the force recovered to its original steady-state value. As described previously (23), the apparent rate constant of force redevelopment (ktr) was estimated by linear transformation of the half-time of force redevelopment (t1/2) as follows (63):
| (3) |
For all the above measurements, strict acceptance criteria were applied. A fiber was included in the analyses only if (i) the sarcomere length of the fiber changed by <0.10 µm between relaxation and maximum activation, and (ii) maximal force changed by <10% between the initial and final activation. To avoid any age-, sex- or biopsy-related effects, for each individual parameter (force, stiffness, ktr, myosin to actin ratio), we compared data from the patients with pooled values of at least five different CTL subjects (with distinct sex, age and biopsy location).
Measurement of protein levels
After the mechanical measurements, each fiber was placed in urea buffer in a plastic microcentrifuge tube and stored at −160°C. MyHC isoform composition was determined by 6% SDS–PAGE. The acrylamide concentration (w/v) was 4% in the stacking gel and 6% in the resolving gel, and the gel matrix included 30% glycerol. Sample loads were kept small (equivalent to ∼0.05 mm of fiber segment) to improve the resolution of the MyHC bands (types I, IIa and IIx). Electrophoresis was performed at 120 V for 24 h with a Tris–glycine electrode buffer (pH 8.3) at 15°C (SE 600 vertical slab gel unit, Hoefer Scientific Instruments). The gels were silver-stained and subsequently scanned in a soft laser densitometer (Molecular Dynamics) with a high spatial resolution (50-μm pixel spacing) and 4096 optical density levels.
In addition, MyHC, actin and tropomyosin isoform contents were quantified by 12% SDS–PAGE. The acrylamide concentration (w/v) was 4% in the stacking gel and 12% in the resolving gel, and the gel matrix included 10% glycerol. The gels were stained with Coomassie blue. The relative protein contents were then calculated from the densitometric scans (see above).
Immunofluorescence staining and confocal imaging
Methods for tissue processing, immunostaining and imaging of human skeletal muscle fiber bundles were adapted from those described previously (9). Cryoprotected fiber bundles were shipped from Uppsala University (Uppsala, Sweden) to The Scripps Research Institute (La Jolla, CA, USA) on dry ice via 2-day delivery and stored at −80°C until use. Bundles were thawed in ice-cold relaxing solution, stretched to resolve Tmod doublets at the pointed ends of thin filaments that extend from neighboring Z-lines, secured onto wooden dowels using 3-0 silk sutures and immersed in ice-cold fixation solution (relaxing solution containing 4% paraformaldehyde). Fiber bundles were fixed overnight at 4°C, re-cryoprotected in 30% sucrose followed by 60% sucrose in relaxing solution, embedded in OCT medium, frozen on a metal block chilled in liquid N2 and sectioned into 12-μm-thick cryosections. Sections were mounted on slides and stored at −20°C for up to 2 weeks.
For immunostaining, sections were washed for 20 min in PBS + 0.1% Triton X-100 (PBST), permeabilized for 20 min in PBS + 0.3% Triton X-100 and blocked overnight at 4°C in 4% BSA + 1% goat serum in PBST. Sections were labeled with the following primary antibodies diluted in blocking solution overnight at 4°C: mouse monoclonal anti-α-actinin (EA53, 1:100; Sigma-Aldrich, St. Louis, MO, USA) and either a rabbit polyclonal antiserum to chicken Tmod4 pre-adsorbed by passage through a Tmod1 Sepharose column (R3577bl3c, 1:25) (10) or an affinity-purified rabbit polyclonal antibody to the nebulin M1M2M3 domain (R1357L, 9.3 µg/ml). After washing in PBST, sections were labeled with Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:200, Invitrogen, Carlsbad, CA, USA) and Alexa Fluor 647-conjugated goat anti-mouse IgG (1:200, Invitrogen) in blocking buffer for 2 h at room temperature. The secondary antibody mixture was supplemented with rhodamine phalloidin (1:100, Invitrogen) to stain F-actin. Tissues were then washed again in PBST, preserved in Gel/Mount™ aqueous mounting medium (Sigma-Aldrich) and cover-slipped. Images of single optical sections were collected on a Bio-Rad Radiance 2100 laser-scanning confocal microscope mounted on a Nikon TE2000-U microscope using a 100×/1.4 NA-oil objective lens (zoom 3). Image sampling was performed randomly throughout the entire longitudinal view of the fiber bundle. Bio-Rad LaserSharp 2000 software was used for image collection. Images were processed with Adobe Photoshop (version CS5).
Distributed deconvolution
DDecon is a super-resolution light microscopy technique that computes thin filament lengths with a precision of 10–20 nm by directly measuring the peak positions of fluorescently labeled Tmod1 or Tmod4 with respect to α-actinin at the Z-line, from line scans of fluorescence intensities along myofibrils (36). Both Tmod1 and Tmod4 cap the pointed ends of the thin filaments in mammalian skeletal muscle and serve as faithful markers for the thin filament pointed ends at the H-zone periphery (9,10,64,65). In this study, we measured thin filament lengths based only on Tmod4 localization, as previous DDecon-based analyses have not observed systematic differences in average thin filament lengths based on Tmod1 versus Tmod4 localization (9,10). We also used DDecon to measure the position of the N-terminal M1M2M3 domain of nebulin, which is located slightly proximal to the Z-line with respect to Tmod4 at the thin filament pointed ends (8–10). These applications of DDecon were previously validated for use in immunostained cryosections of human skeletal muscle fiber bundles (9). Each DDecon analysis was performed on a skeletal muscle fiber bundle and reflects the average thin filament length within the bundle, regardless of the bundle's fiber type composition. Average thin filament length thus incorporates contributions from both type I and type II fibers, which have longer and shorter thin filament lengths, respectively (9).
In this study, we used a DDecon plugin for ImageJ that generates the best fit of a model intensity distribution function for a given thin filament component (Tmod4, nebulin M1M2M3 domain) to an experimental one-dimensional myofibril fluorescence intensity profile (line scan) obtained for each fluorescent probe (anti-Tmod4, anti-nebulin M1M2M3) (36). Each probe-specific model distribution is applied to a line scan of three thin filament arrays along an individual myofibril to calculate the probe's average distance from the Z-line within that myofibril. Model fitting is optimized by an iterative fitting procedure that minimizes the error between the observed line scan intensities and the modeled intensities, as described previously (36). Image regions containing adequately stretched sarcomeres were identified based on the presence of Tmod4 or nebulin M1M2M3 doublets and clearly discernable H-zones. All line scans were background-corrected, as described previously (36). Distances were calculated by converting pixel sizes into micrometer using the magnification factor for each image.
Statistical analysis
Data are presented as the mean ± standard error of the mean (SEM) or mean ± standard deviation (SD), where appropriate. SigmaStat (Jandel Scientific) and Microsoft Excel were used to generate descriptive statistics. Where appropriate, either an unpaired t-test or ANOVAs followed by post hoc tests was used. Otherwise, regression analyses were applied, and relationships were considered to be significantly different from zero at a significance level of P < 0.05.
FUNDING
This study was supported by grants from the Swedish Research Council, Association Française contre les Myopathies, Tore Nilson Stiftelse and Stiftelsen Apotekare Hedbergs fond för Medicinsk Forskning (to J.O.). This work was assisted by the Core Imaging Facility of the San Diego Skeletal Muscle Research Center, supported by NIH grant P30-AR061303 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases at the National Institutes of Health (to V.M.F.). Additional support was provided by a training grant from the National Heart, Lung, and Blood Institute at the National Institutes of Health (T32-HL007195 to D.S.G.).
ACKNOWLEDGMENTS
CK-1909178 was a gift from Dr Alan Russell (Cytokinetics Inc., South San Francisco, CA, USA), EMD 57033 was a gift from Dr Norbert Beier (Merck KGaA, Darmstadt, Germany), the anti-nebulin M1M2M3 antibody was a gift from Dr Carol C. Gregorio (University of Arizona, Tucson, AZ, USA) and NEM-S1 was a gift from Dr Darl R. Swartz (Purdue University, West Lafayette, IN, USA). We are grateful to Nancy E. Kim, Yvette Hedström and Ann-Marie Gustafsson for excellent technical assistance.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Lehman W., Craig R. Tropomyosin and the steric mechanism of muscle regulation. Adv. Exp. Med. Biol. 2008;644:95–109. doi: 10.1007/978-0-387-85766-4_8. doi:10.1007/978-0-387-85766-4_8. [DOI] [PubMed] [Google Scholar]
- 2.Ochala J. Thin filament proteins mutations associated with skeletal myopathies: defective regulation of muscle contraction. J. Mol. Med. 2008;86:1197–1204. doi: 10.1007/s00109-008-0380-9. doi:10.1007/s00109-008-0380-9. [DOI] [PubMed] [Google Scholar]
- 3.Ochala J., Li M., Ohlsson M., Oldfors A., Larsson L. Defective regulation of contractile function in muscle fibres carrying an E41K beta-tropomyosin mutation. J. Physiol. 2008;586:2993–3004. doi: 10.1113/jphysiol.2008.153650. doi:10.1113/jphysiol.2008.153650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ottenheijm C.A., Lawlor M.W., Stienen G.J., Granzier H., Beggs A.H. Changes in cross-bridge cycling underlie muscle weakness in patients with tropomyosin 3-based myopathy. Hum. Mol. Genet. 2011;20:2015–2025. doi: 10.1093/hmg/ddr084. doi:10.1093/hmg/ddr084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanoudou D., Beggs A.H. Clinical and genetic heterogeneity in nemaline myopathy—a disease of skeletal muscle thin filaments. Trends Mol. Med. 2001;7:362–368. doi: 10.1016/s1471-4914(01)02089-5. doi:10.1016/S1471-4914(01)02089-5. [DOI] [PubMed] [Google Scholar]
- 6.Clarke N.F., Kolski H., Dye D.E., Lim E., Smith R.L., Patel R., Fahey M.C., Bellance R., Romero N.B., Johnson E.S., et al. Mutations in TPM3 are a common cause of congenital fiber type disproportion. Ann. Neurol. 2008;63:329–337. doi: 10.1002/ana.21308. doi:10.1002/ana.21308. [DOI] [PubMed] [Google Scholar]
- 7.Michele D.E., Albayya F.P., Metzger J.M. A nemaline myopathy mutation in alpha-tropomyosin causes defective regulation of striated muscle force production. J. Clin. Invest. 1999;104:1575–1581. doi: 10.1172/JCI7842. doi:10.1172/JCI7842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castillo A., Nowak R., Littlefield K.P., Fowler V.M., Littlefield R.S. A nebulin ruler does not dictate thin filament lengths. Biophys. J. 2009;96:1856–1865. doi: 10.1016/j.bpj.2008.10.053. doi:10.1016/j.bpj.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gokhin D.S., Kim N.E., Lewis S.A., Hoenecke H.R., D'Lima D.D., Fowler V.M. Thin-filament length correlates with fiber type in human skeletal muscle. Am. J. Physiol. Cell. Physiol. 2012;302:C555–565. doi: 10.1152/ajpcell.00299.2011. doi:10.1152/ajpcell.00299.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gokhin D.S., Lewis R.A., McKeown C.R., Nowak R.B., Kim N.E., Littlefield R.S., Lieber R.L., Fowler V.M. Tropomodulin isoforms regulate thin filament pointed-end capping and skeletal muscle physiology. J. Cell Biol. 2010;189:95–109. doi: 10.1083/jcb.201001125. doi:10.1083/jcb.201001125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Littlefield R.S., Fowler V.M. Thin filament length regulation in striated muscle sarcomeres: pointed-end dynamics go beyond a nebulin ruler. Semin. Cell Dev. Biol. 2008;19:511–519. doi: 10.1016/j.semcdb.2008.08.009. doi:10.1016/j.semcdb.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ottenheijm C.A., Witt C.C., Stienen G.J., Labeit S., Beggs A.H., Granzier H. Thin filament length dysregulation contributes to muscle weakness in nemaline myopathy patients with nebulin deficiency. Hum. Mol. Genet. 2009;18:2359–2369. doi: 10.1093/hmg/ddp168. doi:10.1093/hmg/ddp168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broschat K.O. Tropomyosin prevents depolymerization of actin filaments from the pointed end. J. Biol. Chem. 1990;265:21323–21329. [PubMed] [Google Scholar]
- 14.Broschat K.O., Weber A., Burgess D.R. Tropomyosin stabilizes the pointed end of actin filaments by slowing depolymerization. Biochemistry. 1989;28:8501–8506. doi: 10.1021/bi00447a035. doi:10.1021/bi00447a035. [DOI] [PubMed] [Google Scholar]
- 15.Endoh M. Cardiac ca(2+) signaling and ca(2+) sensitizers. Circ. J. 2008;72:1915–1925. doi: 10.1253/circj.cj-08-0838. doi:10.1253/circj.CJ-08-0838. [DOI] [PubMed] [Google Scholar]
- 16.Ochala J. Ca(2+) sensitizers: An emerging class of agents for counterbalancing weakness in skeletal muscle diseases? Neuromuscul. Disord. 2009;20:98–101. doi: 10.1016/j.nmd.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 17.van Hees H.W., Dekhuijzen P.N., Heunks L.M. Levosimendan enhances force generation of diaphragm muscle from patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2009;179:41–47. doi: 10.1164/rccm.200805-732OC. doi:10.1164/rccm.200805-732OC. [DOI] [PubMed] [Google Scholar]
- 18.Ochala J., Iwamoto H., Larsson L., Yagi N. A myopathy-linked tropomyosin mutation severely alters thin filament conformational changes during activation. Proc. Natl Acad. Sci. USA. 2010;107:9807–9812. doi: 10.1073/pnas.1001733107. doi:10.1073/pnas.1001733107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ochala J., Lehtokari V.L., Iwamoto H., Li M., Feng H.Z., Jin J.P., Yagi N., Wallgren-Pettersson C., Penisson-Besnier I., Larsson L. Disrupted myosin cross-bridge cycling kinetics triggers muscle weakness in nebulin-related myopathy. FASEB J. 2011;25:1903–1913. doi: 10.1096/fj.10-176727. doi:10.1096/fj.10-176727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ochala J., Li M., Tajsharghi H., Kimber E., Tulinius M., Oldfors A., Larsson L. Effects of a R133W beta-tropomyosin mutation on regulation of muscle contraction in single human muscle fibres. J. Physiol. 2007;581:1283–1292. doi: 10.1113/jphysiol.2007.129759. doi:10.1113/jphysiol.2007.129759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Penisson-Besnier I., Monnier N., Toutain A., Dubas F., Laing N. A second pedigree with autosomal dominant nemaline myopathy caused by TPM3 mutation: a clinical and pathological study. Neuromuscul. Disord. 2007;17:330–337. doi: 10.1016/j.nmd.2007.01.017. doi:10.1016/j.nmd.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 22.Huxley A.F. Muscle structure and theories of contraction. Prog. Biophys. Biophys. Chem. 1957;7:255–318. [PubMed] [Google Scholar]
- 23.Brenner B., Eisenberg E. Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc. Natl Acad. Sci. USA. 1986;83:3542–3546. doi: 10.1073/pnas.83.10.3542. doi:10.1073/pnas.83.10.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kreutziger K.L., Piroddi N., Scellini B., Tesi C., Poggesi C., Regnier M. Thin filament Ca2+ binding properties and regulatory unit interactions alter kinetics of tension development and relaxation in rabbit skeletal muscle. J. Physiol. 2008;586:3683–3700. doi: 10.1113/jphysiol.2008.152181. doi:10.1113/jphysiol.2008.152181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fitzsimons D.P., Patel J.R., Campbell K.S., Moss R.L. Cooperative mechanisms in the activation dependence of the rate of force development in rabbit skinned skeletal muscle fibers. J. Gen. Physiol. 2001;117:133–148. doi: 10.1085/jgp.117.2.133. doi:10.1085/jgp.117.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swartz D.R., Moss R.L. Influence of a strong-binding myosin analogue on calcium-sensitive mechanical properties of skinned skeletal muscle fibers. J. Biol. Chem. 1992;267:20497–20506. [PubMed] [Google Scholar]
- 27.Ottenheijm C.A., Hooijman P., Dechene E.T., Stienen G.J., Beggs A.H., Granzier H. Altered myofilament function depresses force generation in patients with nebulin-based nemaline myopathy (NEM2) J. Struct. Biol. 2010;170:334–343. doi: 10.1016/j.jsb.2009.11.013. doi:10.1016/j.jsb.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ochala J., Radell P.J., Eriksson L.I., Larsson L. EMD 57033 partially reverses ventilator-induced diaphragm muscle fibre calcium desensitisation. Pflugers Arch. 2009;459:475–483. doi: 10.1007/s00424-009-0744-1. doi:10.1007/s00424-009-0744-1. [DOI] [PubMed] [Google Scholar]
- 29.Li Y., Mui S., Brown J.H., Strand J., Reshetnikova L., Tobacman L.S., Cohen C. The crystal structure of the C-terminal fragment of striated-muscle alpha-tropomyosin reveals a key troponin T recognition site. Proc. Natl Acad. Sci. USA. 2002;99:7378–7383. doi: 10.1073/pnas.102179999. doi:10.1073/pnas.102179999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hitchcock-DeGregori S.E., Song Y., Greenfield N.J. Functions of tropomyosin's periodic repeats. Biochemistry. 2002;41:15036–15044. doi: 10.1021/bi026519k. doi:10.1021/bi026519k. [DOI] [PubMed] [Google Scholar]
- 31.Li X.E., Tobacman L.S., Mun J.Y., Craig R., Fischer S., Lehman W. Tropomyosin position on F-actin revealed by EM reconstruction and computational chemistry. Biophys. J. 2011;100:1005–1013. doi: 10.1016/j.bpj.2010.12.3697. doi:10.1016/j.bpj.2010.12.3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marttila M., Lemola E., Wallefeld W., Memo M., Donner K., Laing N.G., Marston S., Gronholm M., Wallgren-Pettersson C. Abnormal actin binding of aberrant beta-tropomyosins is a molecular cause of muscle weakness in TPM2-related nemaline and cap myopathy. Biochem. J. 2012;442:231–239. doi: 10.1042/BJ20111030. doi:10.1042/BJ20111030. [DOI] [PubMed] [Google Scholar]
- 33.Granzier H.L., Akster H.A., Ter Keurs H.E. Effect of thin filament length on the force-sarcomere length relation of skeletal muscle. Am. J. Physiol. 1991;260:C1060–1070. doi: 10.1152/ajpcell.1991.260.5.C1060. [DOI] [PubMed] [Google Scholar]
- 34.Gokhin D.S., Bang M.L., Zhang J., Chen J., Lieber R.L. Reduced thin filament length in nebulin-knockout skeletal muscle alters isometric contractile properties. Am. J. Physiol. Cell Physiol. 2009;296:C1123–1132. doi: 10.1152/ajpcell.00503.2008. doi:10.1152/ajpcell.00503.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walker S.M., Schrodt G.R. I segment lengths and thin filament periods in skeletal muscle fibers of the Rhesus monkey and the human. Anat. Rec. 1974;178:63–81. doi: 10.1002/ar.1091780107. doi:10.1002/ar.1091780107. [DOI] [PubMed] [Google Scholar]
- 36.Littlefield R., Fowler V.M. Measurement of thin filament lengths by distributed deconvolution analysis of fluorescence images. Biophys. J. 2002;82:2548–2564. doi: 10.1016/S0006-3495(02)75598-7. doi:10.1016/S0006-3495(02)75598-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kojima H., Ishijima A., Yanagida T. Direct measurement of stiffness of single actin filaments with and without tropomyosin by in vitro nanomanipulation. Proc. Natl Acad. Sci. USA. 1994;91:12962–12966. doi: 10.1073/pnas.91.26.12962. doi:10.1073/pnas.91.26.12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adami R., Cintio O., Trombetta G., Choquet D., Grazi E. On the stiffness of the natural actin filament decorated with alexa fluor tropomyosin. Biophys. Chem. 2003;104:469–476. doi: 10.1016/s0301-4622(03)00036-x. doi:10.1016/S0301-4622(03)00036-X. [DOI] [PubMed] [Google Scholar]
- 39.Haviv L., Gillo D., Backouche F., Bernheim-Groswasser A. A cytoskeletal demolition worker: myosin II acts as an actin depolymerization agent. J. Mol. Biol. 2008;375:325–330. doi: 10.1016/j.jmb.2007.09.066. doi:10.1016/j.jmb.2007.09.066. [DOI] [PubMed] [Google Scholar]
- 40.Bang M.L., Li X., Littlefield R., Bremner S., Thor A., Knowlton K.U., Lieber R.L., Chen J. Nebulin-deficient mice exhibit shorter thin filament lengths and reduced contractile function in skeletal muscle. J. Cell Biol. 2006;173:905–916. doi: 10.1083/jcb.200603119. doi:10.1083/jcb.200603119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pappas C.T., Bliss K.T., Zieseniss A., Gregorio C.C. The nebulin family: an actin support group. Trends Cell. Biol. 2011;21:29–37. doi: 10.1016/j.tcb.2010.09.005. doi:10.1016/j.tcb.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pappas C.T., Krieg P.A., Gregorio C.C. Nebulin regulates actin filament lengths by a stabilization mechanism. J. Cell Biol. 2010;189:859–870. doi: 10.1083/jcb.201001043. doi:10.1083/jcb.201001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monnier N., Lunardi J., Marty I., Mezin P., Labarre-Vila A., Dieterich K., Jouk P.S. Absence of beta-tropomyosin is a new cause of Escobar syndrome associated with nemaline myopathy. Neuromuscul. Disord. 2009;19:118–123. doi: 10.1016/j.nmd.2008.11.009. doi:10.1016/j.nmd.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 44.Jagatheesan G., Rajan S., Wieczorek D.F. Investigations into tropomyosin function using mouse models. J. Mol. Cell Cardiol. 2009;48:893–898. doi: 10.1016/j.yjmcc.2009.10.003. doi:10.1016/j.yjmcc.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pieples K., Arteaga G., Solaro R.J., Grupp I., Lorenz J.N., Boivin G.P., Jagatheesan G., Labitzke E., DeTombe P.P., Konhilas J.P., et al. Tropomyosin 3 expression leads to hypercontractility and attenuates myofilament length-dependent Ca(2+) activation. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H1344–1353. doi: 10.1152/ajpheart.00351.2002. [DOI] [PubMed] [Google Scholar]
- 46.Muthuchamy M., Boivin G.P., Grupp I.L., Wieczorek D.F. Beta-tropomyosin overexpression induces severe cardiac abnormalities. J. Mol. Cell Cardiol. 1998;30:1545–1557. doi: 10.1006/jmcc.1998.0720. doi:10.1006/jmcc.1998.0720. [DOI] [PubMed] [Google Scholar]
- 47.Muthuchamy M., Grupp I.L., Grupp G., O'Toole B.A., Kier A.B., Boivin G.P., Neumann J., Wieczorek D.F. Molecular and physiological effects of overexpressing striated muscle beta-tropomyosin in the adult murine heart. J. Biol. Chem. 1995;270:30593–30603. doi: 10.1074/jbc.270.51.30593. doi:10.1074/jbc.270.51.30593. [DOI] [PubMed] [Google Scholar]
- 48.Lehman W., Hatch V., Korman V., Rosol M., Thomas L., Maytum R., Geeves M.A., Van Eyk J.E., Tobacman L.S., Craig R. Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. J. Mol. Biol. 2000;302:593–606. doi: 10.1006/jmbi.2000.4080. doi:10.1006/jmbi.2000.4080. [DOI] [PubMed] [Google Scholar]
- 49.Bai F., Weis A., Takeda A.K., Chase P.B., Kawai M. Enhanced active cross-bridges during diastole: molecular pathogenesis of tropomyosin's HCM mutations. Biophys. J. 2011;100:1014–1023. doi: 10.1016/j.bpj.2011.01.001. doi:10.1016/j.bpj.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mathur M.C., Chase P.B., Chalovich J.M. Several cardiomyopathy causing mutations on tropomyosin either destabilize the active state of actomyosin or alter the binding properties of tropomyosin. Biochem. Biophys. Res. Commun. 2011;406:74–78. doi: 10.1016/j.bbrc.2011.01.112. doi:10.1016/j.bbrc.2011.01.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang F., Brunet N.M., Grubich J.R., Bienkiewicz E.A., Asbury T.M., Compton L.A., Mihajlovic G., Miller V.F., Chase P.B. Facilitated cross-bridge interactions with thin filaments by familial hypertrophic cardiomyopathy mutations in alpha-tropomyosin. J. Biomed. Biotechnol. 2011;2011:435271. doi: 10.1155/2011/435271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barclay C.J., Woledge R.C., Curtin N.A. Energy turnover for Ca2+ cycling in skeletal muscle. J. Muscle Res. Cell Motil. 2007;28:259–274. doi: 10.1007/s10974-007-9116-7. doi:10.1007/s10974-007-9116-7. [DOI] [PubMed] [Google Scholar]
- 53.Kass D.A., Solaro R.J. Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation. 2006;113:305–315. doi: 10.1161/CIRCULATIONAHA.105.542407. doi:10.1161/CIRCULATIONAHA.105.542407. [DOI] [PubMed] [Google Scholar]
- 54.Pan B.S., Johnson R.G., Jr Interaction of cardiotonic thiadiazinone derivatives with cardiac troponin C. J. Biol. Chem. 1996;271:817–823. doi:10.1074/jbc.271.2.817. [PubMed] [Google Scholar]
- 55.Wang X., Li M.X., Spyracopoulos L., Beier N., Chandra M., Solaro R.J., Sykes B.D. Structure of the C-domain of human cardiac troponin C in complex with the Ca2+ sensitizing drug EMD 57033. J. Biol. Chem. 2001;276:25456–25466. doi: 10.1074/jbc.M102418200. doi:10.1074/jbc.M102418200. [DOI] [PubMed] [Google Scholar]
- 56.Mohamed J., Quijano-Roy S., Monnier N., Béhin A., Avila-Smirnov D., Romero N.B., Allamand V., Richard P., Barois A., May A., et al. Neuromuscul. Disord. in press: 2012. Whole-Body muscle MRI in a series of patients with congenital myopathy related to TPM2 gene mutations. [DOI] [PubMed] [Google Scholar]
- 57.Frontera W.R., Larsson L. Contractile studies of single human skeletal muscle fibers: a comparison of different muscles, permeabilization procedures, and storage techniques. Muscle Nerve. 1997;20:948–952. doi: 10.1002/(sici)1097-4598(199708)20:8<948::aid-mus3>3.0.co;2-6. doi:10.1002/(SICI)1097-4598(199708)20:8<948::AID-MUS3>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 58.Fabiato A. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 1988;157:378–417. doi: 10.1016/0076-6879(88)57093-3. doi:10.1016/0076-6879(88)57093-3. [DOI] [PubMed] [Google Scholar]
- 59.Moss R.L. Sarcomere length-tension relations of frog skinned muscle fibres during calcium activation at short lengths. J. Physiol. 1979;292:177–192. doi: 10.1113/jphysiol.1979.sp012845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Larsson L., Moss R.L. Maximum velocity of shortening in relation to myosin isoform composition in single fibres from human skeletal muscles. J. Physiol. 1993;472:595–614. doi: 10.1113/jphysiol.1993.sp019964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martyn D.A., Smith L., Kreutziger K.L., Xu S., Yu L.C., Regnier M. The effects of force inhibition by sodium vanadate on cross-bridge binding, force redevelopment, and Ca2+ activation in cardiac muscle. Biophys. J. 2007;92:4379–4390. doi: 10.1529/biophysj.106.096768. doi:10.1529/biophysj.106.096768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McDonald K.S., Fitts R.H. Effect of hindlimb unloading on rat soleus fiber force, stiffness, and calcium sensitivity. J. Appl. Physiol. 1995;79:1796–1802. doi: 10.1152/jappl.1995.79.5.1796. [DOI] [PubMed] [Google Scholar]
- 63.Regnier M., Martyn D.A., Chase P.B. Calcium regulation of tension redevelopment kinetics with 2-deoxy-ATP or low [ATP] in rabbit skeletal muscle. Biophys. J. 1998;74:2005–2015. doi: 10.1016/S0006-3495(98)77907-X. doi:10.1016/S0006-3495(98)77907-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Almenar-Queralt A., Lee A., Conley C.A., Ribas de Pouplana L., Fowler V.M. Identification of a novel tropomodulin isoform, skeletal tropomodulin, that caps actin filament pointed ends in fast skeletal muscle. J. Biol. Chem. 1999;274:28466–28475. doi: 10.1074/jbc.274.40.28466. doi:10.1074/jbc.274.40.28466. [DOI] [PubMed] [Google Scholar]
- 65.Fowler V.M., Sussmann M.A., Miller P.G., Flucher B.E., Daniels M.P. Tropomodulin is associated with the free (pointed) ends of the thin filaments in rat skeletal muscle. J. Cell Biol. 1993;120:411–420. doi: 10.1083/jcb.120.2.411. doi:10.1083/jcb.120.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]