

Abstract
Recent studies indicate that toll-like receptors (TLRs) are expressed on T cells and that these receptors directly or indirectly activate the adaptive immune system. We have shown previously that acute alcohol/ethanol (EtOH) intoxication combined with burn injury suppresses mesenteric lymph node (MLN) T-cell interleukin-2 (IL-2) and interferon γ (IFN-γ) production. We examined whether direct stimulation of T cells with TLR2, 4, 5 and 7 agonists modulates CD3-mediated T-cell IL-2/IFN-γ release following EtOH and burn injury. Male mice were gavaged with EtOH (2.9 gm/kg) 4 h prior to receiving an ~12.5% total body surface area sham or full-thickness burn injury. Animals were killed on d 1 after injury and T cells were purified from MLN and spleens. T cells were cultured with plate-bound anti-CD3 in the presence or absence of various TLR ligands. Although TLR2, 4 and 5 agonists potentiate anti-CD3–dependent IFN-γ by T cells, the TLR2 agonist alone induced IFN-γ production independent of CD3 stimulation. Furthermore, T cells were treated with inhibitors of myeloid differentiation primary response protein 88 (MyD88), TIR domain-containing adaptor protein (TIRAP), p38 and/or extracellular signal-regulated kinase (ERK) to determine the mechanism by which TLR2 mediates IL-2/IFN-γ production. IL-2 was not influenced by TLR agonists. MyD88 and TIRAP inhibitory peptides dose-dependently diminished the ability of T cells to release IFN-γ. p38 and ERK inhibitors also abolished TLR2-mediated T-cell IFN-γ. Together, our findings suggest that TLR2 directly modulates T-cell IFN-γ production following EtOH and burn injury, independent of antigen-presenting cells. Furthermore, we demonstrated that MyD88/TIRAP-dependent p38/ERK activation is critical to TLR2-mediated T-cell IFN-γ release following EtOH and burn injury.
INTRODUCTION
Alcohol remains the most abused substance worldwide. It is a high risk factor for traumatic injury, including burns (1–3). Nearly one million burn injuries are reported in the United States every year, and 50% of these injuries occur in individuals under the influence of alcohol/ethanol (EtOH) (4–8). Studies indicate that intoxicated patients have higher rates of septic complications, longer hospital stays and increased mortality compared with patients who have a similar extent of burn injury but did not consume EtOH before injury (7–11). There is evidence that EtOH intoxication combined with burn injury abrogates the host immune system. Specifically, the combined insult of EtOH and burn injury suppresses T-cell responses, potentiates inflammatory cytokine and chemokine production and induces neutrophil recruitment to the intestine and other organs (12–14). Studies from our laboratory suggest that acute EtOH intoxication combined with burn injury suppresses mesenteric lymph node (MLN) T-cell proliferation as well as interleukin-2 (IL-2) and interferon γ (IFN-γ) production. This effect is accompanied by an increase in bacterial translocation to MLN. We have also demonstrated a role for p38 and extra-cellular signal-regulated kinase (ERK) activation in T-cell suppression following EtOH and burn injury (15–17).
Toll-like receptors (TLRs) are known to play a critical role in host immunity. Classically, TLRs were believed to be expressed only on cells of the innate immune system, including neutrophils, dendritic cells and macrophages, where they function as first-line sensors of invading pathogens (18). TLRs recognize highly conserved molecules derived from microbes. Upon activation, TLRs induce the release of inflammatory mediators and cytokines to initiate adaptive immune responses against the invading pathogens. To date, at least 13 TLRs (TLR1 to TLR13) have been identified in mice and humans (19,20), each recognizing a distinct conserved pathogen-associated molecular pattern (PAMP) (19). Pathogenic microorganisms contain multiple PAMPs that act as TLR agonists: peptidoglycan (TLR2), lipopolysaccharide (LPS) (TLR4), flagellin (TLR5) and single-stranded RNA (ssRNA) (TLR7). The TLR signaling pathway has been widely investigated in the innate immune system. TLR signal transduction associates with Toll/IL-1 receptor (TIR) domain–containing adaptor molecules, such as myeloid differentiation primary response protein 88 (MyD88), TIR domain-containing adaptor protein (TIRAP), Toll-receptor-associated activator of interferon (TRIF) and Toll-receptor-associated molecule (TRAM). Except for TLR3, all the TLR proteins use the MyD88 adaptor protein to activate the mitogen activated protein kinase (MAPK) pathway, and subsequently, the translocation of nuclear factor-κB (NF-κB) to nucleus (21,22).
Although most TLR studies have focused on cells of the innate immune system, recent studies have indicated that TLRs directly or indirectly activate the adaptive immune system (23–29). Some of these studies further demonstrated that T cells express certain TLRs and that activation of these proteins can directly promote T-cell survival and proliferation, as well as IL-2 and IFN-γ production, independently of antigen presenting cells (APCs) (23–25). Yet, the mechanism of TLR intracellular signaling within T cells remains unclear. Moreover, the majority of data describing TLR expression and function within T cells is limited to cell lines or T cells from healthy animals. In this study, we used a mouse model of acute EtOH intoxication and burn injury to determine the effect of TLR2, 4, 5 and 7 agonists on T-cell effector functions in healthy and injury conditions. Among these, only the TLR2 agonist was found to directly modulate T-cell cytokine production. Furthermore, activation of TLR2 on T cells prevents suppression of IFN-γ production following EtOH and burn injury. Lastly, our findings suggest a role for p38 and ERK in TLR2-mediated modulation of T-cell IFN-γ production.
MATERIALS AND METHODS
Animals and Reagents
Male C57BL/6 mice (22–25 g) were obtained from Harlan Laboratories (Indianapolis, IN, USA). IL-2 and IFN-γ enzyme-linked immunosorbent assay (ELISA) kits were obtained from BD Biosciences (San Diego, CA, USA). A mouse TLR 1–9 agonist kit was obtained from InvivoGen (San Diego, CA, USA), Alexa Fluor-647 conjugated anti-TLR2 and phycoerythrin (PE)-Cy5–conjugated anti-CD3e antibodies were obtained from eBioscience (San Diego, CA, USA). A MyD88 homodimerization inhibitory peptide set and TIRAP inhibitory peptide set were obtained from IMGENEX (San Diego, CA, USA). p38 inhibitor SB 203580 and ERK inhibitor PD 98059 were obtained from EMD Chemicals (Gibbstown, NJ, USA). Antibodies to p38, phospho-p38 (Thr180/ Tyr182), ERK 1/2 and phospho-ERK 1/2 (Thr202/Tyr204) were obtained from Cell Signaling Technology (Danvers, MA, USA). Rabbit polyclonal antibody to β-actin was obtained from Abcam (Cambridge, MA, USA).
Mouse Model of Acute EtOH Intoxication and Burn Injury
As described previously (14), mice were randomly divided into four groups: sham vehicle, sham EtOH, burn vehicle and burn EtOH. In the EtOH-treated groups, mice were gavaged with 0.4 mL of 25% EtOH in water (2.9 gm/kg). In the vehicle groups, mice were gavaged with 0.4 mL of water. At 4 h after gavage, mice were anesthetized with a mixture of ketamine and xylazine at a dose of 79.7 mg/kg and 1.18 mg/kg, respectively, by intraperitoneal injection. The dorsal surface was shaved and mice were transferred into a template fabricated to expose ~12.5% of the total body surface area (TBSA). TBSA was calculated by using Meeh’s formula as described by Walker and Mason (30). For burn injury, mice were immersed into hot water maintained at 85°C–90°C for 7 s. For sham injury, mice received identical anesthesia and were shaved, but were immersed in lukewarm water for 7 s. All animals were dried immediately and resuscitated with 1.0 mL physiological saline by intraperitoneal injection. All the animal procedures were carried out in adherence to the National Institutes of Health 2011 Guide for the Care and Use of Laboratory Animals, 8th edition (31) and were approved by the Loyola University Chicago Health Sciences Division Institutional Animal Care and Use Committee.
T-Cell Isolation
One day after injury, mice were anesthetized and the abdominal cavity exposed via midline incision. MLN and spleens were removed aseptically and crushed to prepare single-cell suspensions in Hanks balanced salt solution (HBSS, Fisher Scientific, Pittsburgh, PA, USA) supplemented with 10 mmol/L 4-(2-hydroxyethyl)-1-piperazineethane-sulfonic acid (HEPES), 50 μg/mL gentamicin and 100 U/mL penicillin with 100 μg/mL streptomycin (16,17). Cell suspensions were centrifuged at 290g for 15 min at 10°C. Supernatant was discarded and the cells were reconstituted in phosphate-buffered saline (PBS). For splenic T cells, red blood cells were lysed by adding sterile-distilled H2O followed by 10× PBS and centrifuged at 290g for 15 min at 10°C. Cells prepared from MLN and spleen (106–107 total cells) were resuspended in 90 μL of staining buffer (PBS containing 0.5% bovine serum albumin and 2 mmol/L EDTA) and incubated with 10 μL of CD90 (Thy1.2) MicroBeads (Miltenyi Biotec, Auburn, CA, USA) for 15 min at 4°C. The cells were washed with staining buffer and run through separation columns (Miltenyi Biotec) in a magnetic field. Purified T cells were obtained by flushing out magnetic labeled cells from the columns, and purity was confirmed by flow cytometry. Briefly, isolated cells from MLN and spleen were incubated with PE-Cy5 conjugated anti-CD3e on ice, in the dark for 30 min. After two washes with PBS containing 5% fetal calf serum (FCS), cells were analyzed by flow cytometry. CD3e-positive cells were considered as T cells. As shown in Figure 1, the MLN T-cell purity was over 99% and spleen T-cell purity was over 98%.
Figure 1.

MLN and spleen T-cell purity. MLN and spleen T cells were isolated with CD90 (Thy1.2) MicroBeads in a magnetic field. Isolated cells (106/100 μL) were stained with isotype control (solid black line) or PE-Cy5–conjugated anti-CD3e (shaded histogram). CD3e+ cells were detected by flow cytometry and considered as T cells.
Measurement of T-Cell IL-2 and IFN-γ Levels
To determine whether TLRs modulate T-cell responses following EtOH and burn injury, we designed the following series of experiments.
Experiment 1: To confirm whether EtOH intoxication combined with burn injury suppresses T-cell responses, MicroBeads-purified T cells were resuspended at a density of 5 × 106 cells/mL in RPMI 1640 supplemented with 2 mmol/L l-glutamine, 10 mmol/L HEPES, 50 μg/mL gentamicin, 100 U/mL penicillin with 100 μg/mL streptomycin and 10% FCS. As previously described (16,17), T cells (5 × 105 cells/well) were cultured in 96-well plates in the presence of plate-bound anti-CD3 (2 μg/mL) at 37°C and 5% CO2 for 48 h. Following culture, supernatants were harvested and tested for IL-2 and IFN-γ levels, by use of respective ELISA kits.
Experiment 2: To determine whether TLR agonists directly modulate T-cell responses following EtOH and burn injury, T cells were cultured with/without plate-bound anti-CD3 antibody in the presence or absence of TLR agonists. Specific agonists include: TLR2, heat-killed preparation of Listeria monocytogenes (HKLM; 108 cells/mL); TLR4, LPS (1 μg/mL); TLR5, flagellin from Salmonella typhimurium (ST-FLA; 1 μg/mL) and TLR7, ssRNA40 (1 μg/mL). After 48 h of culture, supernatants were tested for IL-2 and IFN-γ production by ELISA.
Experiment 3: To assess whether TLR2 agonist modulates T-cell responses after EtOH intoxication and burn injury through the MyD88 and TIRAP pathway, T cells were cultured with plate-bound anti-CD3 in the presence or absence of TLR2 agonist (HKLM, 108 cells/mL), MyD88 homodimerization inhibitory peptide (25, 50 and 100 μmol/L) and/or TIRAP inhibitory peptide (25, 50 and 100 μmol/L). After 48 h, cell supernatants were harvested and analyzed for IFN-γ by ELISA.
Experiment 4: To further determine whether p38 and ERK1/2 play any role in the TLR2-mediated T-cell responses following EtOH and burn injury, T cells were cultured with/without plate-bound anti-CD3 and TLR2 agonist (HKLM, 108 cells/mL) in the presence or absence of p38 inhibitor (SB 203580 10 μmol/L) and ERK inhibitor (PD 98059 50 μmol/L). p38 and ERK inhibitor doses were selected from our previous studies (17). After 48 h, IFN-γ was tested in cell supernatant.
T-Cell Staining and Flow Cytometry
Spleen T cells were cultured in the presence and absence of plate-bound anti-CD3 (2 μg/mL) in RPMI 1640 supplemented with 2 mmol/L l-glutamine, 10 mmol/L HEPES, 50 μg/mL gentamicin and 100 U/mL penicillin with 100 μg/mL streptomycin for 16 h. Fresh T cells or cultured T cells (106 cells/ 100 μL) were incubated with PE-Cy5–conjugated anti-mouse CD3e and Alexa Fluor-647 conjugated anti-mouse TLR2 in PBS containing 5% FCS on ice for 30 min in the dark. The cell suspensions were washed two times and resuspended in 0.5 mL PBS. Expression of CD3 and TLR2 were determined at the Loyola University Chicago Health Sciences Division FACS Core Facility using a six-color flow cytometer (BD FACSCanto); data were analyzed with the flow cytometry analysis software FlowJo 7.5 (Tree Star, Inc., Ashland, OR, USA).
Western Blot
For the analysis of p38 and ERK1/2 protein and phosphorylation, T cells were cultured in RPMI 1640 supplemented with 2 mmol/L l-glutamine, 10 mmol/L HEPES, 50 μg/mL gentamicin, 100 U/mL penicillin and 100 μg/mL streptomycin in the presence or absence of MyD88 inhibitor peptide (25 μmol/L) and TIRAP inhibitor peptide (25 μmol/L) for 3 h. T cells were subsequently stimulated with TLR2 agonist, HKLM (108 cells/mL), for 5 min followed by anti-CD3 (1 μg/mL) for 10 min. Following stimulation, T cells were lysed in lysis buffer containing 50 mmol/L HEPES, 150 mmol/L NaCl, 1 mmol/L EDTA, 100 mmol/L NaF, 1 mmol/L MgCl2, 10 mmol/L Na4P2O7, 200 μmol/L Na3VO4, 0.5% Triton X-100 and 10% glycerol on ice for 45 min to 1 h, as previously described (16,17). Lysates were centrifuged and supernatants were harvested and stored at −70°C until analysis. For the analysis of p38 and ERK protein and phosphorylation, an equal amount of protein from each T-cell lysate preparation was separated on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to Immobilon polyvinylidine fluoride membranes by using a semidry Trans-Blot system (Bio-Rad, Hercules, CA, USA), as previously described (16,17). The membranes were saturated with blocking buffer (10 mmol/L Tris and 150 mmol/L NaCl [TBS], 0.05% Tween 20 supplemented with 5% dry milk) for 2 h at room temperature and incubated with the desired primary antibody (1/1000 dilution) at 4°C overnight. The membranes were washed five times with TBS supplemented with 0.05% Tween 20 (TBST) and then incubated with a secondary antibody conjugated with horseradish peroxidase (1/2000 dilution) for 1 h at room temperature. The membranes were washed five times with TBST, probed using Western Lightning Plus-ECL (PerkinElmer Inc., Waltham, MA, USA), and autoradiographed (16,17). Membranes were stripped with Western blot Stripping Buffer (Fisher Scientific, Waltham, MA, USA) for 30 min at room temperature. After five washes with TBST, the membranes were reprobed with desired antibodies or β-actin antibody for loading control. Representative blots shown in the Results section come from the same membranes, which may have multiple samples from the various experimental groups.
Statistical Analysis
The data, wherever applicable, are presented as means ± standard error of the mean (SEM) and were analyzed with analysis of variance (ANOVA) with Tukey post hoc test or Student t test (In-Stat; GraphPad Software Inc., La Jolla, CA, USA). P < 0.05 was considered statistically significant.
RESULTS
TLR Agonists Modulate T-Cell IFN-γ Production, but Not IL-2, following EtOH Intoxication Combined with Burn Injury
In previous studies from our laboratory, we have shown that acute EtOH intoxication prior to burn injury suppresses MLN T-cell IL-2 and IFN-γ production in a rat model (15–17). In this study, we used a recently developed mouse model of acute EtOH intoxication and burn injury (14) to determine whether the same response is present in MLN T cells from mice. As shown in Figure 2A, C, we found that there was no significant difference in MLN T-cell IL-2 and IFN-γ production in mice receiving sham injury regardless of EtOH intoxication or burn injury alone. However, a significant decrease in MLN T-cell IL-2 and IFN-γ production was observed in mice receiving EtOH intoxication combined with burn injury compared with sham-injured and burn-injured mice. These results confirm our previous data obtained from a rat model. Because there were no differences in T-cell IL-2 and IFN-γ production in mice receiving EtOH intoxication or burn injury alone, in subsequent experiments we used only T cells from sham vehicle and burn EtOH animals.
Figure 2.

MLN T-cell IL-2 and IFN-γ production. MLN T cells (5 × 106/mL) collected from various experimental groups were cultured with plate-bound anti-CD3 (2 μg/mL) for 48 h, and supernatants were collected to determine IL-2 (A) and IFN-γ (C) production. White bars indicate vehicle treatment, black bars indicate ethanol treatment. To determine the effects of TLR agonists, MLN T cells (5 × 106/mL) were cultured with plate-bound anti-CD3 antibody (2 μg/mL) in the presence or absence of TLR agonists: TLR2, HKLM (108 cells/mL); TLR4, LPS (1 μg/mL); TLR5, ST-FLA (1 μg/mL) and TLR7, ssRNA40 (1 μg/mL) for 48 h. Supernatants were collected to determine IL-2 (B) and IFN-γ (D) production. White bars indicate sham vehicle, black bars indicate burn ethanol. Values are means ± SEM from five to six animals/group, except the CD3+ TLR4 group, which had three animals. *P < 0.05 compared with other groups; †P < 0.05 compared with sham vehicle without TLR agonists; ‡P < 0.05 compared with burn EtOH without TLR agonists.
We determined whether TLR agonists modulate T-cell responses following EtOH and burn injury. To make this determination, we cultured MLN T cells with plate-bound anti-CD3 in the presence or absence of TLR2 (HKLM 108 cells/mL), TLR4 (LPS 1 μg/mL), TLR5 (ST-FLA 1 μg/mL) and TLR7 (ssRNA40 1 μg/mL) agonists. Following 48 h of culture, cell supernatants were harvested, and IL-2 (Figure 2B) and IFN-γ (Figure 2D) were measured. T cells cultured with anti-CD3 in the presence of TLR2, 4, 5 and 7 agonists prevented the decrease in IL-2 and IFN-γ levels following EtOH intoxication and burn injury. Moreover, we isolated T cells from spleen to detect the effect of TLR agonists on splenic T-cell responses after EtOH and burn injury. As with MLN, T cells isolated from spleen demonstrated a decrease in IL-2 (Figure 3A) and IFN-γ (Figure 3C) following combined insult. Similarly, activation of splenic T cells with anti-CD3 and TLR2, 4, 5 and 7 agonists significantly increased IFN-γ production (Figure 3D). In contrast, TLR agonists except the agonist for TLR7 did not significantly affect the release of IL-2 following combined insult of EtOH and burn injury (Figure 3B). Moreover, TLR2, 4, 5 and 7 agonists significantly decreased IL-2 production in sham vehicle.
Figure 3.

Spleen T-cell IL-2 and IFN-γ production. Spleen T cells (5 × 106 cells/mL), collected from various experimental groups, were cultured with plate-bound anti-CD3 (2 μg/mL) for 48 h, and supernatants were collected to determine IL-2 (A) and IFN-γ (C) production. White bars indicate vehicle treatment, black bars indicate ethanol treatment. To determine the effects of TLR agonists, spleen T cells (5 × 106 cells/mL) were cultured with plate-bound anti-CD3 (2 μg/mL) in the presence or absence of TLR agonists: TLR2, HKLM (108 cells/mL); TLR4, LPS (1 μg/mL); TLR5, ST-FLA (1 μg/mL) and TLR7, ssRNA40 (1 μg/mL) for 48 h. Supernatants were collected to determine IL-2 (B) and IFN-γ (D) production. White bars indicate sham vehicle, black bars indicate burn ethanol. Values are means ± SEM from six to seven animals in each group. *P < 0.05 compared with other groups; †P < 0.05 compared with sham vehicle without TLR agonists; ‡P < 0.05 compared with respective group without TLR agonists.
Activation of TLR2 Directly Prevents Suppression of T-Cell IFN-γ Production following EtOH Intoxication and Burn Injury
T-cell activation and proliferation require dimerization of the CD3 molecule with the T-cell receptor (TCR) (32). It is widely believed that the primary T-cell activation signal is generated by this TCR–CD3 complex formation. In the above experiment, we observed that T cells cultured with plate-bound anti-CD3 and TLR agonists modulated T-cell IFN-γ production after EtOH intoxication combined with burn injury. In this experiment, we determined whether TLR- modulated T-cell IFN-γ production is dependent on TCR/CD3-mediated T-cell activation following EtOH and burn injury. Given the limited number of T cells obtainable from MLN and the similar responses noted in MLN and splenic T cells following EtOH intoxication and burn injury, in subsequent experiments we used splenic T cells. T cells were cultured without anti-CD3 in presence or absence of TLR2, 4, 5 and 7 agonists for 48 h, and IFN-γ was determined. As shown in Figure 4, we observed that T cells cultured with TLR2 agonist alone significantly increased IFN-γ production (3.584 ± 0.170 and 3.762 ± 0.065 ng/mL in sham-injured and EtOH plus burn- injured animals, respectively). It should be noted that this increase in IFN-γ was about 13 times lower than in T cells cultured with plate-bound anti-CD3 and TLR2 agonist (50.694 ± 3.915 and 54.62 ± 2.673 ng/mL, respectively). There was a very low or an undetectable level of IFN-γ in supernatants from T cells cultured with media alone or TLR4, 5 and 7 agonists alone.
Figure 4.
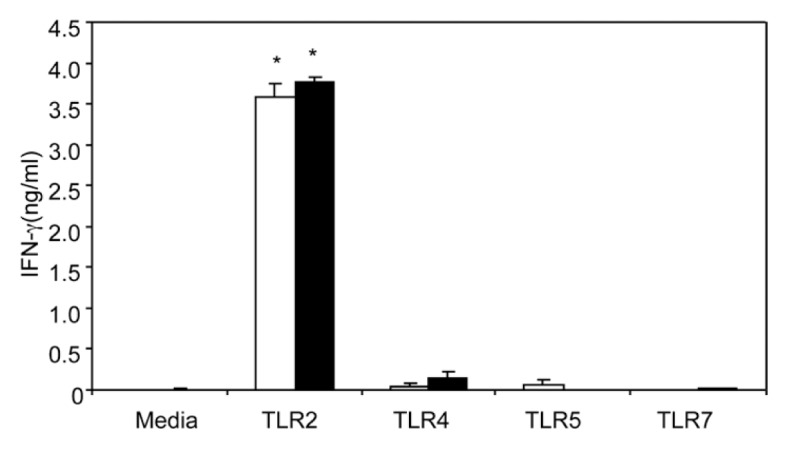
Activation of TLR2 directly prevents suppression of T-cell IFN-γ production following EtOH and burn injury. Spleen T cells (5 × 106/mL) collected from sham vehicle (white bars) and burn ethanol (black bars) groups were cultured without anti-CD3, in the presence or absence of TLR agonists: TLR2, HKLM (108 cells/mL); TLR4, LPS (1 μg/mL); TLR5, ST-FLA (1 μg/mL) and TLR7, ssRNA40 (1 μg/mL) for 48 h. Supernatants were collected to determine IFN-γ production. Values are means ± SEM from six animals/group. *P < 0.05 compared with other groups.
Expression of TLR2 on T Cells following EtOH Intoxication and Burn Injury
Because only TLR2 agonist directly modulated IFN-γ production, we determined whether T cells express the TLR2 receptor. Freshly isolated T cells and T cells cultured in the presence or absence of plate bound anti-CD3 for 16 h were stained with PE-Cy5-conjugated anti-mouse CD3e and Alexa Fluor 647-conjugated anti-mouse TLR2 antibodies, TLR2 expression was determined by flow cytometry (Figure 5). We found that only 1%–2% of freshly isolated T cells express TLR2. Following 16 h of culture conditions, ~30% of T cells were TLR2 positive, regardless of CD3 stimulation. However, there was no significant difference in TLR2 expression following EtOH intoxication and burn injury in freshly isolated or cultured T cells.
Figure 5.
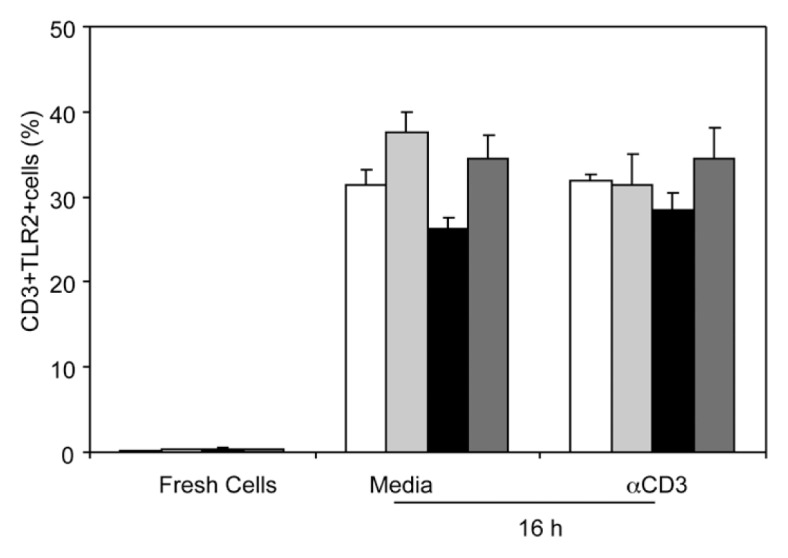
T-cell TLR2 expression. Fresh spleen T cells (106 cells/100 μL) or T cells cultured in the presence or absence of plate-bound anti-CD3 for 16 h were collected and stained with RPE-Cy5–conjugated anti-CD3e and Alexa Fluor-647–conjugated anti-TLR2. Expression of CD3 and TLR2 were determined by flow cytometery. Values are means ± SEM from three to five animals/group. White bars indicate sham vehicle, light gray bars indicate sham ethanol, black bars indicate burn vehicle, dark gray bars indicate burn ethanol.
MyD88 and TIRAP Are Required for TLR2-Mediated T-Cell IFN-γ Production following EtOH Intoxication and Burn Injury
MyD88 and TIRAP are important adaptor molecules for TLR signaling. In this experiment, T cells were cultured with anti-CD3 in the presence of TLR2 agonist with different doses of MyD88 and TIRAP inhibitory peptides (25, 50 and 100 μmol/L) for 48 h, and IFN-γ was measured. When treated with 25 μmol/L MyD88 inhibitor or 25 μmol/L TIRAP inhibitor, the effect of TLR2 on T-cell IFN-γ production was abolished in cells from EtOH and burn-injured mice, but not in cells from sham-injured mice (Figure 6A). In the presence of 50 or 100 μmol/L of MyD88 or TIRAP inhibitor, TLR2-induced T-cell IFN-γ production was completely abolished, particularly at the highest dose of inhibitors (100 μmol/L), in both sham-injured and EtOH plus burn-injured mice (Figures 6B, C).
Figure 6.
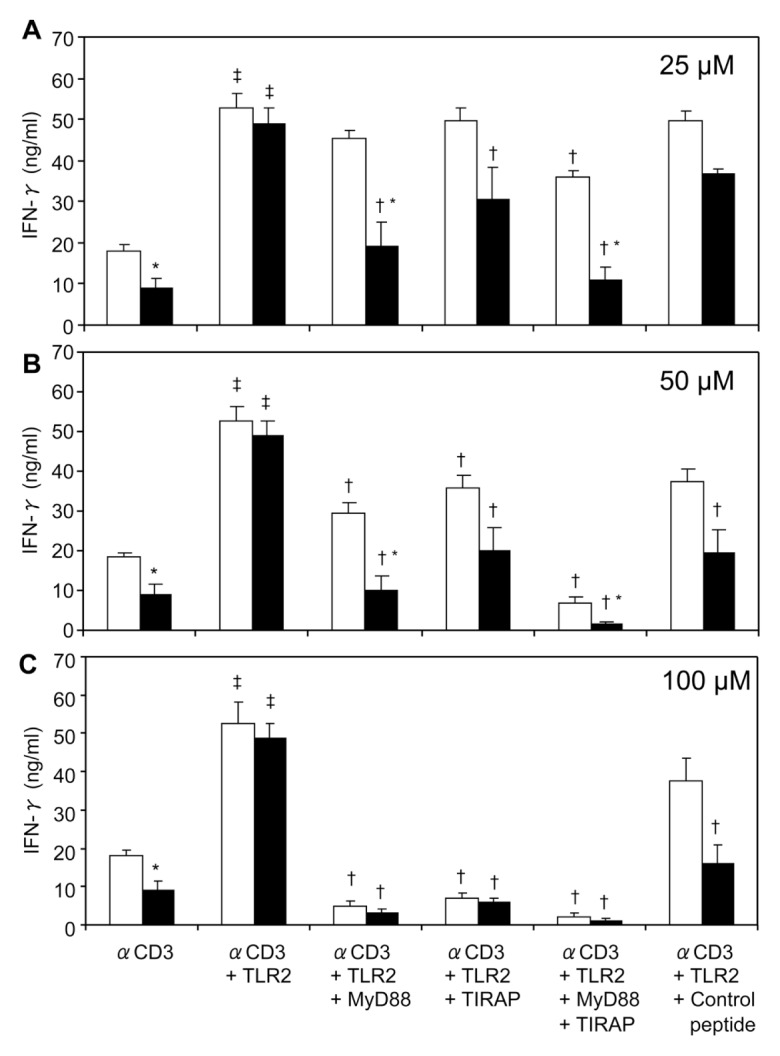
Effect of MyD88 and TIRAP inhibitors on TLR2 modulation of IFN-γ production. Spleen T cells (5 × 106 cells/mL), collected from sham vehicle (white bars) and burn ethanol (black bars) groups, were cultured with plate-bound anti-CD3 (2 μg/mL) in the presence or absence of TLR2 agonist (HKLM 108 cells/mL), and MyD88 inhibitory peptide, TIRAP inhibitory peptide or control peptides, using doses of 25 μmol/L (A), 50 μmol/L (B) and 100 μmol/L (C) for 48 h. Supernatants were collected to determine IFN-γ production. Values are means ± SEM from four to eight animals/group. *P < 0.05 compared with corresponding sham vehicle group; †P < 0.05 compared with respective αCD3 + TLR2 group; ‡P < 0.05 compared with respective αCD3 alone.
TLR2 Modulates T-Cell p38/ERK Signaling following EtOH Intoxication and Burn Injury
Our previous studies have shown that acute EtOH intoxication combined with burn injury suppresses phosphorylation of p38 and ERK1/2 in MLN T cells (15–17). In this study, we determined whether stimulation with TLR2 agonist influences the activation of p38/ERK in T cells following EtOH and burn injury. To make this determination, we first confirmed whether EtOH and burn injury suppresses phosphorylation of p38 and ERK in splenic T cells following EtOH and burn injury. Results summarized in Figure 7 clearly demonstrate that, similarly to MLN T cells (15–17), splenic T cells exhibit a decrease in the phosphorylation of p38 (Figure 7A). Moreover, ERK phosphorylation is also decreased following EtOH and burn injury (Figure 8A). To examine whether TLR2 influences CD3-mediated phosphorylation of p38 and ERK in T cells following EtOH and burn injury, T cells were stimulated with anti-CD3 and TLR2 agonist, and lysed. Because TLR2 utilizes the MyD88/TIRAP pathway to induce T-cell IFN-γ release, we also examined whether these adaptor molecules are critical to p38/ERK phosphorylation. Briefly, T cells were treated with 25 μmol/L MyD88 or TIRAP inhibitory peptide for 3 h and subsequently stimulated with TLR2 agonist for 5 min and anti-CD3 for 10 min. T cells were lysed and phosphorylation of p38 and ERK1/2 was determined by Western blot. As shown in Figure 7B, there was an increase in p38 phosphorylation in T cells stimulated with TLR2 agonist in the presence of anti-CD3, in both sham-injured and EtOH plus burn-injured mice. However, this increase was found to be significant only in T cells isolated from EtOH plus burn-injured mice. Furthermore, this increase in p38 phosphorylation following EtOH and burn injury was abolished in T cells treated with MyD88 or TIRAP inhibitor. Interestingly, this reduction in p38 phosphorylation did not occur in T cells isolated from sham mice. In this setting, T cells stimulated with anti-CD3 alone did not exhibit a decrease in p38/ERK phosphorylation, as we had observed in T cells immediately stimulated for 10 min with anti-CD3 (Figures 7A, 8A). Although the exact reason for such disparities remains to be established, it is likely due to the 3-h delay in stimulation of T cells necessary for treatment of cells with MyD88 or TIRAP inhibitors. Similarly to p38, there was a significant decrease in ERK1/2 phosphorylation, following EtOH plus burn, in splenic T cells stimulated with anti-CD3 alone (Figure 8A). However, when T cells were stimulated with anti-CD3 along with TLR2 agonist, there was a significant increase in ERK phosphorylation, which was abolished in cells treated with MyD88 or TIRAP inhibitors (Figure 8B). Together, these findings suggest that TLR2 agonist indeed modulates CD3-mediated p38 and ERK phosphorylation in T cells following EtOH and burn injury.
Figure 7.
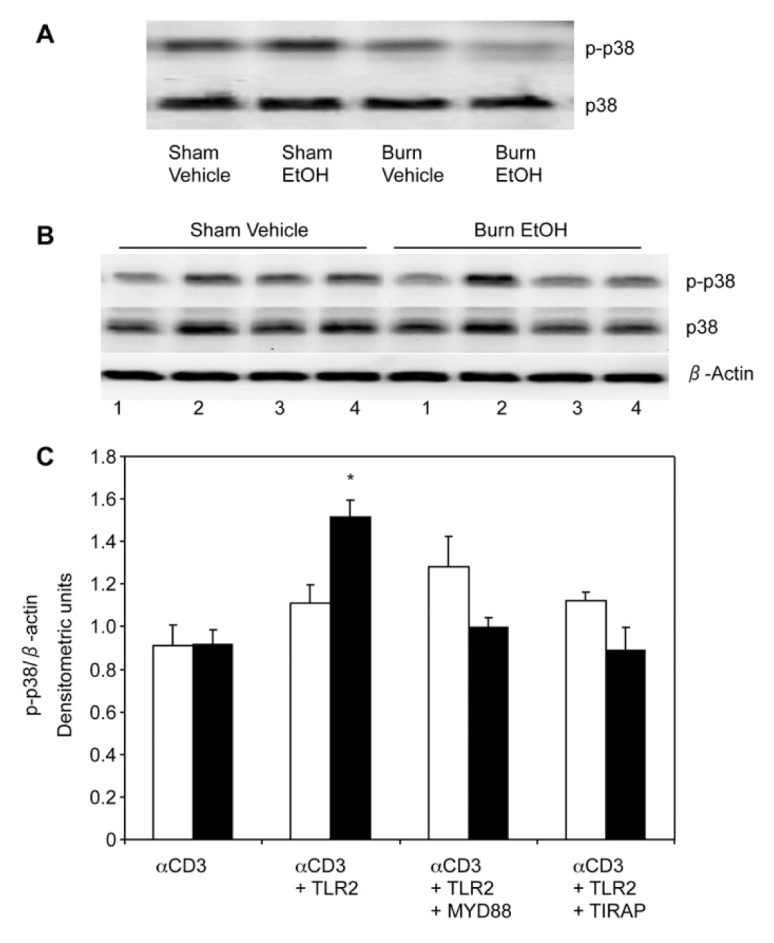
Effect of MyD88 and TIRAP inhibitors on TLR2 modulation of p38 phosphorylation. Spleen T cells (107 cells/mL), collected from various experimental groups, were stimulated with anti-CD3 for 10 min and lysed. Lysates were analyzed for phoso-p38 by Western blot. Blots were stripped and reprobed for p38 (A). T cells (107 cells/mL) were pretreated with 25 μmol/L MyD88 inhibitory peptide or TIRAP inhibitory peptide for 3 h, and treated with TLR2 agonist (HKLM 108 cells/mL) for 5 min following CD3 stimulation for 10 min (1, αCD3; 2, αCD3 + TLR2 agonist; 3, αCD3 + TLR2 agonist + MyD88 inhibitor; 4, αCD3 + TLR2 agonist + TIRAP inhibitor). T cells were lysed and phosphorylation of p38 was determined. Blots were stripped and reprobed for p38 protein levels and β-actin (B). Densitometric values for phosphorylation were normalized to β-actin and are shown in (C) as means ± SEM from three independent animal pools, each pool contained three to four animals; white bars indicate sham vehicle, black bars indicate burn ethanol. *P < 0.05 compared with αCD3 alone and other respective groups.
Figure 8.
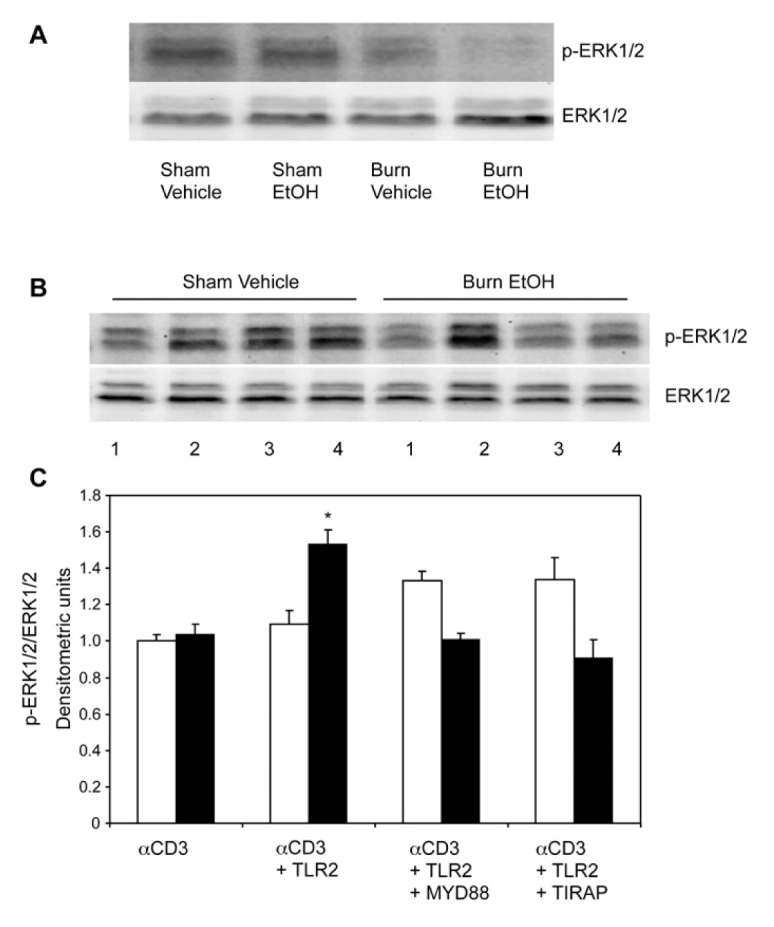
Effect of MyD88 and TIRAP inhibitors on TLR2 modulation of ERK phosphorylation following EtOH and burn injury. Spleen T cells (107 cells/mL), collected from various experimental groups, were stimulated with anti-CD3 antibody for 10 min and lysed. Lysates were analyzed for ERK phosphorylation by Western blot. Blots were stripped and reprobed for ERK (A). T cells (107 cells/mL) were pretreated with 25 μmol/L MyD88 inhibitory peptide or TIRAP inhibitory peptide for 3 h, and treated with TLR2 agonist (HKLM 108 cells/mL) for 5 min following CD3 stimulated for 10 min (1, αCD3; 2, αCD3 + TLR2 agonist; 3, αCD3 + TLR2 agonist + MyD88 inhibitor; 4, αCD3 + TLR2 agonist + TIRAP inhibitor). T cells were lysed and phosphorylation of ERK was determined. Blots were stripped and reprobed for ERK protein levels (B). Densitometric values for phosphorylation were normalized to total ERK protein and are shown in (C) as means ± SEM from three independent animal pools. Each pool contained three to four animals; white bars indicate sham vehicle, black bars indicate burn ethanol. *P < 0.05 compared with αCD3 alone and other respective groups.
TLR2-Mediated T-Cell IFN-γ Production Utilizes p38/ERK Signaling following EtOH Intoxication and Burn Injury
We determined whether the p38/ERK pathway is involved in TLR2-mediated IFN-γ production following EtOH and burn injury. To establish this, we cultured T cells with/without CD3 in the presence or absence of TLR2 agonist, p38 inhibitor (SB 203580 10 μmol/L) and ERK inhibitor (PD 98059 50 μmol/L) for 48 h. We observed that the TLR2-mediated increase in T-cell IFN-γ release was abolished in cells cultured in the presence of anti-CD3 and p38 or ERK inhibitor, both in sham and burn EtOH mice (Figure 9A). Similar results were noted in T cells cultured in the presence of TLR2 agonist, and p38 or ERK inhibitor without CD3 stimulation (Figure 9B).
Figure 9.

Effect of p38 and ERK inhibitors on TLR2 modulation of IFN-γ production. Spleen T cells (5 × 106 cells/mL) collected from sham vehicle (white bars) and burn ethanol (black bars), were cultured with (A) or without (B) plate-bound anti-CD3 (2 μg/mL) in the presence or absence of TLR2 agonist (HKLM 108 cells/mL), p38 inhibitor (SB 203580 10 μmol/L) and/or ERK inhibitor (PD 98059 50 μmol/L) for 48 h. Supernatants were collected and analyzed for IFN-γ production. Values are means ± SEM from four to six animals/group. *P < 0.05 compared with αCD3 sham vehicle. †P < 0.05 compared with other groups.
DISCUSSION
Recent studies demonstrate that certain TLRs are expressed on various T-cell subsets, including memory T cells, natural killer T cells (NKTs) and regulatory T cells (Treg) (33–35). In addition, TLR ligands function as costimulatory factors to induce T-cell activation (36,37). In this study, we determined whether stimulation of TLR2, 4, 5 and 7, with specific TLR agonists, can modulate the suppression of T-cell responses after EtOH intoxication and burn injury. To clearly delineate the role of TLR expression and function on T cells, we isolated T cells from MLN and spleen by using immunomagnetic beads and confirmed T-cell purity by flow cytometry. We observed that T cells cultured in the presence of plate-bound anti-CD3 and TLR2, 4 or 5–specific agonists prevented the decrease in T-cell IFN-γ production in MLN, as well as in splenic T cells obtained from mice receiving EtOH intoxication and burn injury. TLR2 agonist was also found to significantly increase IFN-γ production in the sham vehicle group. Moreover, in the absence of CD3 stimulation, only TLR2 agonist prevented the suppression of IFN-γ production in splenic T cells following EtOH intoxication and burn injury. In contrast to IFN-γ, the effect of TLR ligands on T-cell IL-2 release was found to be organ specific. In MLN T cells, agonists for TLR2, 4, 5 and 7 prevented EtOH-plus-burn–induced suppression of IL-2 secretion in MLN. Similar treatment of splenic T cells with TLR agonists (except TLR7 agonist) did not significantly influence the IL-2 secretion following EtOH and burn injury. Moreover, these agonists caused a significant decrease in IL-2 release in the sham vehicle group. TLR7, on the other hand, increased IL-2 secretion by splenic T cells following EtOH and burn injury. The exact mechanism of such dichotomy in IL-2 release by two different lymphoid organs (MLN versus spleen) in healthy and injured conditions remains unknown at this time and is a limitation of this study.
We further explored the relationship between CD3 stimulation and TLR2 activation in the modulation of IFN-γ production. We observed that in the absence of CD3 stimulation, T cells cultured with TLR2 ligand demonstrated increased IFN-γ production compared with T cells cultured with media alone, in both sham and injured mice. Moreover, CD3 stimulation greatly augmented this response, increasing IFN-γ levels more than 13-fold compared with T cells cultured with TLR2 ligand alone, suggesting that TLR2 activation directly modulates T-cell IFN-γ production and that activation of CD3 has a synergic effect on this response. Furthermore, in the absence of CD3 stimulation, TLR 4, 5 and 7 agonists fail to induce IFN-γ production. Consistent with our findings, Xu et al. observed that activated CD4+ T cells express similar levels of TLR2 and TLR4. Yet, when cultured with anti-CD3 with/without Pam3Cys-SK (TLR2 ligand) or LPS (TLR4 ligand), only Pam3Cys-SK increased T-cell proliferation and IFN-γ production (20). Similarly, recent studies have indicated that CD3/CD28-activated CD8+ T cells are functionally responsive to direct stimulation by TLR2 ligand Pam3CYs (38). In the absence of specific antigen, TLR2 acts as a costimulatory receptor on CD8+ T cells to directly control memory CD8+ T-cell proliferation and IFN-γ production (39). Moreover, T cells isolated from TLR2-deficient mice failed to respond to Lip-Ospa, a prototypic lipoprotein known to induce T-cell proliferation and IFN-γ production (40). Anti-CD3–activated T cells from TLR2−/− mice also did not respond to Pam3CYs, even in the presence of 10% of wild-type APCs, emphasizing the role of TLR2 signaling on T-cell immunity (20). TLR2 activation on T cells was further validated when Imanishi et al. indicated that TLR2 directly induces T-helper 1 (Th1) IFN-γ production in the absence of TCR stimulation. Treatment of Th1 cells with cyclosporine A (CsA), an inhibitor of TCR-mediated calcineurin activation, did not affect the TLR2-induced IFN-γ production (41). Together, these findings suggest that TLR2 modulates T-cell IFN-γ production independent of TCR activation. Our data fall in line with these findings, as we demonstrate that TLR2 activation not only modulates, but also prevents suppression of T-cell IFN-γ production following EtOH and burn injury. Our findings further demonstrate a role for TCR activation, because CD3 stimulation acted in synergy with TLR2 to modulate T-cell IFN-γ release.
In determining the mechanism by which TLR2 activation modulates IFN-γ production, we determined the levels of TLR2 expression on isolated T cells, and analyzed whether this expression was altered following EtOH and burn injury. We found that only ~1% of freshly isolated CD3+ T cells express TLR2. Interestingly, TLR2 expression increases to ~30% of CD3+ T cells following 16 h in culture conditions, irrespective of CD3 stimulation or injury. Lee et al. previously reported that TLR2 expression on Listeria-specific CD8+ memory T cells did not require CD3 prestimulation (42). Because EtOH and burn injury did not influence TLR2 expression on T cells, we analyzed the effect of combined insult on the TLR2 signaling pathway.
Previous studies have demonstrated that T cells can be activated by PAMPs, and that TLR2 recognizes the largest spectrum of PAMPs among the TLRs (18,43,44). TLR-mediated responses are controlled mainly by the MyD88-dependent signaling pathway, which has been conserved by all described TLR members, except TLR3. The stimulation of cells with TLR ligands recruits adaptor proteins containing a TIR domain, such as MyD88 and TIRAP, to cell surface receptors. MyD88 contains a death domain, which allows it to recruit IL-1R– associated kinase 4 (IRAK4) and IRAK1 through a homophilic interaction of the death domains. The interaction between the two death domains results in phosphorylation of IRAKs, and formation of a complex with TNFR-associated factor 6 (TRAF 6). TRAF 6 then recruits TGF-β–activated kinase 1 (TAK1) and TAK1 binding proteins (TAB1, TAB2 and TAB3 complex). Activated TAK1 induces the activation of Iκ kinase α (IKK-α), IKK-β, IKK-γ and NF-κB, which subsequently results in the translocation of NF-κB into nucleus to induce target gene expression. In addition, activated TAK1 also induces activation of MAPK kinase 6 (MKK6), which modulates activation of the MAPK pathway (18,44). Thus, MyD88 and TIRAP are key adaptor molecules in the classically described TLR pathway. However, the majority of TLR signaling studies have focused on the role of this pathway in innate immune cell responses. In this study, we determined whether similar pathways are involved in adaptive T-cell responses following EtOH intoxication and burn injury. We observed that purified T cells cultured with anti-CD3 and TLR2 ligand in the presence of high doses (100 μmol/L) of MyD88 or TIRAP inhibitory peptides completely diminished TLR2-mediated IFN-γ production both in sham and burn-injured mice. Culturing T cells in the presence of anti-CD3, TLR2 ligand and a low dose of MyD88 (25 μmol/L) or TIRAP (25 μmol/L) inhibitory peptides was not sufficient to diminish TLR2-mediated IFN-γ production in sham mice, but did diminish TLR2-mediated IFN-γ production in EtOH and burn-injured mice. This finding suggests that T cells from the EtOH burn group are more sensitive to inhibition of MyD88 and TIRAP than cells from the sham vehicle group because the effect of MyD88/TIRAP inhibitors at 25 μmol/L is significant in the EtOH burn group compared with the sham vehicle group. Moreover, higher concentrations of MyD88/TIRAP inhibitors are required to inhibit cells from the sham vehicle group. Although the exact cause for increased sensitivity of T cells from the EtOH burn group to MyD88/TIRAP inhibitors remains to be established, it is likely that the combined insult perturbs TLR2 signaling. Consistent with our finding, Tomita et al. reported that CD4+ T cells from MyD88−/− animals showed lower proliferation and less IFN-γ production compared with wild-type CD4+ T cells in a murine model of chronic colitis (45).
Several lines of evidence indicate that MAPKs (for example, p38 and ERK) play a central role in T-cell activation, proliferation and subsequent differentiation into Th cells. A recent study from our laboratory indicated that acute EtOH intoxication combined with burn injury suppresses T-cell IL-2 and IFN-γ production by inhibiting p38 and ERK phosphorylation. Moreover, our study demonstrates that IL-12 modulates T-cell IL-2 and IFN-γ production by utilizing the p38 and ERK pathways, following EtOH and burn injury. In this current study, we observed that treatment of T cells with MyD88 or TIRAP inhibitor, in the presence of TLR2 agonist and anti-CD3, significantly decreased p38 and ERK activation, compared with T cells stimulated with TLR2 agonist and anti-CD3, following EtOH and burn injury. Moreover, treatment of T cells with p38 or ERK inhibitor, and subsequent stimulation with TLR2 agonist, abolished TLR2-mediated IFN-γ production in both sham and burn-injured mice, irrespective of CD3 activation. Consistent with our findings, Imanishi et al. demonstrated that IFN-γ production was severely impaired in MyD88−/− and IRAK4−/− Th1 cells stimulated with TLR2 agonist Pam3, even in the presence of IL-2. IL-2 and IL-12 enhance TLR2-mediated IFN-γ production in Th1 cells by activation of MAPKs. Yet, Pam3-induced activation of p38 and ERK was also impaired in MyD88−/− and IRAK4−/− Th1 cells (41). Because TLR2 and CD3-induced TCR activation in T cells are p38/ERK-dependent, there may be crosstalk between the TLR2 and TCR signaling pathways. TLR2 has a synergistic effect with CD3 activation and may play a critical role in T-cell IFN-γ production following EtOH and burn injury.
CONCLUSION
In this study, our results indicate that in conjunction with CD3 stimulation, several TLR ligands enhance T-cell IFN-γ production following EtOH intoxication and burn injury, a response that does not require the presence of APCs. Our results also show that the TLR2 agonist directly prevents the suppression of T-cell IFN-γ production following injury, irrespective of CD3 stimulation. However, the TLR2 agonist has a synergistic effect with CD3 activation on T-cell IFN-γ release. It might be that the activated T cells, such as those from EtOH and burn-injured mice, are more sensitive to PAMPs. Furthermore, we demonstrate that MyD88/TIRAP-dependent pathways play a critical role in TLR2-mediated T-cell activation, through their modulation of p38 and ERK, following EtOH and burn injury. Our findings provide a novel role for TLR2 in T-cell activation and function, and provide an understanding of TLR2-mediated T-cell responses following EtOH intoxication combined with burn injury.
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health (NIH) grants R01AA015731, R01AA015731-04S1 and in part by the Dr. Ralph and Marian C. Falk Medical Research Trust. JL Rendon was supported by NIH grants F30AA020167, T32AA013527 and the Loyola University Chicago Stritch School of Medicine Combined MD/PhD Program.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Germann G, Barthold U, Lefering R, Raff T, Hartmann B. The impact of risk factors and pre-existing conditions on the mortality of burn patients and the precision of predictive admission-scoring systems. Burns. 1997;23:195–203. doi: 10.1016/s0305-4179(96)00112-x. [DOI] [PubMed] [Google Scholar]
- 2.McGill V, Kowal-Vern A, Fisher SG, Kahn S, Gamelli RL. The impact of substance use on mortality and morbidity from thermal injury. J Trauma. 1995;38:931–34. doi: 10.1097/00005373-199506000-00019. [DOI] [PubMed] [Google Scholar]
- 3.Messingham KA, Faunce DE, Kovacs EJ. Alcohol, injury, and cellular immunity. Alcohol. 2002;28:137–49. doi: 10.1016/s0741-8329(02)00278-1. [DOI] [PubMed] [Google Scholar]
- 4.American Burn Association. Burn incidence and treatment in the US: 2000 fact sheet. 2000. [Google Scholar]
- 5.Choudhry MA, Gamelli RL, Chaudry IH. Alcohol Abuse: a Major Contributing Factor to Post-Burn/Trauma Immune Complications. In: Vincent JL, editor. 2004 Yearbook of Intensive Care and Emergency Medicine. Springer-Verlag; Berlin, Heidelberg: 2004. pp. 15–26. [Google Scholar]
- 6.Grobmyer SR, Maniscalco SP, Purdue GF, Hunt JL. Alcohol, drug intoxication, or both at the time of burn injury as a predictor of complications and mortality in hospitalized patients with burns. J Burn Care Rehabil. 1996;17:532–9. doi: 10.1097/00004630-199611000-00010. [DOI] [PubMed] [Google Scholar]
- 7.Haum A, et al. Alcohol and drug abuse in burn injuries. Burns. 1995;21:194–9. doi: 10.1016/0305-4179(95)80008-c. [DOI] [PubMed] [Google Scholar]
- 8.Jones JD, Barber B, Engrav L, Heimbach D. Alcohol use and burn injury. J Burn Care Rehabil. 1991;12:148–52. doi: 10.1097/00004630-199103000-00012. [DOI] [PubMed] [Google Scholar]
- 9.Choudhry MA, et al. Impaired intestinal immunity and barrier function: a cause for enhanced bacterial translocation in alcohol intoxication and burn injury. Alcohol. 2004;33:199–208. doi: 10.1016/j.alcohol.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Marshall SW, et al. Fatal residential fires: who dies and who survives? JAMA. 1998;279:1633–7. doi: 10.1001/jama.279.20.1633. [DOI] [PubMed] [Google Scholar]
- 11.Silver GM, et al. Adverse clinical outcomes associated with elevated blood alcohol levels at the time of burn injury. J Burn Care Res. 2008;29:784–9. doi: 10.1097/BCR.0b013e31818481bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faunce DE, Gregory MS, Kovacs EJ. Acute ethanol exposure prior to thermal injury results in decreased T-cell responses mediated in part by increased production of IL-6. Shock. 1998;10:135–40. doi: 10.1097/00024382-199808000-00009. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Rana SN, Schwacha MG, Chaudry IH, Choudhry MA. A novel role for IL-18 in corticosterone-mediated intestinal damage in a two-hit rodent model of alcohol intoxication and injury. J Leukoc Biol. 2006;80:367–5. doi: 10.1189/jlb.1205745. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Akhtar S, Kovacs EJ, Gamelli RL, Choudhry MA. Inflammatory response in multiple organs in a mouse model of acute alcohol intoxication and burn injury. J Burn Care Res. 2011;32:489–97. doi: 10.1097/BCR.0b013e3182223c9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, et al. Corticosterone suppresses mesenteric lymph node T cells by inhibiting p38/ERK pathway and promotes bacterial translocation after alcohol and burn injury. Am J Physiol Regul Integr Comp Physiol. 2005;289:R37–44. doi: 10.1152/ajpregu.00782.2004. [DOI] [PubMed] [Google Scholar]
- 16.Li X, Schwacha MG, Chaudry IH, Choudhry MA. A role of PP1/PP2A in mesenteric lymph node T cell suppression in a two-hit rodent model of alcohol intoxication and injury. J Leukoc Biol. 2006;79:453–62. doi: 10.1189/jlb.0705369. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Chaudry IH, Choudhry MA. ERK and not p38 pathway is required for IL-12 restoration of T cell IL-2 and IFN-gamma in a rodent model of alcohol intoxication and burn injury. J Immunol. 2009;183:3955–62. doi: 10.4049/jimmunol.0804103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–7. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 19.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 20.Xu D, Komai-Koma M, Liew FY. Expression and function of Toll-like receptor on T cells. Cell Immunol. 2005;233:85–9. doi: 10.1016/j.cellimm.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 21.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 22.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–22. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 23.Bendigs S, Salzer U, Lipford GB, Wagner H, Heeg K. CpG-oligodeoxynucleotides co-stimulate primary T cells in the absence of antigen-presenting cells. Eur J Immunol. 1999;29:1209–18. doi: 10.1002/(SICI)1521-4141(199904)29:04<1209::AID-IMMU1209>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 24.Caron G, et al. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J Immunol. 2005;175:1551–7. doi: 10.4049/jimmunol.175.3.1551. [DOI] [PubMed] [Google Scholar]
- 25.Gelman AE, Zhang J, Choi Y, Turka LA. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J Immunol. 2004;172:6065–73. doi: 10.4049/jimmunol.172.10.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cairns BA, Barnes CM, Mlot S, Meyer AA, Maile R. Toll-like receptor 2 and 4 ligation results in complex altered cytokine profiles early and late after burn injury. J Trauma. 2008;64:1069–77. doi: 10.1097/TA.0b013e318166b7d9. [DOI] [PubMed] [Google Scholar]
- 27.Maung AA, et al. Enhanced TLR4 reactivity following injury is mediated by increased p38 activation. J Leukoc Biol. 2005;78:565–73. doi: 10.1189/jlb.1204698. [DOI] [PubMed] [Google Scholar]
- 28.Paterson HM, et al. Injury primes the innate immune system for enhanced Toll-like receptor reactivity. J Immunol. 2003;171:1473–83. doi: 10.4049/jimmunol.171.3.1473. [DOI] [PubMed] [Google Scholar]
- 29.Schwacha MG, Daniel T. Up-regulation of cell surface Toll-like receptors on circulating gammadelta T-cells following burn injury. Cytokine. 2008;44:328–34. doi: 10.1016/j.cyto.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walker HL, Mason AD., Jr A standard animal burn. J Trauma. 1968;8:1049–51. doi: 10.1097/00005373-196811000-00006. [DOI] [PubMed] [Google Scholar]
- 31.Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research, Division on Earth and Life Studies, National Research Council of the National Academies. Guide for the Care and Use of Laboratory Animals. 8th edition. Washington (DC): National Academies Press; 2011. [cited 2012 Aug 8]. Available from: http://oacu.od.nih.gov/regs/ [Google Scholar]
- 32.Thomas S, Stauss HJ, Morris EC. Molecular immunology lessons from therapeutic T-cell receptor gene transfer. Immunology. 2010;129:170–7. doi: 10.1111/j.1365-2567.2009.03227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hiromatsu T, et al. NK T cells stimulated with a ligand for TLR2 at least partly contribute to liver injury caused by Escherichia coli infection in mice. Eur J Immunol. 2003;33:2511–9. doi: 10.1002/eji.200324077. [DOI] [PubMed] [Google Scholar]
- 34.Kabelitz D. Expression and function of Toll-like receptors in T lymphocytes. Curr Opin Immunol. 2007;19:39–45. doi: 10.1016/j.coi.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 35.Crellin NK, et al. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol. 2005;175:8051–9. doi: 10.4049/jimmunol.175.12.8051. [DOI] [PubMed] [Google Scholar]
- 36.Ghosh TK, et al. Toll-like receptor (TLR) 2–9 agonists-induced cytokines and chemokines: I. Comparison with T cell receptor-induced responses. Cell Immunol. 2006;243:48–57. doi: 10.1016/j.cellimm.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Schwarz K, et al. Role of Toll-like receptors in costimulating cytotoxic T cell responses. Eur J Immunol. 2003;33:1465–70. doi: 10.1002/eji.200323919. [DOI] [PubMed] [Google Scholar]
- 38.McCarron M, Reen DJ. Activated human neonatal CD8+ T cells are subject to immuno-modulation by direct TLR2 or TLR5 stimulation. J Immunol. 2009;182:55–62. doi: 10.4049/jimmunol.182.1.55. [DOI] [PubMed] [Google Scholar]
- 39.Cottalorda A, et al. TLR2 engagement on memory CD8(+) T cells improves their cytokine-mediated proliferation and IFN-gamma secretion in the absence of Ag. Eur J Immunol. 2009;39:2673–81. doi: 10.1002/eji.200939627. [DOI] [PubMed] [Google Scholar]
- 40.Sobek V, et al. Direct Toll-like receptor 2 mediated co-stimulation of T cells in the mouse system as a basis for chronic inflammatory joint disease. Arthritis Res Ther. 2004;6:R433–46. doi: 10.1186/ar1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imanishi T, et al. Cutting edge: TLR2 directly triggers Th1 effector functions. J Immunol. 2007;178:6715–9. doi: 10.4049/jimmunol.178.11.6715. [DOI] [PubMed] [Google Scholar]
- 42.Lee SM, Joo YD, Seo SK. Expression and function of TLR2 on CD4 versus CD8 T cells. Immune Netw. 2009;9:127–32. doi: 10.4110/in.2009.9.4.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–80. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 44.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 45.Tomita T, et al. MyD88-dependent pathway in T cells directly modulates the expansion of colitogenic CD4+ T cells in chronic colitis. J Immunol. 2008;180:5291–9. doi: 10.4049/jimmunol.180.8.5291. [DOI] [PubMed] [Google Scholar]