

Abstract
Hyperimmunoglobulin E syndrome (HIES), also known as Job's syndrome, is a rare primary immunodeficiency characterized by eczema, recurrent skin and lung infections, elevated serum IgE, and connective tissue and skeletal abnormalities. Individuals with HIES share a characteristic facial appearance and many oral manifestations including retained primary dentition, a high-arched palate, variations of the oral mucosa and gingiva, and recurrent oral candidiasis. An 18-year-old lady presented with gingival swelling, bleeding from the gums, recurrent skin infections, and recurrent respiratory infections with intermittent fever. After thorough extra oral, intra oral and radiographic examination, serological investigations were performed. Growth of candida hyphae in the biopsy specimen of gingiva and increased levels of serum IgE with typical extra oral findings established the diagnosis as Job's syndrome (hyper IgE syndrome). Treatment with anti-fungal antibiotics and phase-I therapy including scaling and root planing followed by gingivoplasty using diode laser (980 nm) was performed. HIES was previously defined on the basis of clinical manifestations and laboratory markers that were not specific to the disease. With the identification of STAT3 mutations as the cause of HIES, we can definitively characterize the disease at molecular and immunologic levels. This case emphasizes the role of the dentist in the diagnosis of rare syndromes which alters the treatment plan.
Keywords: Candidiasis, fungal infection, hyper IgE disorder, hyperimmunoglobulin E syndrome, immunodeficiency, Job's syndrome
INTRODUCTION
Hyperimmunoglobulin E syndrome (HIES), also known as Job's syndrome or Buckley's syndrome, is a primary immunological disorder that affects the immune system, connective tissues, skeleton, and the dentition. Job's syndrome was the name given to describe the patients based on the book of Job 2:7, “so went satan forth from the presence of the Lord, and smote Job with some boils from the sole of his feet unto his crown.” The condition of recurring skin abscess was first described by Davis in 1966.[1] This rare immunodeficiency familial disorder was subsequently delineated by Buckley to elevated levels of IgE, which resulted in the additional identification of the condition as Buckley's syndrome.[2] Since then, HIES has been increasingly recognized as a multisystem immune system deficiency characterized not only by the classic triad of high serum IgE levels, eczema, skin and lung infections, but also by retained primary dentition, variations of oral mucosa and gingiva, osteopenia, minimal trauma fractures, scoliosis, a characteristic facial appearance, central nervous system abnormalities, and arterial aneurysms (Freeman AF 2009).[3]
The exact incidence of the disease is not known due to its rarity, and therefore few reports are available in literature. However, both autosomal recessive and dominant HIES affect males and females equally. Patients with autosomal dominant HIES range in age from birth to 60 years and not all patients have the same range of infection, facial features, and skeletal anomalies. It has been reported that individuals with autosomal dominant form of HIES have skeletal and dental abnormalities, as opposed to the recessive form.[4] Recently, missense or in-frame deletions resulting in one amino acid change or loss in STAT3 have been identified as the etiology for the majority of the cases of the autosomal dominant form of this disease.[5] However, pathogenesis of many manifestations remains poorly understood. The HIES diagnosis is still based on the patient's clinical and laboratory features, as no specific diagnostic test is available.
There is a paucity of literature describing oral findings in HIES patients. A few reports have described oral ulcerations, gingivitis, and prolonged oral and cervicofacial infections in these patients.[6–8]
Fungal infection of gingiva in HIES patients, however, is rarely reported. We present an 18-year-old girl with autosomal dominant HIES with Candida albicans infection of the gingiva, along with other oro-facial findings typical of the disease.
CASE REPORT
An 18-year-old female patient reported to the Department of Periodontics, Subharti Dental College, Meerut (Uttar Pradesh), India, with a chief complaint of swelling and bleeding from gums while brushing since 3 years. None of her siblings or immediate family suffered from periodontal disease. She was undernourished and of below average height and weight. Her medical history revealed the occurrence of multiple episodes of respiratory infection, intermittent fever, and repeated skin infections for which she had been investigated earlier. Her previous laboratory reports were unremarkable except significantly increased IgE levels (839 IU/mL).
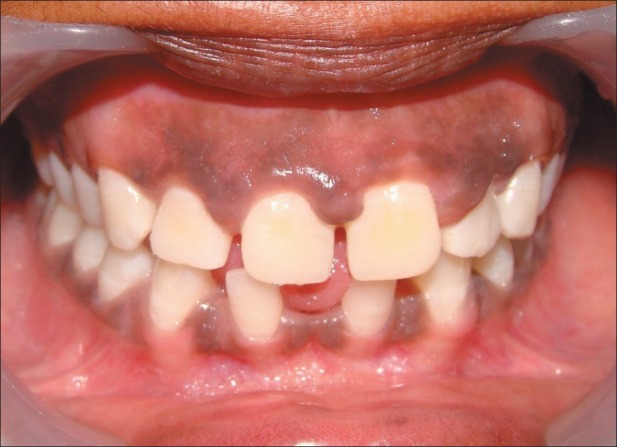
On examination, the patient had dry, crusted lips, a broad nasal bridge and prominent chin [Figures 1 and 2]. Intraorally, there was significant gingival enlargement in maxillary anterior region. Although nodular and fibrous in consistency; there was bleeding on probing of the gingiva. However, no ulcerations were seen to be present. She also had dental abnormalities like congenitally missing mandibular permanent lateral incisors, with retained mandibular primary central incisors [Figure 3a and b].
Figure 1.
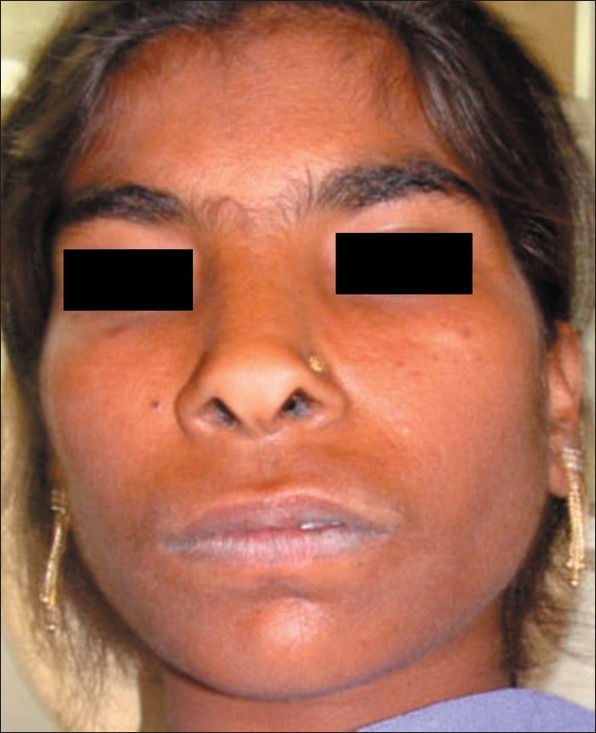
Extra oral photograph showing characteristic appearance
Figure 2.
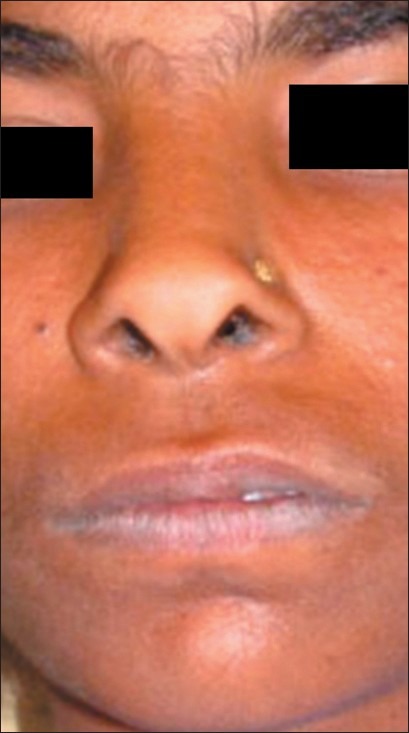
Extra oral photograph showing broad nasal bridge
Figure 3.
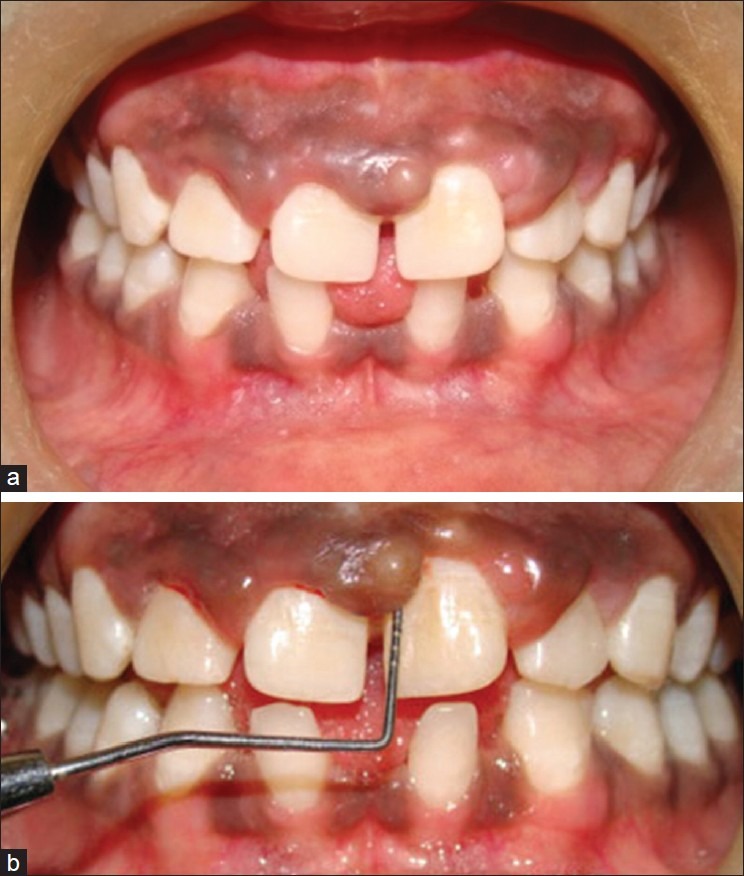
(a, b) Clinical presentation of spacing in lower anteriors and nodular gingival enlargement in maxillary anterior region
Orthopantomogram (OPG) confirmed congenitally missing mandibular permanent lateral incisors [Figure 4]. On chest X-ray, haziness on the right side indicative of pulmonary pneumonia was seen [Figure 5].
Figure 4.
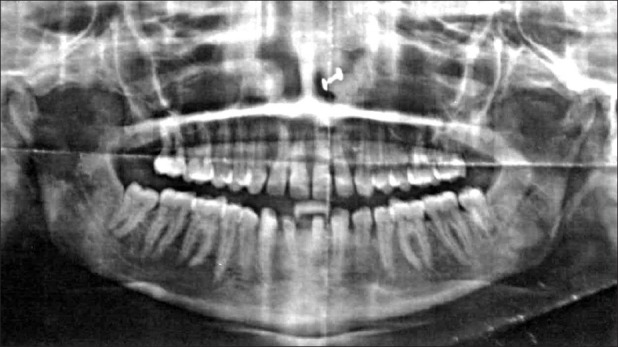
Orthopantomograph showing congenitally missing mandibular lateral incisors
Figure 5.
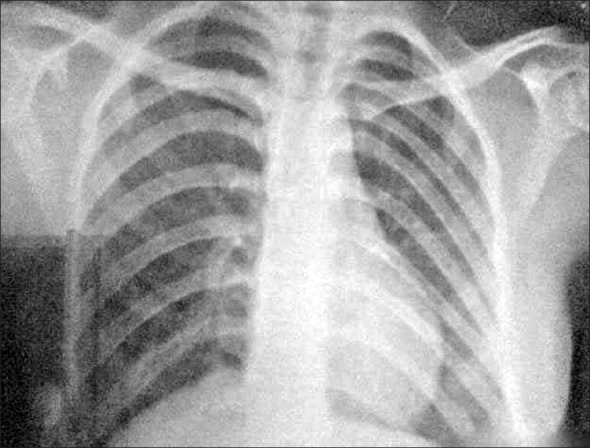
Chest radiograph showing haziness on the right side
Microbiologic analysis performed on the representative gingival lesions showed the growth of Candida albicans hyphae suggestive of an immunodeficient state [Figure 6]. The patient was thus further investigated for the cause of immunodeficiency.
Figure 6.
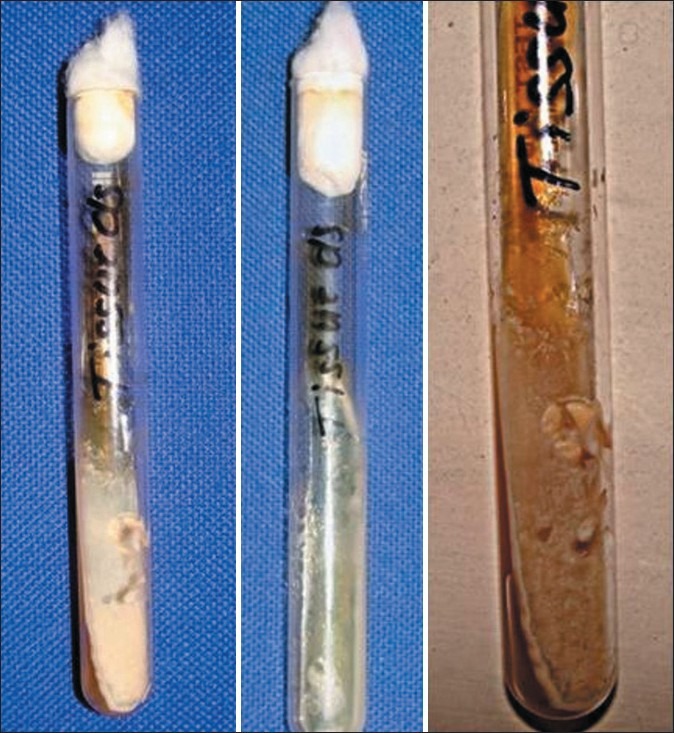
Microbiologic test showing the growth of the candida hyphae
Sputum and Mantoux test were negative for mycobacterium. Serological tests for HIV and HBsAg were negative. Immunofluorescent test showed increased levels of serum IgE (839 IU/mL).
Thus, based on the medical history, clinical examination, and the laboratory features, the diagnosis was confirmed as HIES with C. albicans infection of the gingiva.
Medical treatment
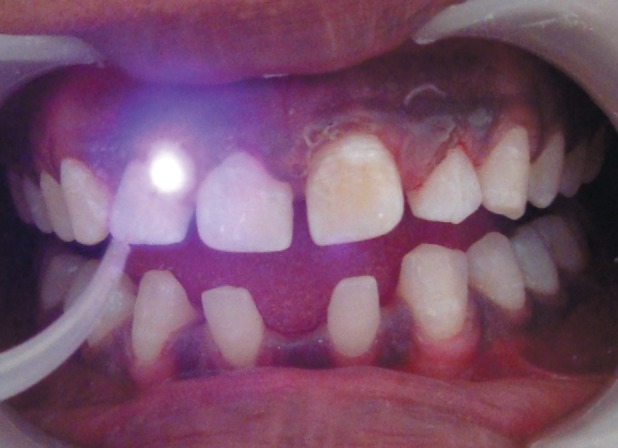
Patient was put on ketaconazole 100 mg twice a day for 4 weeks followed by ketaconazole 100 mg once a day for the next 2 months [Figure 7]. Respiratory infections were treated with the administration of antibiotics. Nutritional supplements were prescribed to improve the general health of the patient.
Figure 7.
After treatment with anti-fungal antibiotics
Periodontal treatment
Scaling and root planing were performed. Gingival recontouring in the maxillary anterior region was performed by using diode laser 980 nm [Figures 8 and 9]. Healing was uneventful, and the patient was advised to use chlorhexidine mouth rinses with regular oral hygiene maintenance.
Figure 8.
Gingivoplasty done after 1 year to recontour the gingiva by diode laser 980 nm
Figure 9.
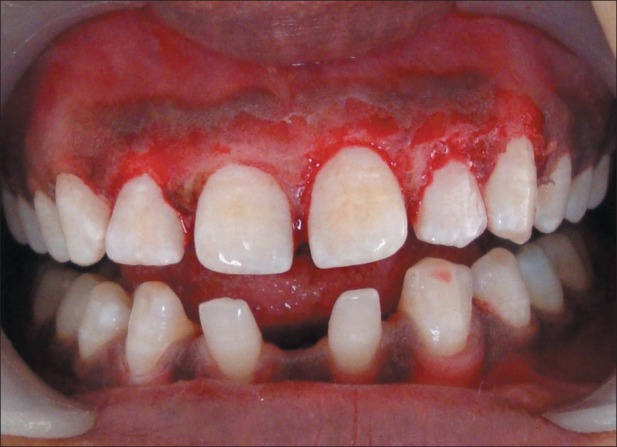
Immediate postoperative photograph after gingivoplasty
Follow-up observations
Gingival health was satisfactory in the subsequent follow-up visits [Figure 10]. The patient was satisfied and more comfortable with the overall general health too.
Figure 10.
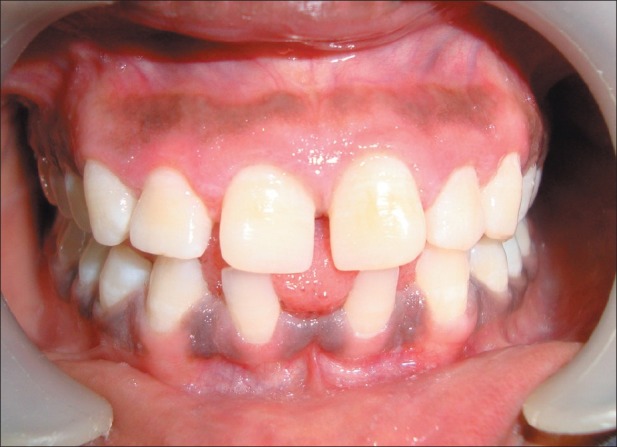
Two weeks postoperative photograph after gingivoplasty
DISCUSSION
This illustrated case demonstrates both clinical and laboratory features consistent with HIES. Autosomal dominant hyper IgE (HIES or Job′s) syndrome is a rare primary immune deficiency characterized by eczema, recurrent skin and lung infections, extremely elevated serum IgE, and a variety of connective tissue and skeletal abnormalities. Individuals with HIES share a characteristic facial appearance (coarse facies with prominent nose, broad nasal bridge, dryness of the face with prominent pores) and many oral manifestations including retained primary dentition, a high-arched palate, variations of oral mucosa and gingiva, and recurrent oral candidiasis since 3 years.
Oral and gingival manifestations of our case included nodular gingival enlargement in maxillary anterior teeth region, retained primary teeth (lower central incisors), and congenitally missing lower incisors with confirmation of the growth of candida hyphae by microbiological examination of the excised biopsy specimen. Aldous et al. (2007) have reported that dental abnormalities are variable and include retention of primary teeth which sometimes results in double rows of teeth upon the eruption of the permanent teeth, and high-arched palates.[9] Charon (1985) reported a higher incidence of gingivitis, thrush, and plaque in a group of patients with Job's syndrome.[7] Remmer et al. (2004) reported that individuals with the autosomal dominant form of HIES have skeletal and dental abnormalities, whereas those with the recessive form do not have these.[4] O’Connell (2000) reported that 44% of individuals with the syndrome presented with elevated palates,[10] while Grimbacher (1999) reported the palates to be higher in 71% of the individuals in one article and in 65% of the patients in another article.[11]
The majority of HIES individuals (64%) exhibit failure of primary teeth exfoliation, often preventing the eruption of succedaneous teeth.[10,12] This prolonged retention of primary teeth can lead to permanent teeth impaction or formation of double rows, in which succedaneous teeth erupt lingual to the deciduous teeth and predispose to malocclusion. Lesions of the oral mucosa and gingiva, involving the hard palate, dorsal tongue, buccal mucosa, and lip mucosa, have been identified in over 75% of the patients.[12] The oral lesions in HIES may represent developmental abnormalities, reactive lesions arising from chronic infections associated with the syndrome, or manifestations of the role of the HIES gene, STAT3, in epithelial development. Oral candidiasis (pseudomembranous, erythematous, median rhomboid glossitis and angular cheilitis) is also common.[3]
Earlier the diagnosis was solely based on the clinical and laboratory features. Since the finding of STAT3 mutations, the diagnosis may be based on a high level of clinical suspicion and confirmed through STAT3 mutational analysis.[5,13] Although STAT3 is the only gene so far identified to cause autosomal dominant HIES, it remains possible that more than one genetic etiology exists.[3] The mutations in STAT3 have been described predominantly in two regions of the gene: the DNA binding domain, which mediates DNA–protein interactions, and the SH2 domain, which mediates protein–protein interactions.[5]
Mutations in STAT3 account for the majority of the cases of autosomal dominant HIES, but the pathogenesis of many varied features remains poorly understood. There have been multiple reports with small numbers of patient and conflicting data as to whether a chemotactic defect is present and whether there is a T helper Th1/Th2 cytokine imbalance.[14] Recent data suggest that pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α and interferon-γ are elevated at rest and after stimulation, consistent with mouse models of STAT3 immune regulation.[5] Naïve T cells have been shown to be unable to differentiate into IL-17 producing T helper cells (Th 17 cells), thus resulting in no IL-17 production.[15] Th 17 cells and IL-17 appear to be critical in control of both extracellular bacterial and fungal infections; their absence may help explain some of the infections in HIES.
STAT3-deficient HIES must be distinguished from the distinct syndrome of autosomal recessive HIES,[4] which is characterized by extremely elevated serum IgE and severe eczema, often complicated by bacterial and viral super infections (herpes simplex virus, molluscum contagiosum). Autosomal recessive HIES does not share the musculoskeletal and dental manifestations of the autosomal dominant disease and has a high incidence of neurologic complications from either infection or vasculitis.
CONCLUSION
An awareness of the diverse features of the hyper IgE syndrome will facilitate the recognition and early diagnosis, make genetic counseling possible, and improve patient care. Patients benefit from aggressive antibiotic treatment to minimize infectious complications. Future study of HIES and STAT3 will help us understand eczema, IgE regulation, infection susceptibility, scoliosis, and bronchiectasis, as well as provide mechanistic insights into treatment.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Davis SD, Schaller J, Wedgwood RJ. Job's syndrome: recurrent, “cold”, staphylococcal abscesses. Lancet. 1966;1:1013–15. doi: 10.1016/s0140-6736(66)90119-x. [DOI] [PubMed] [Google Scholar]
- 2.Buckley RH, Wray BB, Belmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics. 1972;49:59–70. [PubMed] [Google Scholar]
- 3.Freeman AF, Domingo DL, Holland SM. Hyper IgE (Job′s) syndrome: A primary immune deficiency with oral manifestations. Oral diseases. 2009;15:2–7. doi: 10.1111/j.1601-0825.2008.01463.x. [DOI] [PubMed] [Google Scholar]
- 4.Remmer ED, Puck JM, Holland SM, Schmitt M, Weiss M, Frosch M, et al. Autosomal recessive hyperimmunoglobulin E syndrome.A distinct disease entity. J Pediatr. 2004;144:93–9. doi: 10.1016/S0022-3476(03)00449-9. [DOI] [PubMed] [Google Scholar]
- 5.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;18:1608–19. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 6.Vigliante CE, Costello BJ, Quinn PD. Life-threatening cervicofacial infection in a child with Hyperimmunoglobulin E syndrome. J Oral Maxillofac Surg. 2001;59:561–5. doi: 10.1053/joms.2001.22689. [DOI] [PubMed] [Google Scholar]
- 7.Charon JA, Mergenhagen SE, Gallin JI. Gingivitis and oral ulceration in patients with neutrophil dysfunction. J Oral Pathol. 1985;14:150–5. doi: 10.1111/j.1600-0714.1985.tb00478.x. [DOI] [PubMed] [Google Scholar]
- 8.Hatori M, Yoshiya M, Kurachi Y, Nagumo M. Prolonged infection of the floor of the mouth in hyperimmunoglobulinemiaE (Buckley's syndrome). Report of a case. Oral Surg Oral Med Oral Pathol. 1993;76:289–93. doi: 10.1016/0030-4220(93)90255-3. [DOI] [PubMed] [Google Scholar]
- 9.Aldous JA, Olson GJ, Parkin MJ. Dental observations of hyper IgE disorder. J Clin Pediatr Dent. 2007;32:69–72. doi: 10.17796/jcpd.32.1.044778063546u638. [DOI] [PubMed] [Google Scholar]
- 10.O’Connell AC, Puck JM, Grimbacher B, Facchetti F, Majorana A, Gallin JI, et al. Delayed eruption of permanent teeth in hyperimmunoglobulinemia E recurrent infection syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;89:177–85. doi: 10.1067/moe.2000.103129. [DOI] [PubMed] [Google Scholar]
- 11.Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, et al. Hyper IgE syndrome with recurrent infections-an autosomal dominant multisystem disorder. N Engl J Med. 1999;340:692–702. doi: 10.1056/NEJM199903043400904. [DOI] [PubMed] [Google Scholar]
- 12.Domingo DL, Freeman AF, Davis J, Puck JM, Tianxia W, Holland SM, et al. Novel intraoral phenotypes in Hyperimmunoglobulin E syndrome. Oral Dis. 2008;14:73–81. doi: 10.1111/j.1601-0825.2007.01363.x. [DOI] [PubMed] [Google Scholar]
- 13.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–62. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 14.Hill HR, Ochs HD, Quie PG, Clark RA, Pabst HF, Klebanoff SJ, et al. Defect in neutrophil granulocyte chemotaxis in Job's syndrome of recurrent “cold” staphylococcal abscesses. Lancet. 1974;14:617–9. doi: 10.1016/s0140-6736(74)91942-4. [DOI] [PubMed] [Google Scholar]
- 15.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T (H) 17 cell differential in subjects with autosomal dominant hyper IgE syndrome. Nature. 2008;452:773–6. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]