

Abstract
Diffuse mesangial sclerosis (DMS) is a rare cause of nephrotic syndrome in the infantile and childhood period. DMS is a phenotypic expression of syndromic entities such as WAGR syndrome (Wilms’ tumor, aniridia, genitourinary anomalies and mental retardation), Denys Drash syndrome, Pierson syndrome, Frasier syndrome, or Galloway–Mowat syndrome. We report two cases of DMS, one presenting in first year of life and another in second decade of life. Both of them had fatal outcome. Recognition of the disease is very important in modifying the management of patient and active surveillance of family members.
Keywords: Diffuse mesangial sclerosis, India, nephrotic syndrome in childhood
Introduction
Diffuse mesangial sclerosis (DMS) is one of the rarer causes of nephrotic syndrome in infantile and childhood period. It is characterized by progressive sclerosis of the mesangial matrix with minimal or absent mesangial cell proliferation, hypertrophy of podocytes in early disease, thickened basement membranes, diminished patency of capillary lumen and, crown-like appearance of vacuolated podocytes in advanced disease. Electron microscopy shows mesangial collagen fibrils along with wavy and split basement membranes. Genetic forms of nephropathy including DMS is usually resistant to immunosuppressive therapy and rapidly progresses to end-stage kidney disease (ESKD).[1] Genetic mutation in genes WT1 and PLCE1 are found in of DMS.[2–4] On pubmed search, we found only four cases of DMS reported from India.[5–7] We report two cases of DMS, one presenting in the first year of life and another in second decade of life. Both of them had fatal outcome.
Case Reports
Case 1
An 8-month-old male child presented to a local practitioner with facial puffiness of 2 weeks duration. Child weighed 7 kg and was a full-term product of an uncomplicated pregnancy, born to a 33-year-old G3P3L3 mother. Milestones were normal for age. Blood pressure was normal for age. Physical examination was essentially normal except for edema. External genitalia were normal. Systemic examination was unremarkable. Routine urine examination showed 3+ albuminuria and inactive sediments. Serum creatinine was 0.5 mg/dL. The child was empirically treated with steroids (60 mg/m2 OD). However, after 4 months child was referred to a nephrologist with symptoms of anasarca, vomiting, and oliguria. Urine examination at this time showed 4+ albuminuria, inactive sediments, and granular casts. Urine protein to creatinine ratio (PCR) was 56.5 g/g. Hemogram revealed Hb14.3 gm/dL, total leukocyte count 14,500 cells/mm3 and platelets 800 × 103/μL. Serum albumin and total cholesterol were 0.7 gm/dL and 3.9.75 mg/dL, respectively. Serum creatinine was 2.4 mg/dL. Viral markers (HIV, HbsAg, anti-HCV) were negative. Ultrasound showed normal sized kidneys with increased cortical echotexture and loss of corticomedullary differentiation. Kidney biopsy revealed 19 normal-sized glomeruli showing global and segmental mesangial sclerosis with obliteration of capillary lumina. Prominence of podocytes was seen with focal hyperplasia. Occasional tufts showed patent capillaries with variably thickened basement membranes. Bowman's space was unremarkable. Tubulointerstitial compartment showed marked chronicity diffusely containing granular, leukocytic casts in the dilated lumen [Figure 1]. Arteries appeared unremarkable. Immunohistochemically, podocytes did not express Ki-67. Immunofluorescence showed trapping of IgM (2+) in the sclerotic area. Rest of the panel (IgG, IgA, C3, C1q, and fibrin) were negative. Child was put on supportive hemodialysis (twice a week) and treated with low-dose prednisolone 40 mg/m2, nutritional supplements. However, child succumbed later to severe sepsis at the age of 17 months.
Figure 1 a–d.
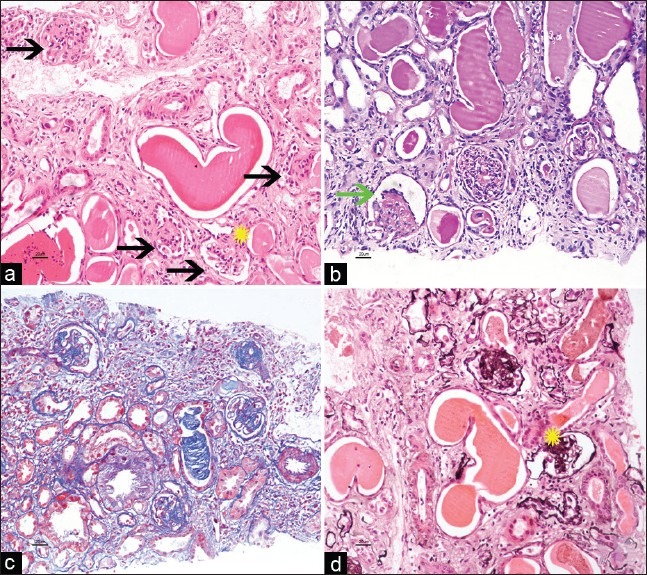
Four glomeruli displaying global mesangial sclerosis with variably increased mesangial cells, obliteration of capillary lumen, and patent bowmans’ space (black arrows, a). Crowning of podocytes is noted (green arrow, b). Few capillaries had patent lumen with slightly thickened basement membranes (yellow star, a and d). Tubules are dilated and contain Tamm–Horsfall protein casts. Interstitial space reveals lymphomononuclear infiltrates (a-H and E ×20, b-PAS, c-Masson Trichrome, d- PASM stain)
Case 2
A 16-year-old female presented with pedal edema and facial puffiness of 2 months duration. She had developed fever, vomiting, and loose stools since 3 days. There were no similar complaints in the past. She was eighth of nine siblings born out of a nonconsanguinous marriage. Ninth sibling expired at the age of 6 month due to suspected renal disease. Hemogram revealed Hb 15.6 gm/dL, total leukocyte count 27,000 cells/mm3 and platelets 600 × 103/μL. Urine examination showed 3+ albumin, inactive sediments, and granular casts. Urine PCR was 7 g/g. Serum creatinine was 2 mg/dL at presentation. Serum albumin and cholesterol was 0.5 gm/dL and 346.87 mg/dL, respectively. Complements were normal. ANA and anti-dsDNA were negative. Viral markers (HIV/HBsAg/antiHCV) were negative. Stool examination showed cysts of cryptosporidium. Ultrasound examination showed normal-sized kidneys with increased echotexture and maintained cortico-medullary differentiation. Kidney biopsy revealed eight normal sized glomeruli, all of which had global mesangial sclerosis and crowning of podocytes over the sclerosed tufts. Capillaries were compromised diffusely. There was moderate interstitial fibrosis and tubular atrophy [Figure 2]. Arteries showed hyperplasia of the media. Immunohistochemically, podocytes did not express Ki-67. Immunofluorescence was negative for the panel (IgG, IgA, C3, C1q, and fibrin). She was treated with oral nitazoxanide 500 mg BD and intravenous piperacillin-tazobactam 2.25 gm TID. However, six days later she succumbed to severe sepsis despite active treatment.
Figure 2.
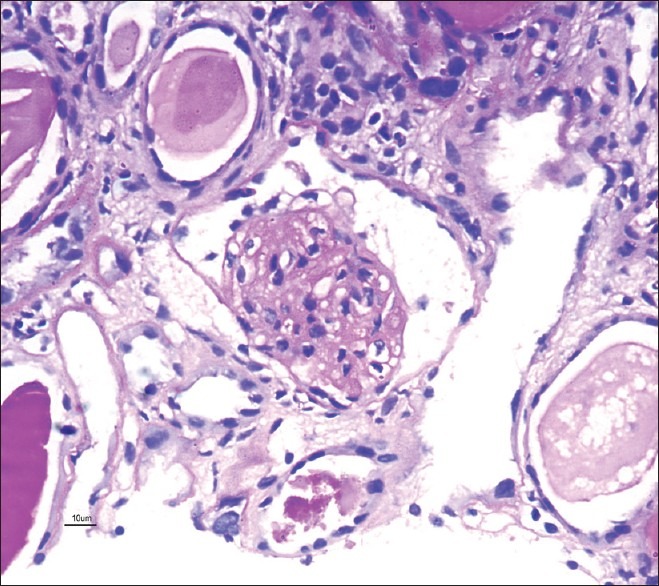
Glomerulus showing mesangial sclerosis globally with obliteration of capillary lumen and patent bowmans’ space (×40, PAS stain)
Discussion
Diffuse mesangial sclerosis (DMS) was first described by Habib and Bios in 1973.[8] DMS is one of the causes for nephrotic syndrome (NS) that occurs more commonly in first two decades of life. It has a fatal course. Although rare cases of spontaneous remission in other causes of congenital nephrotic syndrome have been described,[9] it is unheard of in patients with DMS. Management of such cases is a gigantic task. This includes antiproteinuric measures and medical or surgical nephrectomy in nonresponders to mitigate proteinuria, nephrotic syndrome, and its complications. Continuous ambulatory peritoneal dialysis is the usual standard modality of dialysis in such patients. This is followed by renal allograft transplant as soon as the child attains 10 kg of weight. Gonadectomy and/or nephrectomy is also advised prophylactically for prevention of tumor development.[10]
The phenotypic finding of a DMS is not a single entity but is associated with many genotypic disorders including the WAGR syndrome (Wilms’ tumor, aniridia, genitourinary anomalies and mental retardation), Denys Drash syndrome (NS, male gonadal dysgenesis and Wilms tumor), Frasier syndrome (NS, normal female external genitalia, streak gonads, XY karyotype), Pierson syndrome (NS with microconia), and Galloway–Mowat syndrome (NS, microcephaly, gyral abnormalities, and hiatal hernia) or an isolated Diffuse mesangial sclerosis.[2–4] Acquired cytomegalovirus infection is also linked to causation of DMS.[11]
Two genes are implicated in DMS – WT1 and PLCE1. WT1 gene is located on chromosome 11p13 and is composed of ten exons.[12] PLCE1 gene is located on chromosome 10q23, encodes for a protein PLCε1 that is a member of phospholipase family of enzymes.[3] Benoit et al. advocated in their review article about WT1 and PLCE1 gene mutational study in infantile or childhood patients developing NS, and DMS as the histologic lesion.[13] Chernin et al. concluded that patients with KTS mutations presented at a significantly older age and with a slower progression toward ESKD compared with missense mutations.[14] Gbadegesin et al. analyzed a worldwide cohort of 1368 children with NS, and detected PLCE1 mutation as the most frequent (28.6%) and WT1 mutation as the second most frequent (8.5%) cause of isolated DMS.[3]
On PubMed search, there are only four cases reported from India. Three of these cases are reported from a single center with an incidence of 1% over a period of 10 years, studying 290 biopsied cases of the idiopathic nephrotic syndrome under the age of 16 years.[5,6] The patients were residents of Northern and Eastern part of India. Another case is from Southern India. We found similar incidence (1%) over a period of 28 months evaluating 196 cases of adequate biopsies under the age of 16 years with similar presentation. Demography details on previous cases are available in only three of them (age range 3 to 9 months; male to female – 3:0).[5–7] Among those, three cases were in the infantile age group. One of our cases presented in second decade of life. This could probably be due to a mutation that presents at an older age than usual. Clinical details and histologic findings are available in only one case[7] wherein the 9-month-old child had nephrotic syndrome, hypertension, and normal renal function at presentation. External genitalia were normal and USG showed increased echotexture. Kidney biopsy had revealed 44 glomeruli showing variable degree of mesangial sclerosis in majority of them. The child was treated with captopril and indomethacin. Follow-up information on this case though incomplete in duration, mentions complete remission.[7] Family history and screening of family members is not available in any of the cases reported. Both of our cases presented with nephrotic syndrome and renal insufficiency. Biopsy demonstrated hallmark lesions of DMS. Case 2 had a sibling with history of renal disease. The presentation with nephrotic syndrome in Case 1 and Case 2 was at the age of 8 months and 15 years, respectively. Case 1 became dialysis dependant within 4 months of first presentation. He did not have any abnormality of either external genitalia or eyes. Renal ultrasound did not reveal kidney mass lesion. The siblings and family members of Case 1 were screened for renal abnormalities and found to be normal. Case 2 did not have other organ involvement. There are no set guidelines for surveillance of family members of affected patients.
The histologic appearance is pathognomonic. Differential diagnosis of DMS includes collapsing variant of focal segmental glomerulosclerosis (FSGS-Col). The latter is usually a focal process possessing diagnostic features such as de-differentiation of podocytes in the form of hyperplasia (>2 cell layer), proliferative marker Ki-67 nuclear positive podocytes, and loss of WT1 nuclear expression; and accompanied with collapse of underlying tufts.[15] Tubular microcyst formation with or without simplified epithelial lining is more commonly seen with FSGS-Col than DMS. Ki-67 was negative in both cases. Electron microscopy of kidney tissue from paraffin block was tried; however, the attempt was unsuccessful. As there were no extra-renal features associated with various syndromes, it is wise enough to diagnose “isolated” DMS; however, genetic analysis for at least WT1 and PLCE1 genes would have given more in depth knowledge of the disease.
Recognition of this entity is very important in modifying the management of patient. DMS must be suspected in a child that presents with NS and/or rapidly progressive renal insufficiency, especially if there is family history of renal disease. There is an immense need for development of genetic analysis in this part of the world, especially for pediatric patients presenting with nephrotic syndrome and renal insufficiency, and their asymptomatic family members.
Acknowledgments
We sincerely thank Mrs. Tulasi Kumari, Mrs. Hema Nagaraj, Mr. Nagaraj, and rest for their outstanding technical support in Histopathology section.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Büscher AK, Kranz B, Büscher R, Hildebrandt F, Dworniczak B, Pennekamp P, et al. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2010;5:2075–84. doi: 10.2215/CJN.01190210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen AH, Turner MC. Kidney in Galloway-Mowat syndrome: Clinical spectrum with description of pathology. Kidney Int. 1994;45:1407–15. doi: 10.1038/ki.1994.184. [DOI] [PubMed] [Google Scholar]
- 3.Gbadegesin R, Hinkes BG, Hoskins BE, Vlangos CN, Heeringa SF, Liu J, et al. Mutations in PLCE1 are a major cause of isolated diffuse mesangial sclerosis (IDMS) Nephrol Dial Transplant. 2008;23:1291–7. doi: 10.1093/ndt/gfm759. [DOI] [PubMed] [Google Scholar]
- 4.Koziell A, Grundy R. Frasier and Denys-Drash syndromes: Different disorders or part of a spectrum? Arch Dis Child. 1999;81:365–9. doi: 10.1136/adc.81.4.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gulati S, Sharma AP, Sharma RK, Gupta A, Gupta RK. Do current recommendations for kidney biopsy in nephrotic syndrome need modifications? Pediatr Nephrol. 2002;17:404–8. doi: 10.1007/s00467-002-0840-3. [DOI] [PubMed] [Google Scholar]
- 6.Kumar J, Gulati S, Sharma AP, Sharma RK, Gupta RK. Histopathological spectrum of childhood nephrotic syndrome in Indian children. Pediatr Nephrol. 2003;18:657–60. doi: 10.1007/s00467-003-1154-9. [DOI] [PubMed] [Google Scholar]
- 7.Umesh L, Prashanth A, Benakappa DG, Govindaraj M, Benakappa N. Diffuse mesangial sclerosis presenting as infantile nephrotic syndrome. Indian Pediatr. 2001;38:663–4. [PubMed] [Google Scholar]
- 8.Habib R, Bois E. Heterogeneity of early onset nephrotic syndromes in infants (nephrotic syndrome “in infants”). Anatomical, clinical and genetic study of 37 cases. Helv Paediatr Acta. 1973;28:91–107. [PubMed] [Google Scholar]
- 9.Haws RM, Weinberg AG, Baum M. Spontaneous remission of congenital nephrotic syndrome: A case report and review of the literature. Pediatr Nephrol. 1992;6:82–4. doi: 10.1007/BF00856846. [DOI] [PubMed] [Google Scholar]
- 10.Hu M, Zhang GY, Arbuckle S, Graf N, Shun A, Silink M, et al. Prophylactic bilateral nephrectomies in two paediatric patients with missense mutations in the WT1 gene. Nephrol Dial Transplant. 2004;19:223–6. doi: 10.1093/ndt/gfg473. [DOI] [PubMed] [Google Scholar]
- 11.Besbas N, Bayrakci US, Kale G, Cengiz AB, Akcoren Z, Akinci D, et al. Cytomegalovirus-related congenital nephrotic syndrome with diffuse mesangial sclerosis. Pediatr Nephrol. 2006;21:740–2. doi: 10.1007/s00467-006-0051-4. [DOI] [PubMed] [Google Scholar]
- 12.Niaudet P, Gubler MC. WT1 and glomerular diseases. Pediatr Nephrol. 2006;21:1653–60. doi: 10.1007/s00467-006-0208-1. [DOI] [PubMed] [Google Scholar]
- 13.Benoit G, Machuca E, Antignac C. Hereditary nephrotic syndrome: A systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr Nephrol. 2010;2:1621–32. doi: 10.1007/s00467-010-1495-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chernin G, Vega-Warner V, Schoeb DS, Heeringa SF, Ovunc B, Saisawat P, et al. Genotype/phenotype correlation in nephrotic syndrome caused by WT1 mutations. Clin J Am Soc Nephrol. 2010;5:1655–62. doi: 10.2215/CJN.09351209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Albaqumi M, Barisoni L. Current views on collapsing glomerulopathy. J Am Soc Nephrol. 2008;19:1276–81. doi: 10.1681/ASN.2007080926. [DOI] [PubMed] [Google Scholar]