

Abstract
Trichothiodystrophy (TTD) is a rare, autosomal recessive disease, characterised by brittle, sulfur deficient hair and multisystem abnormalities. A systematic literature review identified 112 patients ranging from 12 weeks to 47 years of age (median 6 years). In addition to hair abnormalities, common features reported were developmental delay/intellectual impairment (86%), short stature (73%), ichthyosis (65%), abnormal characteristics at birth (55%), ocular abnormalities (51%), infections (46%), photosensitivity (42%), maternal pregnancy complications (28%) and defective DNA repair (37%). There was high mortality, with 19 deaths under the age of 10 years (13 infection related), which is 20-fold higher compared to the US population. The spectrum of clinical features varied from mild disease with only hair involvement to severe disease with profound developmental defects, recurrent infections and a high mortality at a young age. Abnormal characteristics at birth and pregnancy complications, unrecognised but common features of TTD, suggest a role for DNA repair genes in normal fetal development.
Trichothiodystrophy (TTD) is a rare, autosomal recessive disease, in which patients have brittle, sulphur deficient hair.1,2 When the hair from TTD patients is observed under polarising microscopy, it displays a diagnostic alternating light and dark banding pattern, called “tiger tail banding”3,4 (fig 1). TTD results from mutations in one of several different DNA repair genes (XPB, XPD or TTDA)5,6 and TTDN1, a gene of unknown function.7 Although XPB and XPD mutations are also seen in xeroderma pigmentosum, a disease with a 1000-fold increase in skin cancer,6,8–11 TTD patients have not been reported to have an increase in cancer.
Figure 1.

Microscopic diagnosis of trichothiodystrophy. (Left) Routine microscopic examination of hair from a patient with trichothiodystrophy shows shaft abnormalities of trichoschisis (arrows) and trichorrhexis nodosa-like fraying (arrows). (Right) Under polarising microscopy the hair shafts show a striking alternating dark and light (tiger tail) banding pattern.
TTD patients display a wide variety of clinical features, including cutaneous, neurological, and growth abnormalities. As a result, a variety of names have been used to describe the disease. In 1979, Price coined the term “trichothiodystrophy,” which encompasses a wide spectrum of neurocutaneous findings, to describe the unifying feature.12 The name reflects the brittle, sulfur deficient hair seen in all TTD patients (from Greek, tricho-meaning hair; -thio-, sulfur; -dys-, faulty; -trophy, nourishment). Several acronyms have been used to describe the clinical features of these patients. PIBIDS,13 IBIDS14,15 and BIDS16 describe six features of TTD: Photosensitivity, Ichthyosis, Brittle hair, Intellectual impairment, Decreased fertility, and Short stature. In order to assess the prevalence of the reported clinical features of TTD, we performed an extensive literature review to find all published case reports of patients with TTD. We analysed the frequency of the clinical findings described in an effort to characterise the spectrum of the disease better.
We modelled this review after a similar study on xeroderma pigmentosum.9
METHODS
We developed a standard Excel spreadsheet listing more than 200 clinical and laboratory characteristics. The search was restricted to published information in reports, and no effort was made to obtain unpublished data on the reported patients. This approach results in underreporting of characteristics not noted at the time of publication. However, when reported patients were identifiable as being the same individual in a subsequently reported paper, the data were consolidated. We searched PubMed/Medline, Web of Science, and the references cited in retrieved articles. Search terms were trichothiodystrophy, TTD, Tay syndrome, Pollitt syndrome, PIBIDS, IBIDS, and BIDS.
The most definitive clinical criteria include microscopic examination of hair shafts for tiger tail banding and structural abnormalities and the analysis of hair shaft sulfur content. However, diagnostic criteria for TTD have evolved over the decades since these reports have been published. As a result, some reports included patients with convincing clinical features of TTD and a confirmed DNA repair abnormality, but the clinical workup did not include hair analysis. In order to standardise selection of patients, we chose criteria, which determined whether or not a case report was included. Inclusion criteria were based on having at least two of the four following clinical or laboratory abnormalities: (1) presence of brittle hair and/or hair shaft abnormalities; (2) tiger tail banding with polarised microscopy; (3) decreased sulfur or cystine content of hair; and (4) DNA repair abnormality. While any one of these features is highly suggestive of TTD,3,11,17 we required a minimum of two features to confirm the diagnosis. We chose criteria which we reasoned would allow us to capture reports of most patients with TTD and which were important in forming the basis for the various subtypes which have led to our current understanding of the disease. These criteria were developed in order to provide a uniform approach to inclusion of case reports with varied amounts of information and published over more than 40 years and not to be used as criteria for clinical diagnosis of new patients.3,4
In the case of reported siblings with similar clinical features, if one sibling qualified according to the above criteria, then both siblings were included. Patients described collectively as a group were not included when the clinical features could not be traced to individual patients. We did not include cases reported only in meeting proceedings, where the report was not indexed.
We considered intrauterine growth restriction (IUGR) as fetuses specified to have intrauterine growth retardation or intrauterine growth restriction in the report. If this was not stated we used the standard of <10th centile for gestational age at the time of birth based on the criteria specified in Lubchenco.18 Low birth weight was stated in the report or was defined as infants or who were <2500 g at birth.
RESULTS
History of trichothiodystrophy
Vera Price first proposed the name “trichothiodystrophy” in 1979 in the book Haar und Haarkrankheiten.12 In 1980, Price17 reported two patients with a wide range of clinical features, and associated the low sulfur (cystine) content of the hair with the alternating bright and dark banding with polarised microscopy, now known as tiger tailed banding. This work established specific hair findings as the unifying marker for this neuroectodermal symptom complex, which we now know as TTD.
Before the name TTD was coined in 1979, several papers were published describing cases that are today considered to be the earliest reports of TTD. Some of these papers, however, did not have sufficient hair analysis to meet the inclusion criteria that we chose for this paper.19–21 The earliest paper that we included in this study is from Pollitt22 in 1968, which described two severely affected siblings with brittle, sulphur deficient hair, as well as intellectual and growth retardation. This report led to the name Pollitt syndrome. In 1970, Brown23 described alternating birefringence in the hair viewed under polarised microscopy in a 4-year-old girl with brittle hair and normal intelligence. Tay,24 in 1971, reported three siblings in Singapore with brittle hair, mental deficiency and growth retardation, who also had non-bullous congenital ichthyosiform erythroderma. Tay suggested an autosomal recessive pattern of inheritance. Tay’s 1971 report did not, however, include sufficient hair analysis to meet the inclusion criteria for this analysis. In 1974, Jackson25 described decreased fertility and autosomal recessive inheritance in an Amish kindred with brittle hair, intellectual impairment and short stature. Two index cases were sufficiently described to be included in this analysis.
Jackson’s report led to the name “Amish brittle hair brain syndrome”. As a result of these similar clinical descriptions, the acronym BIDS (Brittle hair, Intellectual impairment, Decreased fertility and Short stature) was suggested in 1976.16 Subsequently, the additional presence of ichthyosis led to the acronym IBIDS.14,15 Unlike some later cases of TTD with ichthyosis, it has been suggested that Tay syndrome specifically refers to the presence of congenital ichthyosis in addition to BIDS.26
Two siblings from Sabinas, Mexico were reported in 197627 as having brittle hair, developmental delay, and normal stature. This report, in conjunction with a report28 of a group of 11 additional TTD cases from Sabinas, led to the name Sabinas syndrome in 1981, which refers to the presence of hair and nail abnormalities in association with mental retardation. The 1981 report28 was not included in this review because the patients were described as a general group and not individually.
The addition of photosensitivity to the acronym IBIDS (resulting in PIBIDS) was recommended in 1983 by Crovato.13 There was also some debate as to whether TTD was in fact a single entity, due to the various presentations of this neuroectodermal disorder.29 In 1988, Chapmann reported a patient and recommended the addition of skeletal abnormalities instead of photosensitivity to the acronym, resulting in SIBIDS.30 The patient described in this report did not meet our inclusion criteria for this analysis.
In 1985, Van Neste31 reported defective DNA excision repair in ultraviolet (UV) exposed lymphocytes from a TTD patient. The first such gene mutation was identified the next year,32 when cells from four patients with TTD were found to have cellular UV hypersensitivity, very low levels of unscheduled DNA synthesis, and characteristics of the XP-D complementation group. Despite some patients having the same gene defects seen in xeroderma pigmentosum (XPB and XPD), patients with TTD do not have an increased incidence of skin cancers.2,11 In 1993, patient TTD1BR was reported to have a new DNA repair complementation group, called TTD-A.33 A second patient with TTD-A has since been reported.34 The TTD-A gene (called GTF2H5) was identified in 2004.35 The recently discovered TTDN1 gene with unknown function was described in association with non-photosensitive patients.7
To date, four genes have been identified as causing TTD: XPD, XPB, TTDA, and TTDN1.7,32,33,36
Cases reports included in this study
A total of 94 articles were found that met our inclusion criteria for TTD case reports. The articles were published from 1968 to 2005.7,13–17,22,23,25–27,31–34,36–114 They contained data on 112 patients. The reported cases met at least two of the four entry criteria, as follows: 96% (108 cases) had brittle hair or hair shaft abnormalities; there were 73% (82 cases) with tiger tail banding of the hair with polarised microscopy; 70% (78 cases) had decreased sulfur or cystine content of their hair; 37% (41 cases) had a DNA repair abnormality reported; four patients were included based on having brittle hair and a diagnosed sibling with TTD.85,90,98
Table 1 shows patient location and origin. As these data were only reported for 50 patients, the author’s location was used for the remaining 62 patients, assuming that the patients were from the same location as the author. Patients/authors were reported from 20 countries from all over the world, including Europe, North and South America, Africa, Asia and Australia. The greatest numbers of reports were from Italy (23%), the USA (16%), and the UK (16%).
Table 1.
Distribution of patient location or origin for reported trichothiodystrophy patients (n=112)
| Author location* | Patients No. (%) |
|---|---|
| Italy | 27 (23) |
| USA | 19 (16) |
| UK | 18 (16) |
| France | 12 (11) |
| Germany | 3 (3) |
| Canada | 4 (4) |
| Morocco | 4 (4) |
| Turkey | 4 (4) |
| Switzerland | 4 (4) |
| Other† | 17 (15) |
| Total | 112 (100) |
Author location used as surrogate for 62 patients whose location/origin was not reported.
“Other” refers to one or two patients from each of the following countries: Australia, Belgium, Denmark, Mexico, Austria, India, Spain, Poland, Finland, Czechoslovakia, and the Netherlands
Mode of inheritance, demographics, age, and survival
Gender was reported for 105 patients in this review, and consisted of 54 males (51%) and 51 females (49%). TTD is an autosomal recessive disease, and is therefore expected to have an equal distribution between males and females. There was one report65 suggesting the possibility of X-linked inheritance in a TTD patient with urea cycle dysfunction.
Age was reported for 110 of the 112 patients included in this study (fig 2). The age at last report ranged from 12 weeks to 47 years, with a median of 6 years. There was a median age of 6.5 years for males and 6 years for females. The ages ranged from 12 weeks to 44 years for males and 5 months to 47 years for females.
Figure 2.
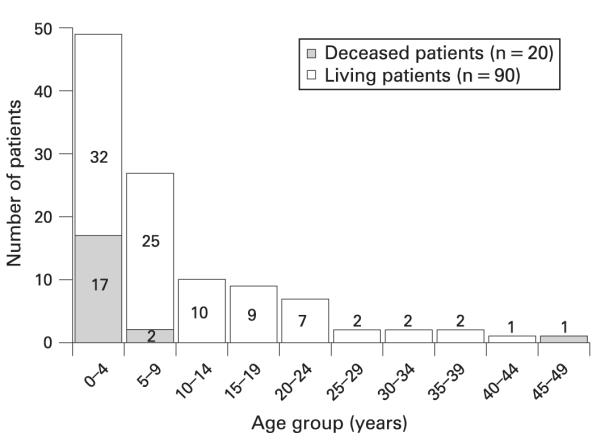
Age at last report in trichothiodystrophy (TTD) patients (n = 110). The number in each bar indicates the number of patients reported in the indicated age group among the 90 reported living TTD patients. The shaded portion of the bar indicates the number of patients who died in the indicated age range among the 20 reported deceased TTD patients.
Twenty patients were reported as deceased, ranging in age from 12 weeks to 47 years (fig 2). All but one of these patients were under 10 years old and the median age at death was 3 years. The cause of death was pneumonia or other infection (especially sepsis) for 13 of these patients (fig 3).40,41,63,74,85,90,91,94,98,99,107 One additional patient85 died at 12 months after developing a fever, despite antibiotic use. The remaining patients died of drowning,62 cachexia and dehydration,98 respiratory failure,46 or a sudden or unexpected death.32,68 One patient32 who died suddenly had a history of frequent hospitalisations for respiratory and gastrointestinal illnesses, and thus may have also died from infectious complications. The oldest patient56 died at 47 years after presenting with generalised oedema and urinary retention, which progressed to coma. Cause of death was not reported for one patient.111
Figure 3.
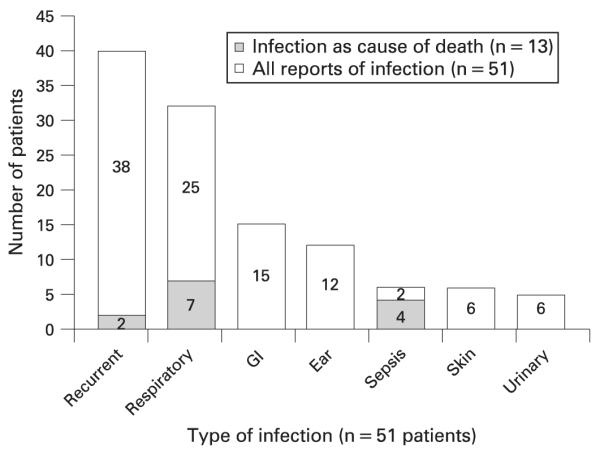
Infections reported (n = 51) in trichothiodystrophy patients. The number in each bar indicates the number of patients reported with the indicated type of infection among the 51 reported patients. A patient may have more than one reported infection. The shaded portion of the bar indicates the number of patients with the indicated type of infection as the cause of death among the 13 cases who died of infection. Aetiologies of infections included bacterial, fungal and viral. GI, gastrointestinal.
Kaplan–Meier analysis of the reported deaths indicated that at age 3 years there was 10.7% probability of reported death, and by age 9 years the probability had increased to 21.3%. This represents an approximately 20-fold higher mortality compared to the US general population. While we would expect that unusual and severe outcomes might be preferentially reported, this large number of deaths at a young age highlights the potential severity of TTD in the neonatal and early childhood period.
Recessively inherited disorders occur more commonly in populations where consanguinity is frequent. This literature review revealed 17% consanguinity, 49% non-consanguinity and 36% unreported. None of the parents were reported to have TTD. Data were given on the presence or absence of siblings in 80 patients. Thirty-seven patients have a sibling who is also described as a patient in this review (represents 18 families for these 37 patients). The total number of siblings ranged from 0 to 9, with a median of 1.
Spectrum of clinical abnormalities reported in TTD patients
Figure 4 displays the common clinical features reported for each patient in the format of a clinical array. Each column represents one patient, with each clinical feature indicated as present, absent, or not mentioned in the report. The columns are grouped to facilitate identification of patients by gender with features of PIBI(D)S (28%), IBI(D)S (20%), BI(D)S (16%) and those that did not fit these categories (36%). Because decreased fertility (D) is age dependent and difficult to quantify from these reports, this feature was not included in this figure. Instead, we have indicated patients with gonadal dysgenesis. Arraying patients in this format highlights those who fit the clinical criteria defined by these acronyms and those where the acronym inadequately describes the clinical presentation. Other commonly reported clinical features are also shown in this figure, including abnormal characteristics at birth, pregnancy complications, ocular abnormalities, and infections. DNA repair abnormalities that were identified are also shown.
Figure 4.

A clinical array of features reported in the literature on 112 trichothiodystrophy patients. Each column of rectangles represents clinical features of one reported patient. Presence or absence of each feature is indicated in each rectangle of a column. Abnormal clinical features reported are indicated by “1” in a coloured rectangle. Normal reported features are indicated by “0” in a tan rectangle. Unreported features are blank. The rows represent P (yellow)—photosensitivity (n = 47 cases); I (orange)—ichthyosis (n = 73 cases); B (powder blue)—brittle hair or hair shaft abnormality (n = 108); II (pink) —intellectual impairment (n = 96 cases); GD (grey) —gonadal dysgenesis (n = 16 cases); BC (light green) —abnormal birth characteristics (n = 62 cases); PC (dark green) —pregnancy complications (n = 31 pregnancies); O (maroon)—ocular abnormality (n = 57 cases); IN (royal blue) —infections (n = 51 patients); D (red)—DNA repair abnormality (n = 41); D in red rectangle—XPD (n = 32 cases); B in striped red rectangle—XPB (n = 2 cases); A in striped red rectangle—TTDA (n = 2); N in striped pink rectangle—TTDN1 (n = 6 cases); I in red rectangle—cellular ultraviolet (UV) hypersensitivity, gene not determined (n = 5); G—gender (n = 105 patients); blue rectangles—males (n = 54 cases); pink rectangles—females (n = 51 cases). Patients whose clinical features fulfil the criteria for PIBIDS (28%), IBIDS (20%), BIDS (16%) and those that do not (OTHER) (36%), are grouped by bold outline (decreased fertility is ignored in this grouping due to inability to assess in children.)
Skin findings
Seventy-nine per cent of the 112 reported TTD patients had skin abnormalities (table 2). Seven patients were reported to have normal skin and 16 reports did not include any skin descriptions. The most frequently reported skin finding was ichthyosis (65%) (table 2, fig 4). Of the 73 patients with ichthyosis, 27 had collodion membrane at birth.
Table 2.
Frequency of skin abnormalities reported (n=89) in trichothiodystrophy patients
| Patients No. (%) |
|
|---|---|
| Skin abnormality | 89 (79) |
| Ichthyosis (all forms) | 73 (65) |
| Lamellar ichthyosis | 10 (9) |
| Photosensitivity | 47 (42) |
| Eczema | 9 (8) |
| Freckles | 8 (7) |
| Dry skin, NOS | 7 (6) |
| Erythroderma | 5 (4) |
| Other* | 20 (18) |
| Normal skin; not reported | 7 (6); 16 (14) |
| Total | 112 (100) |
NOS, not otherwise specified.
“Other” refers to telangiectasia (5 patients), pruritis (4), folliculitis (3), cheilitis (3), follicular keratosis (3), hypohidrosis (3), haemangioma of skin (2), poikiloderma (2).
Ten patients were reported to have lamellar ichthyosis and six of these had collodion membrane at birth. Ichthyosis was seen in almost all age groups.
The second most frequent skin finding reported was photosensitivity (42%).
Seventy per cent of the 112 patients in this study had photosensitivity and/or ichthyosis (28% of all patients had both photosensitivity and ichthyosis) (fig 4). Twenty-seven of the 47 patients with photosensitivity were reported as having mutations in XPD, one in XPB, and one in TTDA (table 3, fig 4). Four patients were not assigned to a complementation group, but were found to have cellular UV hypersensitivity.31,59,100 Although the XPD gene mutation has been reported to be associated with the clinical finding of photosensitivity, there was one case of a TTD patient112 with an XPD gene mutation reported as non-photosensitive. Other cutaneous findings include dry skin (21%), eczema (8%), and freckles (7%). Freckling is generally associated with XP and not TTD. Two of the TTD patients45 with freckles were reported to also have XP (the XP/TTD complex), and one of them had a skin cancer. Six other patients with freckles were not reported to have skin cancer.32,41,58,81 One additional TTD patient54 had a well differentiated, invasive squamous carcinoma on his nose, but was not reported to have freckles. Other more rare skin findings include two reports of haemangioma84,105 and three reports of cheilitis.13,22,65
Table 3.
Cellular studies reported (n=58) in trichothiodystrophy patients
| Gene defect | Patients No. (%) |
|---|---|
| Reduced DNA repair | 41 (37) |
| XPD | 32 (29) |
| XPB | 2 (2) |
| TTD-A | 2 (2) |
| TTDN1 | 6 (5) |
| Normal DNA repair; not reported | 11 (10); 54 (48) |
| Total | 112 (100) |
TTD, trichothiodystrophy; XP, xeroderma pigmentosum.
Nail abnormalities (table 4) were reported in 70 patients (63%). The most frequent nail abnormality reported was onychodystrophy in 41 patients (37%), which included dysplasia, dystrophic nails, thickening or yellow discoloration. Other common nail findings were brittle nails (14%), hypoplasia (13%), and koilonychia (12%).
Table 4.
Frequency of hair and nail features (n=112) in reported trichothiodystrophy patients
| Patients No. (%) |
|
|---|---|
| Hair feature | 112 (100) |
| Brittle hair or hair shaft abnormality | 108 (96) |
| Brittle hair | 98 (88) |
| Hair shaft abnormality | 76 (68) |
| Tiger tail | 82 (73) |
| Decreased sulfur or cystine | 79 (71) |
| Sparse hair | 54 (48) |
| Alopecia | 44 (39) |
| Hair loss with fever or infection | 8 (7) |
| Dry hair | 24 (21) |
| Fine hair | 16 (14) |
| Slow growing hair | 8 (7) |
| Long hair | 5 (4) |
| Nail feature | 70 (63) |
| Onychodystrophy* | 41 (37) |
| Brittle nails | 16 (14) |
| Hypoplasia | 15 (13) |
| Koilonychia | 13 (12) |
| Splitting (onychoschizia); peeling | 8 (7) |
| Ridging | 7 (6) |
| Other† | 3 (3) |
| Normal nails; not reported | 14 (13); 28 (25) |
| Total | 112 (100) |
We collated reports of dysplasia, dystrophic nails, thickening and yellow discoloration as onychodystrophy.
Slow growing nails (2 patients) and soft nails (1).
Hair and nail findings
Hair abnormalities were reported in all 112 patients (table 4, fig 4). They are a defining feature of TTD, and were part of the inclusion criteria. The most frequent hair findings were brittle hair or hair shaft abnormalities (96%), tiger tail banding (73%), and decreased sulfur or cystine (71%). Two patients with the XP/TTD complex were reported not to have brittle hair, but did have decreased sulfur, tiger tail banding with polarised microscopy, and XPD mutations.45 Sparse hair (48%) and alopecia (39%) were also commonly reported. Eight patients had hair loss with fever or infection.50,58,59,62,68,87,107 Although TTD is usually associated with short hair (due to its brittle and easily breakable nature), there were also five reports of patients with long or normal length hair,36,38,45,78,115 including 1 XP/TTD patient.45
Neurologic findings
Neurologic abnormalities were reported in 100 patients (table 5, fig 4). Developmental delay or intellectual impairment was reported in 86% of patients, and spanned all age groups. These usually presented as failing to achieve developmental milestones, such as sitting, walking, or talking, on time.112 Eleven of the 16 patients who were not reported to have developmental delay or intellectual impairment were <5 years old.23,38,46,61,78,84,90,107 Of the patients with developmental delay or intellectual impairment, 41 also had impaired motor control or psychomotor retardation. Seventeen of the patients with neurologic abnormalities were also described as having notably sociable or outgoing behaviour. This outgoing, sociable interaction is also a feature of patients with Cockayne syndrome (CS).11
Table 5.
Frequency of neurologic features reported (n=100) in trichothiodystrophy patients
| Patients No. (%) |
|
|---|---|
| Developmental delay or intellectual impairment | 96 (86%) |
| Intellectual impairment | 84 (75%) |
| Developmental delay | 76 (68%) |
| Impaired motor control/psychomotor retardation | 41 (37%) |
| Sociable/outgoing behaviour | 17 (15%) |
| Clinical neurologic findings | 84 (75%) |
| Microcephaly | 46 (50%) |
| Abnormal gait/ataxia | 29 (26%) |
| Audiologic exam performed: normal hearing; sensorineural hearing loss |
20 (18%); 5 (4%) |
| Abnormal deep tendon reflex: increased; decreased | 15 (13%); 1 (1%) |
| Electroencephalogram: normal; abnormal | 14 (13%); 13 (12%) |
| Abnormal muscle tone: increased; diminished | 8 (7%); 11 (10%) |
| Nerve conduction velocity performed: normal; slow | 9 (8%); 3 (3%) |
| Spasticity | 11 (10%) |
| Intention tremor | 8 (7%) |
| Seizure | 7 (6%) |
| Paresis/plegia | 6 (5%) |
| Dysarthria | 5 (4%) |
| Pyramidal signs | 5 (4%) |
| Peripheral neuropathy | 2 (2%) |
| Neuroimaging abnormality | 26 (23%) |
| Dysmyelination | 16 (14%) |
| Atrophy: cerebellar; cortical | 5 (4%); 3 (3%) |
| Dilated ventricles | 4 (4%) |
| Calcifications | 2 (2%) |
| Other* | 5 (4%) |
| No abnormality reported | 12 (11%) |
| Total | 112 (100%) |
“Other” refers to partial agenesis of corpus callosum (1 patient), slight widening of subarachnoid spaces (1), thin corpus callosum (1), cerebral infarction (1), focal grey matter heterotopia and acute necrotising encephalopathy (1).
Intelligence quotient (IQ) was given for 21 patients. These tests included Terman–Merrill (three patients, IQ range 25– 40),89 Wechsler Intelligence Scale for Children (two patients, IQ range 45–89),25,39 Stanford–Binet (three patients, IQ range 32– 79)14,15,93 and not specified or other exam (such as Leiter scale and Ruth Griffiths test) (13 patients, IQ range 34– 88).17,27,37,60,64,69,71,76,77,79,83 As a result, an average value could not be determined. Ichthyosis was closely linked to developmental delay since 67 (92%) of 73 patients reported with ichthyosis were also reported to have developmental delay.
Other abnormal neurologic findings described include microcephaly (50%), abnormal gait (26%), and increased deep tendon reflexes (13%). Audiologic examination was performed in 25 patients, and found normal hearing in 20 and sensorineural hearing loss in the other five. Nine patients were reported to have high pitched/raspy voice14,17,57,73,76,77,82 and one patient had dysphonia.40 Six patients were reported to have attention deficit or hyper-activity22,27,50,69,81,89 and three patients as autistic-like.75,81,101
Neuroimaging abnormalities were given in 23% of patients. The most common findings were dysmyelination (14%), cerebellar atrophy (4%), and dilated ventricles (4%), which are similar to features found in CS.11 One patient had a progressive encephalopathy with ataxia and a gradual deterioration of previously acquired skills.63 In one patient,70 an attack of measles at age 4 years was reported to be followed by general disability, and according to his mother a regression of development, but subsequent to that had slow progress with no further degeneration. Another patient,56 however, did not change during a 30 year period. The electroencephalogram (EEG) findings were reported in 27 patients, of which 14 were normal and 13 were abnormal (four of these patients had seizures).25,60,113
Five cases reported “mild TTD”, in which the patients had involvement of only hair, skin or nails.23,37,38,84 Two of these patients38 had abnormal nails and one84 had dry skin, but none had the neurologic abnormalities seen in many TTD patients. No gene defect was reported for these patients.
Facial dysmorphism was reported in 66% of patients (table 6). These included microcephaly (50%), large or protruding ears (30%), and micrognathia (29%). As in CS, there have been descriptions of TTD patients with aged (9%) or “bird-like” appearances (8%).
Table 6.
Facial dysmorphism reported (n=74) in trichothiodystrophy patients
| Patients No. (%) |
|
|---|---|
| Facial dysmorphism | 74 (66) |
| Microcephaly | 56 (50) |
| Large or protruding ears | 34 (30) |
| Micrognathia | 33 (29) |
| Aged appearance | 10 (9) |
| “Bird-like” | 9 (8) |
| High arched palate | 8 (7) |
| Epicanthal fold | 8 (7) |
| Frontal bossing | 5 (4) |
| Hypotelorism; hypertelorism | 6 (5); 3 (3) |
| Craniosynostosis | 1 (1) |
| Not reported | 38 (34) |
| Total | 112 (100) |
Growth abnormalities
Eighty-one per cent of patients were reported to have either low height and/or weight (which includes six patients described as having “growth retardation”) (fig 4). Sixty-one per cent of patients had both short stature and low weight or poor weight gain. An additional 13 patients27,45,56,60 had short stature with either normal or unreported weight. Six patients had normal height and weight.27,38,62,84,97,113
Gonadal dysgenesis
Sixteen patients (14%) had sexual/reproductive abnormalities reported (fig 4). Thirteen of these patients had hypogonadism15,26,40,45,56,71,76,77,86,89,101 of which two were females40,56 and nine had cryptorchidism.15,25,26,40,58,73,76,77,86 Two cases reported delayed pubertal development.70,111 Additional genitourinary abnormalities in females included poor sexual maturation14,22 and partial panhypopituitarism.114
Pregnancy and birth characteristics
Thirty-four patients overall were reported to have parents in good health. When the TTD patients were born, the median reported maternal age was 25 years (based on 16 patients) and the median reported paternal age was 27 years (14 patients).
Two previously unrecognised findings not commonly associated with TTD are abnormal characteristics at birth (fig 5) and pregnancy complications (fig 6). Abnormal characteristics at birth were reported in 62 patients (55%) (fig 5). The most common finding was low birth weight (defined as birth weight <2500 g or specified as being low) which was reported in 41 patients (37%). In the USA for the year 2005 8.2% of infants were born with low birth weight (<2500 g)116 The actual birth weight was specified for 53 patients and ranged from 0.94–4 kg, with a median of 2.2 kg. This is much smaller than the median birth weight of 3.0–3.5 kg of infants born between 37–40 weeks of gestation in the US general population.116 Five additional cases specified low birth weight without giving a value.14,85,98,107 The length of gestation was reported in 53 patients and ranged from 25–42 weeks. The median gestational age for all reported cases was 37 weeks. Thirty-two of these patients (29%) were born prematurely (<37 weeks). Apgar scores were given for eight patients, two of which76,100 were <7 at 5 min, indicating perinatal asphyxia. Twenty-nine of the infants (26%) had a collodion membrane at birth. Twenty-seven of these also were reported to develop ichthyosis (six of which were lamellar ichthyosis). For the two patients with collodion presentation not reported to have icthyosis, one paper had very limited information about the patient,46 and the other paper only reports that the patient later had dry scaly skin.76 Thirteen patients had brittle or abnormal hair. There were 14 patients (13%) reported to have short birth length22,34,40,45,68,70,81,85,98,99,106 and eight patients (7%) to have small birth head circumference.40,85,98,99,106 Values for birth length were given for 18 patients, ranging from 36–51 cm, with a median of 46 cm. Values for birth head circumference were given for 10 patients, ranging from 28–35 cm, with a median of 31 cm. Five additional patients were specified to have low birth length and head circumference, but no value was given.85,98 Cryptorchidism was reported for nine (17%) of the boys. Congenital cataracts were identified in eight (7%) of the reported cases. This is also a feature of CS and is greatly elevated compared to the frequency of 1–2.5 per 10 000 live born infants117 in the general population. In addition, five patients were described as having infections (including respiratory infections) in the neonatal period (fig 3).
Figure 5.
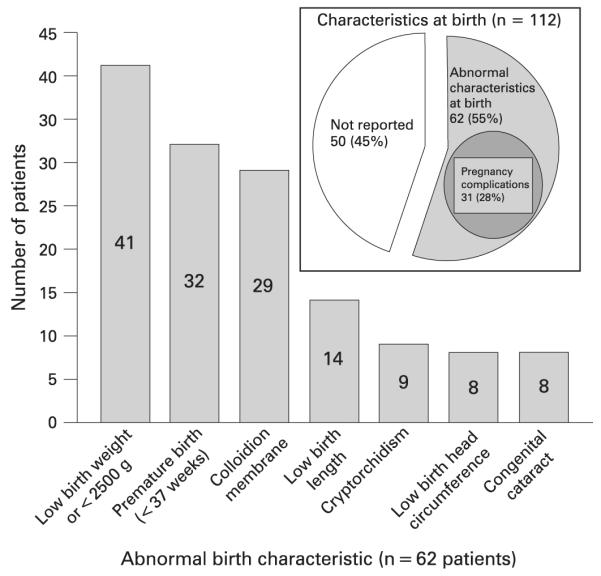
Abnormal characteristics at birth reported (n = 62) in trichothiodystrophy patients. The number in each bar indicates the number of patients reported with the indicated birth characteristic among the 62 reported patients. A patient may have more than one abnormal birth characteristic. The inset shows the proportion of the 112 cases reporting abnormal birth characteristics, pregnancy complications or both.
Figure 6.

Reported course of pregnancy in mothers delivering trichothiodystrophy (TTD) patients (n = 60). The number in each bar indicates the number of TTD pregnancies reported with the indicated pregnancy complication among the 60 pregnancies detailed in the reports. The pregnancies with complications are indicated with shaded bars. A pregnancy may have more than one complication reported. The inset shows the proportion of the 112 cases reporting pregnancy complications. The bar labelled “Other” refers to bleeding (2 patients), oligohydramnios (1), placental abnormalities (1). IUGR, intrauterine growth retardation.
The course of pregnancy refers to the pregnancy of the patient’s mother, when she was pregnant with the reported TTD case (fig 6). There was no information reported for 52 (48%) of the pregnancies. This is not surprising since many of the reports were in the dermatologic literature. In 29 of the 112 cases (26%) the pregnancy was described as uncomplicated; however, eight of these neonates had abnormal characteristics at birth. Pregnancy complications were reported in 31 cases (28%) and the TTD neonates from these pregnancies all had abnormal characteristics at birth (fig 5). The reports contained information on gestational age at birth and birth weight of the newborns. Twenty-three patients (21%) had IUGR stated in the report or alternatively, which we determined as low weight for the gestational age at birth.18 Pre-eclampsia was reported in eight pregnancies (7%)22,32,39,68,94,100,105,115 and eclampsia or seizures in three.17,63,108 There did not seem to be a correlation between IUGR and pre-eclampsia, as only three cases had both.32,68,100 In addition, one pregnancy85 was reported as having an abnormal prenatal screening test (elevated maternal serum α-fetoprotein). There were 13 cases of caesarean section and four cases of breech presentation.57,73,99,108 The caesarean sections were performed secondary to both maternal indications (five cases) and fetal indications (eight cases). The maternal indications were pre-eclampsia (three cases),32,39,105 toxaemia and seizures (one)63 and abruptio placenta (one).26 Fetal indications included IUGR (four cases),40,75,85 breech presentation (two),57,99 fetal distress (one)69 and fetal asphyxia (one).74 Other pregnancy complications reported were bleeding (two cases), oligohydramnios (one) and placental abnormality (one). Pregnancy loss was reported in five pregnancies of mothers who also had a child with TTD. One loss was an intrauterine fetal demise at 19 weeks 4 days of gestation; the other four losses were described as spontaneous abortions or miscarriage with no gestational age provided.32,90,97–99
In addition to the patients described in this review, five additional patients were diagnosed with TTD in utero by prenatal diagnostic methods. Prenatal diagnosis was reported in families with a previous child diagnosed with TTD. None of the pregnancies with the affected fetuses went to term. Methods of prenatal diagnosis reported included fetal hair biopsy and DNA repair measurements.90,98,100,118,119 One study measured UV induced unscheduled DNA synthesis (UDS) in cultivated amniotic fluid cells at 17 weeks gestation. After a therapeutic abortion, the diagnosis was confirmed by severe DNA excision repair defect in fetal skin fibroblasts. While UV induced UDS cannot differentiate among different DNA repair abnormalities,11 this family had a previous child with diagnosed TTD.100 During a later pregnancy in the same reported family, prenatal diagnosis was made by chorionic villus sampling at 9 weeks gestation and finding quantitatively normal DNA excision repair.119 Another study that same year looked at two pregnancies using DNA repair defects in trophoblasts (at 9 weeks gestation) or amniotic cells (at 21 weeks gestation) and then further supported by fetal hair analysis.98 Alkaline comet assay (single cell gel electrophoresis assay) was performed on amniotic or chorionic villus cells to diagnose a fetus as having TTD.118 A later study used endoscopically guided fetal eyebrow biopsy during the second trimester, and found tiger tail banding under polarised light.90
Ocular abnormalities and infection
Ocular abnormalities were reported in 51% of patients (fig 7). Thirty-two of these patients had cataracts, of which 20 were specified as bilateral and eight as congenital (fig 5). The median age of reported patients with cataracts was 7.5 years, and all but one patient were <25 years old. Three patients15,70,99 were reported to have surgery to correct their cataracts. Other ocular findings include nystagmus (14%) and strabismus (10%).
Figure 7.
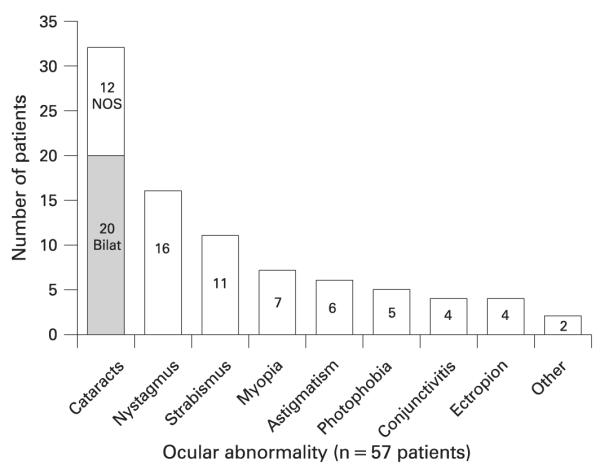
cular abnormalities reported (n = 57) in trichothiodystrophy (TTD) patients. The number in each bar indicates the number of TTD cases reported with the indicated ocular abnormality among the 57 patients detailed in the reports. “Bilat” in shaded bar—bilateral cataracts; of the 32 patients with cataracts, 20 were reported as bilateral and 8 as congenital. NOS—cataracts not otherwise specified. “Other” refers to dry eyes (1 patient) and retinal pigmentation (1). A patient may have more than one ocular abnormality reported.
Infections were described in 51 patients (fig 3). Fourteen patients were described as having infections (especially respiratory infections) within the first year of life, including five in the neonatal period. Forty patients (36%) had recurrent infections. Reported infections were most commonly respiratory (29%), gastrointestinal (13%), and ear (11%). Recurrent urinary infections were reported in five patients, all starting younger than age 5 years.32,66,69,73,83 Aetiologies of infections included bacterial, fungal and viral. Two patients38,91 were reported to have hypogammaglobulinaemia, for which they both received intravenous immunoglobulin. One patient91 was reported to receive prophylactic trimethoprim–sulfamethoxazole, but was also the only patient reported to have combined immunodeficiency. Three patients41 with recurrent infections in childhood were reported in adolescence to no longer be prone to infections. In addition, patients were reported as having asthma or allergies (five patients)15,17,34,51,69 and hypergammaglobulinaemia (two patients).56,66 Thirteen patients40,41,46,63,85,90,91,94,98,99,107 died of infection, which mostly consisted of respiratory infection or sepsis. The immune system of one patient with combined immunodeficiency was studied in two papers. One found the patient to have defective dendritic cell maturation and the second found decreased T cell regulation repertoire complexity suggesting a possible T cell regulation abnormality.49,91
Skeletal and dental abnormalities
Skeletal and dental findings are listed in table 7. All except one47 of these 46 patients also had neurologic abnormalities. Radiographic bone abnormalities were reported in 38% patients. The most common findings were osteosclerosis (14%), delayed bone age (13%), and osteopenia (9%). When specified, the osteosclerosis was usually axial, and the osteopenia distal. Four patients had both osteosclerosis and osteopenia.17,51,63,106 Seven additional patients were reported as having normal bone age, and no radiographic bone abnormalities.
Table 7.
Skeletal and dental abnormalities (n=46) in reported trichothiodystrophy patients
| Patients No. (%) |
|
|---|---|
| Radiographic bone abnormality | 43 (38) |
| Osteosclerosis* | 16 (14) |
| Delayed bone age | 14 (13) |
| Osteopenia | 10 (9) |
| Kyphosis | 7 (6) |
| Coxa valga | 4 (4) |
| Other† | 8 (7) |
| Joint abnormality | 15 (13) |
| Contractures‡ | 8 (7) |
| Joint dislocation | 4 (4) |
| Other§ | 7 (6) |
| Tooth abnormality | 23 (21) |
| Caries | 21 (19) |
| Enamel hypoplasia | 4 (4) |
| Other¶ | 4 (4) |
| Normal teeth | 18 (16) |
| Normal or no skeletal or tooth abnormality reported | 66 (59) |
| Total | 112 (100) |
4 patients had both osteosclerosis and osteopenia.
Pectus excavatum (3 patients), scoliosis (2), hallux valgus (2), bilateral hammer toe deformities (2).
Contractures of hips and knees (3), and hands (3).
Clinodactyly of 5th finger, short limbs, tapering fingers, syndactly, joint hypermobility.
Dystrophic teeth (2) and hypoplastic teeth (enamel not specified) (2).
The most common joint abnormalities were contractures (7%) and joint dislocation (4%). Contractures were of hip and knees (three patients)51,69,82 and hands (three patients).56,71,103 Subluxation was of the hip (three patients)51,56,114 and toes (one patient).76 The most common tooth abnormality was caries (19%). Eleven of these 21 patients had severe caries.13,17,22,47,57,58,69,72,76,77,99
Cardiac and hepatic abnormalities
In addition, cardiac defects were noted in eight patients and included cardiomyopathy, pulmonic stenosis and ventricular septal defect.22,74,75,85,107 Three additional patients were reported to have a murmur, but no cardiac defect was identified.36,63,72 Two patients85 were reported to have multiple liver haemangioendotheliomas.
Haematologic abnormalities
Haematologic abnormalities were reported in 24 patients (table 8). These findings consisted of anaemia (12%), low mean corpuscular volume (MCV) (9%), neutropenia (9%), and elevated haemoglobin A2 (7%). Two cases69,76 of anaemia were due to iron deficiency. Eight TTD patients111 with XPD mutations were reported as having “haematologic features of beta-thalassemia trait, and reduced levels of beta-globin synthesis and beta-globin mRNA”. The cause of anaemia in the remaining three patients38,54,72 was Coombs positive haemolytic anaemia, sideroblastic anaemia, and unspecified. Twenty-one per cent of patients had either a normal complete blood cell count (CBC) or routine blood analysis.
Table 8.
Haematologic abnormalities reported (n=24) in trichothiodystrophy patients
| Patients No. (%) |
|
|---|---|
| Haematologic abnormality | 24 (21) |
| Anaemia* | 13 (12) |
| Low MCV | 10 (9) |
| Neutropenia | 10 (9) |
| Elevated haemoglobin | A2 8 (7) |
| Normal; not reported | 24 (21); 64 (57) |
| Total | 112 (100) |
MCV, mean corpuscular volume.
Two patients reported with iron deficiency.
DNA repair abnormalities and gene defects
DNA repair abnormalities or gene defects were reported in 41 patients. Thirty-two patients were reported as having mutations in XPD, two in XPB and two in TTDA (table 3, fig 4). Five patients,59,80,100,108 were reported to have cellular UV hypersensitivity, with no specific gene defect determined. Six additional patients7 were reported as having mutations in the newly discovered TTDN1 gene with unknown function. Eleven patients were reported to not have a DNA repair abnormality.37,57,67,72,75,85,90,107 Of the 32 patients with mutations in XPD, 27 were reported to have photosensitivity. While most of the cases with a DNA repair abnormality were from patients with photosensitivity, this might be due to ascertainment bias in that photosensitivity is a reason to suspect a DNA repair abnormality. Genotype–phenotype correlation is best studied on a group of patients who are studied in the same manner. It may not be valid to compare phenotypes from reports with different extents of clinical information provided.
DISCUSSION
Birth abnormalities, pregnancy complications and increased mortality in TTD
TTD has substantial morbidity and mortality in the neonatal and childhood years. There was an approximately 20-fold increase in the probability of death in reported TTD children ≤10 years of age compared to the US general population. This increased mortality in TTD is neither widely recognised nor well understood. However, there may be bias in reporting more severe cases, thereby suggesting a worse prognosis for TTD.
This study documents the wide spectrum of severity within TTD. The high frequencies of reported abnormal characteristics at birth and pregnancy abnormalities suggest that childhood and neonatal morbidity can begin in the prenatal period. Fifty-three per cent of reported patients had abnormal characteristics at birth and 28% of pregnancies were abnormal. The relatively young median parental ages (maternal: 25 years; paternal: 27 years) at birth of TTD patients indicates that advanced parental age was a not a factor in the frequency of pregnancy abnormalities or in the development of TTD. This surprisingly large number of reports of pregnancy abnormalities suggests that the pathophysiology of TTD involves a developmental abnormality directly affecting the pregnancy. This adds a complex dimension to understanding the clinical phenotype of TTD. Some of the clinical features may be due to the effect of TTD on the affected patient; in addition, some of the clinical disease may be secondary to compromise resulting from maternal pregnancy abnormality.
Different mutations in the XPD or XPB genes can lead to TTD, XP or a clinical overlap of both, the XP/TTD complex. Genes that are defective in TTD, XPB, XPD and TTDA, are components of the basal transcription factor, TFIIH, as well as the nucleotide excision repair pathway.5,6,120,121 A current theory suggests that mutations in these genes in patients with XP predominantly impede DNA repair, while mutations in the same genes in TTD patients predominantly affect transcription.1,121 Thus, XP is a disease of progressive sunlight induced degeneration of the skin.2,11 In contrast to XP, TTD is primarily a disorder of development which may be the consequence of transcriptional anomalies resulting from different defects in the same DNA repair genes.11,120 Thus the finding of elevated haemoglobin A2 and low red blood cell MCV that mimic thalassaemia without a defect in a haemoglobin gene was interpreted as a transcription defect in TTD patients with mutations in the XPD gene.111 The other developmental features of TTD may represent abnormalities in transcription of genes that are essential for normal pregnancy and fetal development. The high frequency of reported fetal abnormalities and maternal pregnancy complications in mothers of TTD patients suggests a role of the DNA repair/transcription genes in normal pregnancy and fetal development. CS, another rare genetic disease with defective DNA repair, shares some of the same clinical features as TTD including photosensitivity, short stature, developmental delay, IUGR, dysmyelination of the brain, and an outgoing social personality.2,6,122 CS is caused by CSA and CSB genes, which have a role in repair of actively transcribing genes.
Classification
Trichothiodystrophy is a rare multisystem disorder with a wide spectrum of clinical involvement. We were able to identify only 112 patients reported in the world’s literature who fit our criteria for inclusion into this study of TTD reports. These criteria allowed us to capture a large number of TTD cases from the literature where limited information was available on each patient. These criteria were not intended to be used for diagnosis of new patients where more extensive evaluation should be possible.3,4 The goal of this study was to assess the frequency of clinical features in order to better understand the spectrum of manifestations of TTD.1,123 The most common clinical features were brittle hair or hair shaft abnormalities (96%), intellectual impairment or developmental delay (86%), short stature (73%) and ichthyosis (65%). While it is useful to look at the frequency of different features across the broad population of patients, it is also important to know how often a set of clinical features occurs together (fig 4). Sixty-four per cent of patients had the clinical features to fit into the category of either PIBI(D)S, IBI(D)S or BI(D)S; however, the others (36%) did not. Almost all of the patients who had clinical features sufficient to fit into these designations had additional clinical manifestations not specified by the acronyms. In addition, even the broadest acronym, PIBIDS, does not include several major clinical features found to be more common than photosensitivity (42%) and decreased fertility, including abnormal characteristics at birth (53%), ocular abnormalities (51%), and infections (46%), which should be considered major clinical features of TTD. So these acronyms are poor descriptors of TTD patients’ clinical manifestations.
Van Neste115 suggested a classification system in 1989 based on increasing severity beginning with only hair defects. While this schema takes into account additional features of TTD beyond PIBIDS, it intrinsically implies a sequential pattern to the progression of disease severity. As seen in fig 4, not all patients fit into a uniform sequence. For example, although the Van Neste classification lists photosensitivity as a more severe case of TTD, some patients may have photosensitivity without ichthyosis or short stature. Van Neste’s classification was later expanded to include more features, such as chronic neutropenia or immunoglobulin deficiency, severe IUGR and basal ganglia calcifications.1
Multiple reported abnormalities in TTD
Surveys of reported clinical features have several weaknesses. These include ascertainment bias, leading to the reporting of patients who are more interesting and severe and the under-representation of more mildly affected patients. Reports vary with respect to thoroughness of clinical evaluation, leading to the probable underreporting of many features that may not have been evaluated. This suggests that the prevalence of many clinical features summarised here may under-represent their true frequencies. In addition, since we would expect milder phenotypes to be less likely to be reported, TTD may be much more common than the number of reported cases implies.
Neurologic abnormalities (86%) were frequently reported in TTD cases, manifesting most commonly as developmental delay, intellectual impairment, microcephaly, impaired motor control or psychomotor retardation. This high frequency may be an underestimate, since 11 of the 16 patients who were not reported to have developmental delay or intellectual impairment were <5 years old. In general, these findings were not found to be deteriorations in neurologic status, but rather were more suggestive of a chronic non-progressive condition. Two exceptions were reported. One patient63 had progressive encephalopathy and ataxia and a second patient70 had developmental regression after an episode of measles. This further supports an early developmental abnormality being a key factor leading to TTD neurologic involvement. In contrast, about 20% of XP patients, who have different mutations in many of the same genes as TTD patients,121 have neurological abnormalities which manifest as progressive degeneration.2,9 Recent studies have looked at the relationship between DNA repair defects and impaired neurologic development.11 The presence of ichthyosis may be a marker of a systemic developmental abnormality since more than 90% of the TTD patients reported to have ichthyosis also have developmental delay.
Infections were commonly (46%) reported and were often recurrent (36%). Sixty-five per cent of the 20 reported deaths were related to infections. This frequency and severity of infections suggests that the pathophysiology of TTD includes an immunologic abnormality. However, no consistent laboratory abnormality in the immune system has been identified in TTD patients.
TTD involves many medical specialties
Effective management of the multisystem abnormalities of TTD involves a multidisciplinary approach involving many medical specialties. Seventy-seven per cent of patients were (14 years old, and thus it is important for paediatricians to be aware of this disease. Sixty-three per cent of patients had abnormal characteristics at birth, signalling importance for the neonatologist. Twenty-three per cent were from abnormal pregnancies, which would bring these mothers to the attention of obstetricians. Twenty-nine per cent of patients had cataracts (median age 7.5 years), including eight with congenital cataracts, which, if undetected, can lead to vision impairment and interference with early childhood development and learning. The oldest TTD patient in the literature (47 years) was first seen by those researchers at age 17 with pruritis and urticaria. She also had symptoms in her first year of life, consisting of collodion baby, congenital hip subluxation, and psychomotor developmental delay.56
These patients may present to specialists in obstetrics, neonatology, paediatrics, ophthalmology, neurology, orthopaedics, internal medicine, rehabilitation medicine, immunology, infectious disease, haematology, genetics, or radiology in addition to dermatology. If properly aware, any of these specialists can make the diagnosis. Since prenatal diagnosis is possible, establishment of a diagnosis can identify the risk to future pregnancies. It is surprising that a disorder with such a broad range of multisystem abnormalities can be unified by the simple finding of tiger tailed banding under polarised microscopy. This very simple and inexpensive test can reliably establish a diagnosis in both the healthy adult with learning disabilities and the severely ill, collodion baby in the neonatal intensive care unit. This review characterises the wide spectrum of TTD and reinforces the importance of this simple screening test for patients with these multisystem findings. Greater recognition among a broad range of specialists can facilitate early diagnosis and treatment and identification of risk to future pregnancies.
Acknowledgements
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. We would like to thank Philip Rosenberg, PhD, Biostatistics Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute for assistance with the Kaplan Meier analysis and Melissa Meredith, MD, National Human Genome Research Institute for assistance in evaluation of obstetric information. Christine Liang, MD, made the photomicrographs of the TTD hairs while she was a Howard Hughes Medical Institute-NIH Research Scholar in our laboratory. An abstract of this work was presented at the 68th Annual Meeting of the Society for Investigative Dermatology, May 2007.124
Footnotes
Competing interests: None.
REFERENCES
- 1.Itin PH, Sarasin A, Pittelkow MR. Trichothiodystrophy: update on the sulfurdeficient brittle hair syndromes. J Am Acad Dermatol. 2001;44:891–920. doi: 10.1067/mjd.2001.114294. [DOI] [PubMed] [Google Scholar]
- 2.Ruenger TM, DiGiovanna JJ, Kraemer KH. Hereditary Diseases of genome instability and DNA repair. In: Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, editors. Fitzpatrick’s dermatology in general medicine. 7 ed McGraw Hill; New York: 2008. pp. 1311–25. [Google Scholar]
- 3.Liang C, Kraemer KH, Morris A, Schiffmann R, Price VH, Menefee E, DiGiovanna JJ. Characterization of tiger tail banding and hair shaft abnormalities in trichothiodystrophy. J Am Acad Dermatol. 2005;52(2 Pt1):224–32. doi: 10.1016/j.jaad.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 4.Liang C, Morris A, Schlucker S, Imoto K, Price VH, Menefee E, Wincovitch SM, Levin IW, Tamura D, Strehle KR, Kraemer KH, DiGiovanna JJ. Structural and molecular hair abnormalities in trichothiodystrophy. J Invest Dermatol. 2006;126:2210–6. doi: 10.1038/sj.jid.5700384. [DOI] [PubMed] [Google Scholar]
- 5.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA repair and mutagenesis. 2 ed ASM Press; Washington DC: 2006. [Google Scholar]
- 6.Kraemer KH, Ruenger TM. Genome instability, DNA repair and cancer. In: Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, editors. Fitzpatrick’s dermatology in general medicine. 7 ed McGraw Hill; New York: 2008. pp. 977–86. [Google Scholar]
- 7.Nakabayashi K, Amann D, Ren Y, Saarialho-Kere U, Avidan N, Gentles S, MacDonald JR, Puffenberger EG, Christiano AM, Martinez-Mir A, Salas-Alanis JC, Rizzo R, Vamos E, Raams A, Les C, Seboun E, Jaspers NG, Beckmann JS, Jackson CE, Scherer SW. Identification of C7orf11 (TTDN1) gene mutations and genetic heterogeneity in nonphotosensitive trichothiodystrophy. Am J Hum Genet. 2005;76:510–6. doi: 10.1086/428141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kraemer KH, Lee MM, Scotto J. DNA repair protects against cutaneous and internal neoplasia: evidence from xeroderma pigmentosum. Carcinogenesis. 1984;5:511–4. doi: 10.1093/carcin/5.4.511. [DOI] [PubMed] [Google Scholar]
- 9.Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–50. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- 10.Kraemer KH, Lee MM, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol. 1994;130:1018–21. [PubMed] [Google Scholar]
- 11.Kraemer K, Patronas N, Schiffmann R, Brooks B, Tamura D, DiGiovanna J. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience. 2007;145:1388–96. doi: 10.1016/j.neuroscience.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Price VH. Strukturanomalien des Haarschaftes. In: Orfanos CE, editor. Haar und Haarkrankheiten. Gustav Fischer Verlag; Stuttgart: 1979. pp. 387–446. [Google Scholar]
- 13.Crovato F, Borrone C, Rebora A. Trichothiodystrophy—BIDS, IBIDS and PIBIDS? Br J Dermatol. 1983;108:247. doi: 10.1111/j.1365-2133.1983.tb00068.x. [DOI] [PubMed] [Google Scholar]
- 14.Jorizzo JL, Crounse RG, Wheeler CE., Jr Lamellar ichthyosis, dwarfism, mental retardation, and hair shaft abnormalities. A link between the ichthyosis-associated and BIDS syndromes. J Am Acad Dermatol. 1980;2:309–17. doi: 10.1016/s0190-9622(80)80043-0. [DOI] [PubMed] [Google Scholar]
- 15.Jorizzo JL, Atherton DJ, Crounse RG, Wells RS. Ichthyosis, brittle hair, impaired intelligence, decreased fertility and short stature (IBIDS syndrome) Br J Dermatol. 1982;106:705–10. doi: 10.1111/j.1365-2133.1982.tb11687.x. [DOI] [PubMed] [Google Scholar]
- 16.Baden HP, Jackson CE, Weiss L, Jimbow K, Lee L, Kubilus J, Gold RJ. The physicochemical properties of hair in the BIDS syndrome. Am J Hum Genet. 1976;28:514–21. [PMC free article] [PubMed] [Google Scholar]
- 17.Price VH, Odom RB, Ward WH, Jones FT. Trichothiodystrophy: sulfur-deficient brittle hair as a marker for a neuroectodermal symptom complex. Arch Dermatol. 1980;116:1375–84. doi: 10.1001/archderm.116.12.1375. [DOI] [PubMed] [Google Scholar]
- 18.Lubchenco LO, Hansman C, Dressler M, Boyd E. Intrauterine growth as estimated from liveborn birth-weight data at 24 to 42 weeks of gestation. Pediatrics. 1963;32:793–800. [PubMed] [Google Scholar]
- 19.Braun-Falco O, Ring J, Butenandt O, Selzle D, Landthaler M. [Ichthyosis vulgaris, growth retardation, hair dysplasia, tooth abnormalities, immunologic deficiencies, psychomotor retardation and resorption disorders. Case report of 2 siblings] Hautarzt. 1981;32:67–74. [PubMed] [Google Scholar]
- 20.Leupold D. [Ichthyosis congenita, cataract, mental retardation, ataxia, osteosclerosis and immunologic deficiency—a particular syndrome?] Monatsschr Kinderheilkd. 1979;127:307–8. [PubMed] [Google Scholar]
- 21.Salfeld K, Lindley MJ. [On the problem of combination of symptoms in ichthyosis vulgaris with bamboo hair formation and ectodermal dysplasia] Dermatol Wochenschr. 1963;147:118–28. [PubMed] [Google Scholar]
- 22.Pollitt RJ, Jenner FA, Davies M. Sibs with mental and physical retardation and trichorrhexis nodosa with abnormal amino acid composition of the hair. Arch Dis Child. 1968;43:211–6. doi: 10.1136/adc.43.228.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown AC, Belser RB, Crounse RG, Wehr RF. A congenital hair defect: trichoschisis with alternating birefringence and low sulfur content. J Invest Dermatol. 1970;54:496–509. doi: 10.1111/1523-1747.ep12259317. [DOI] [PubMed] [Google Scholar]
- 24.Tay CH. Ichthyosiform erythroderma, hair shaft abnormalities, and mental and growth retardation. A new recessive disorder. Arch Dermatol. 1971;104:4–13. [PubMed] [Google Scholar]
- 25.Jackson CE, Weiss L, Watson JH. “Brittle” hair with short stature, intellectual impairment and decreased fertility: an autosomal recessive syndrome in an Amish kindred. Pediatrics. 1974;54:201–7. [PubMed] [Google Scholar]
- 26.Happle R, Traupe H, Grobe H, Bonsmann G. The Tay syndrome(congenital ichthyosis with trichothiodystrophy) Eur J Pediatr. 1984;141:147–52. doi: 10.1007/BF00443212. [DOI] [PubMed] [Google Scholar]
- 27.Arbisser AI, Scott CI, Jr, Howell RR, Ong PS, Cox HL., Jr A syndrome manifested by brittle hair with morphologic and biochemical abnormalities, developmental delay and normal stature. Birth Defects Orig Artic Ser. 1976;12:219–28. [PubMed] [Google Scholar]
- 28.Howell RR, Arbisser AI, Parsons DS, Scott CI, Fraustadt U, Collie WR, Marshall RN, Ibarra OC. The Sabinas syndrome. Am J Hum Genet. 1981;33:957–67. [PMC free article] [PubMed] [Google Scholar]
- 29.Happle R, Traupe H, Grobe H, Bonsmann G. Author’s reply. Eur J Pediatr. 1984;142:234. doi: 10.1007/BF00443212. [DOI] [PubMed] [Google Scholar]
- 30.Chapman S. The trichothiodystrophy syndrome of Pollitt. Pediatr Radiol. 1988;18:154–6. doi: 10.1007/BF02387560. [DOI] [PubMed] [Google Scholar]
- 31.van Neste D, Caulier B, Thomas P, Vasseur F. PIBIDS: Tay’s syndrome and xeroderma pigmentosum. J Am Acad Dermatol. 1985;12(2 Pt 1):372–3. doi: 10.1016/s0190-9622(85)80062-1. [DOI] [PubMed] [Google Scholar]
- 32.Stefanini M, Lagomarsini P, Arlett CF, Marinoni S, Borrone C, Crovato F, Trevisan G, Cordone G, Nuzzo F. Xeroderma pigmentosum (complementation group D) mutation is present in patients affected by trichothiodystrophy with photosensitivity. Hum Genet. 1986;74:107–12. doi: 10.1007/BF00282072. [DOI] [PubMed] [Google Scholar]
- 33.Stefanini M, Vermeulen W, Weeda G, Giliani S, Nardo T, Mezzina M, Sarasin A, Harper JI, Arlett CF, Hoeijmakers JH. A new nucleotide-excision-repair gene associated with the disorder trichothiodystrophy. Am J Hum Genet. 1993;53:817–21. [PMC free article] [PubMed] [Google Scholar]
- 34.Vandenberghe K, Casteels I, Vandenbussche E, De Zegher F, De Boeck K. Bilateral cataract and high myopia in a child with trichothiodystrophy: a case report. Bull Soc Belge Ophtalmol. 2001;282:15–8. [PubMed] [Google Scholar]
- 35.Giglia-Mari G, Coin F, Ranish JA, Hoogstraten D, Theil A, Wijgers N, Jaspers NG, Raams A, Argentini M, van der Spek PJ, Botta E, Stefanini M, Egly JM, Aebersold R, Hoeijmakers JH, Vermeulen W. A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A. Nat Genet. 2004;36:714–9. doi: 10.1038/ng1387. [DOI] [PubMed] [Google Scholar]
- 36.Weeda G, Eveno E, Donker I, Vermeulen W, Chevallier-Lagente O, Taieb A, Stary A, Hoeijmakers JH, Mezzina M, Sarasin A. A mutation in the XPB/ERCC3 DNA repair transcription gene, associated with trichothiodystrophy. Am J Hum Genet. 1997;60:320–9. [PMC free article] [PubMed] [Google Scholar]
- 37.Alfandari S, Delaporte E, van Neste D, Lucidarme-Delespierre E, Piette F, Bergoend H. A new case of isolated trichothiodystrophy. Dermatology. 1993;186:197–200. doi: 10.1159/000247345. [DOI] [PubMed] [Google Scholar]
- 38.Baden HP, Katz A. Trichothiodystrophy without retardation: one patient exhibiting transient combined immunodeficiency syndrome. Pediatr Dermatol. 1988;5:257–9. doi: 10.1111/j.1525-1470.1988.tb00899.x. [DOI] [PubMed] [Google Scholar]
- 39.Battistella PA, Peserico A. Central nervous system dysmyelination in PIBI(D)S syndrome: a further case. Childs Nerv Syst. 1996;12:110–3. doi: 10.1007/BF00819509. [DOI] [PubMed] [Google Scholar]
- 40.Blomquist HK, Back O, Fagerlund M, Holmgren G, Stecksen-Blicks C. Tay or IBIDS syndrome. A case with growth and mental retardation, congenital ichthyosis and brittle hair. Acta Paediatr Scand. 1991;80:1241–5. doi: 10.1111/j.1651-2227.1991.tb11817.x. [DOI] [PubMed] [Google Scholar]
- 41.Botta E, Nardo T, Broughton BC, Marinoni S, Lehmann AR, Stefanini M. Analysis of mutations in the XPD gene in Italian patients with trichothiodystrophy: site of mutation correlates with repair deficiency, but gene dosage appears to determine clinical severity. Am J Hum Genet. 1998;63:1036–48. doi: 10.1086/302063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bracun R, Hemmer W, Wolf-Abdolvahab S, Focke M, Botzi C, Killian W, Gotz M, Jarisch R. Diagnosis of trichothiodystrophy in 2 siblings. Dermatology. 1997;194:74–6. doi: 10.1159/000246064. [DOI] [PubMed] [Google Scholar]
- 43.Broughton BC, Lehmann AR, Harcourt SA, Arlett CF, Sarasin A, Kleijer WJ, Beemer FA, Nairn R, Mitchell DL. Relationship between pyrimidine dimers, 6-4 photoproducts, repair synthesis and cell survival: studies using cells from patients with trichothiodystrophy. Mutat Res. 1990;235:33–40. doi: 10.1016/0921-8777(90)90020-6. [DOI] [PubMed] [Google Scholar]
- 44.Broughton BC, Steingrimsdottir H, Weber CA, Lehmann AR. Mutations in the xeroderma pigmentosum group D DNA repair/transcription gene in patients with trichothiodystrophy. Nat Genet. 1994;7:189–94. doi: 10.1038/ng0694-189. [DOI] [PubMed] [Google Scholar]
- 45.Broughton BC, Berneburg M, Fawcett H, Taylor EM, Arlett CF, Nardo T, Stefanini M, Menefee E, Price VH, Queille S, Sarasin A, Bohnert E, Krutmann J, Davidson R, Kraemer KH, Lehmann AR. Two individuals with features of both xeroderma pigmentosum and trichothiodystrophy highlight the complexity of the clinical outcomes of mutations in the XPD gene. Hum Mol Genet. 2001;10:2539–47. doi: 10.1093/hmg/10.22.2539. [DOI] [PubMed] [Google Scholar]
- 46.Brusasco A, Restano L. The typical ‘tiger tail’ pattern of the hair shaft in trichothiodystrophy may not be evident at birth. Arch Dermatol. 1997;133:249. doi: 10.1001/archderm.1997.03890380123028. [DOI] [PubMed] [Google Scholar]
- 47.Calvieri S, Rossi A, Amorosi B, Giustini S, Innocenzi D, Micali G, Rizzo R. Trichothiodystrophy: ultrastructural studies of two patients. Pediatr Dermatol. 1993;10:111–6. doi: 10.1111/j.1525-1470.1993.tb00033.x. [DOI] [PubMed] [Google Scholar]
- 48.Calvieri S, Giustini S, Nini G. Trichothiodystrophy: 2 cases. In: Wilkinson DS, editor. Clinical dermatology: The CMD case collection. Schattauer; Stuttgart: 1987. pp. 65–6. [Google Scholar]
- 49.Cancrini C, Romiti ML, Di Cesare S, Angelini F, Gigliotti D, Livadiotti S, Bertini E, Rossi P, Racioppi L. Restriction in T-cell receptor repertoire in a patient affected by trichothiodystrophy and CD4+ lymphopenia. Scand J Immunol. 2002;56:212–6. doi: 10.1046/j.1365-3083.2002.01122.x. [DOI] [PubMed] [Google Scholar]
- 50.Chen E, Cleaver JE, Weber CA, Packman S, Barkovich AJ, Koch TK, Williams ML, Golabi M, Price VH. Trichothiodystrophy: clinical spectrum, central nervous system imaging, and biochemical characterization of two siblings. J Invest Dermatol. 1994;103(5 Suppl):154S–8S. doi: 10.1111/1523-1747.ep12399493. [DOI] [PubMed] [Google Scholar]
- 51.Civitelli R, McAlister WH, Teitelbaum SL, Whyte MP. Central osteosclerosis with ectodermal dysplasia: clinical, laboratory, radiologic, and histopathologic characterization with review of the literature. J Bone Miner Res. 1989;4:863–75. doi: 10.1002/jbmr.5650040611. [DOI] [PubMed] [Google Scholar]
- 52.Coulter DL, Beals TF, Allen RJ. Neurotrichosis: hair-shaft abnormalities associated with neurological diseases. Dev Med Child Neurol. 1982;24:634–44. doi: 10.1111/j.1469-8749.1982.tb13674.x. [DOI] [PubMed] [Google Scholar]
- 53.Crovato F, Borrone C, Rebora A. The Tay syndrome (congenital ichthyosis with trichothiodystrophy) Eur J Pediatr. 1984;142:233–4. doi: 10.1007/BF00442459. [DOI] [PubMed] [Google Scholar]
- 54.Dahbi-Skali H, Benamar L, Benchikhi H, Lakhdar H, Pinel N. PIBIDS syndrome (trichothiodystrophy type F) and skin cancer: an exceptional association. Photodermatol Photoimmunol Photomed. 2004;20:157–8. doi: 10.1111/j.1600-0781.2004.00038.x. [DOI] [PubMed] [Google Scholar]
- 55.de Prost Y, Lemaistre R, Dupre A. Trichothiodystrophie associee a une ichtyose et a un retard statural et psychomoteur. Ann Dermatol Venereol. 1986;113:1016–7. [Google Scholar]
- 56.Feier V, Solovan C. [Trichothiodystrophy and hypereosinophilic syndrome, an unusual association] Ann Dermatol Venereol. 1994;121:151–5. [PubMed] [Google Scholar]
- 57.Fois A, Balestri P, Calvieri S, Zampetti M, Giustini S, Stefanini M, Lagomarsini P. Trichothiodystrophy without photosensitivity. Biochemical, ultrastructural and DNA repair studies. Eur J Pediatr. 1988;147:439–41. doi: 10.1007/BF00496431. [DOI] [PubMed] [Google Scholar]
- 58.Fortina AB, Alaibac M, Piaserico S, Peserico A. PIBI(D)S: clinical and molecular characterization of a new case. J Eur Acad Dermatol Venereol. 2001;15:65–9. doi: 10.1046/j.1468-3083.2001.00212.x. [DOI] [PubMed] [Google Scholar]
- 59.Foulc P, Jumbou O, David A, Sarasin A, Stalder JF. [Trichothiodystrophy: progresssive manifestations] Ann Dermatol Venereol. 1999;126:703–7. [PubMed] [Google Scholar]
- 60.Gillespie JM, Marshall RC, Rogers M. Trichothiodystrophy—biochemical and clinical studies. Australas J Dermatol. 1988;29:85–93. doi: 10.1111/j.1440-0960.1988.tb00369.x. [DOI] [PubMed] [Google Scholar]
- 61.Gummer CL, Dawber RP. Trichothiodystrophy: an ultrastructural study of the hair follicle. Br J Dermatol. 1985;113:273–80. doi: 10.1111/j.1365-2133.1985.tb02078.x. [DOI] [PubMed] [Google Scholar]
- 62.Hansen LK, Wulff K, Brandrup F. [Trichothiodystrophy. Hair examination as a diagnostic tool]. Ugeskr Laeger. 1993;155:1949–52. [PubMed] [Google Scholar]
- 63.Hersh JH, Klein LR, Joyce MR, Hordinsky MK, Tsai MY, Paller A, Hyzer R, Zax RH. Trichothiodystrophy and associated anomalies: a ariant of SIBIDS or new symptom complex? Pediatr Dermatol. 1993;10:117–22. doi: 10.1111/j.1525-1470.1993.tb00034.x. [DOI] [PubMed] [Google Scholar]
- 64.Hora RK, Murthy VS. Mental retardation, short stature and brittle hair (BIDS syndrome; hair brain syndrome) Indian J Pediatr. 1996;63:117–20. doi: 10.1007/BF02823882. [DOI] [PubMed] [Google Scholar]
- 65.Hordinsky MK, Briden B, Berry SA. Friable hair, urea cycle dysfunction, and trichothiodystrophy. A new X-linked genodermatosis. Curr Probl Dermatol. 1987;17:52–60. doi: 10.1159/000413474. [DOI] [PubMed] [Google Scholar]
- 66.Itin PH, Pittelkow MR. Trichothiodystrophy with chronic neutropenia and mild mental retardation. J Am Acad Dermatol. 1991;24(2 Pt 2):356–8. doi: 10.1016/0190-9622(91)70051-3. [DOI] [PubMed] [Google Scholar]
- 67.King MD, Gummer CL, Stephenson JB. Trichothiodystrophy-neurotrichocutaneous syndrome of Pollitt: a report of two unrelated cases. J Med Genet. 1984;21:286–9. doi: 10.1136/jmg.21.4.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kleijer WJ, Beemer FA, Boom BW. Intermittent hair loss in a child with PIBI(D)S syndrome and trichothiodystrophy with defective DNA repair-xeroderma pigmentosum group D. Am J Med Genet. 1994;52:227–30. doi: 10.1002/ajmg.1320520220. [DOI] [PubMed] [Google Scholar]
- 69.Kousseff BG, Esterly NB. Trichothiodystrophy, IBIDS syndrome or Tay syndrome? Birth Defects Orig Artic Ser. 1988;24:169–81. [PubMed] [Google Scholar]
- 70.Lehmann AR, Arlett CF, Broughton BC, Harcourt SA, Steingrimsdottir H, Stefanini M, Malcolm A, Taylor R, Natarajan AT, Green S. Trichothiodystrophy, a human DNA repair disorder with heterogeneity in the cellular response to ultraviolet light. Cancer Res. 1988;48:6090–6. [PubMed] [Google Scholar]
- 71.Lucky PA, Kirsch N, Lucky AW, Carter DM. Low-sulfur hair syndrome associated with UVB photosensitivity and testicular failure. J Am Acad Dermatol. 1984;11(2 Pt 2):340–6. doi: 10.1016/s0190-9622(84)70169-1. [DOI] [PubMed] [Google Scholar]
- 72.Lynch SA, de Berker D, Lehmann AR, Pollitt RJ, Reid MM, Lamb WH. Trichothiodystrophy with sideroblastic anaemia and developmental delay. Arch Dis Child. 1995;73:249–51. doi: 10.1136/adc.73.3.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Malvehy J, Ferrando J, Soler J, Tuneu A, Ballesta F, Estrach T. Trichothiodystrophy associated with urologic malformation and primary hypercalciuria. Pediatr Dermatol. 1997;14:441–5. doi: 10.1111/j.1525-1470.1997.tb00685.x. [DOI] [PubMed] [Google Scholar]
- 74.Marinoni S, Gaeta G, Not T, Freschi P, Trevisan G, Briscik E, Giliani S. Early recognition of trichothiodystrophy with xeroderma pigmentation group D mutation in a collodion baby. In: Panconesi E, editor. Dermatology in Europe. Blackwell Scientific; Oxford: 1991. pp. 632–3. [Google Scholar]
- 75.Mazereeuw-Hautier J, Pech JH, Heitz F, Bonafe JL. Trichothiodystrophy and congenital heart disease in two sisters] Ann Dermatol Venereol. 2002;129(10 Pt 1):1168–71. [PubMed] [Google Scholar]
- 76.McCuaig C, Marcoux D, Rasmussen JE, Werner MM, Gentner NE. Trichothiodystrophy associated with photosensitivity, gonadal failure, and striking osteosclerosis. J Am Acad Dermatol. 1993;28(5 Pt 2):820–6. doi: 10.1016/0190-9622(93)70109-7. [DOI] [PubMed] [Google Scholar]
- 77.Meynadier J, Guillot B, Barneon G, Djian B, Levy A. [Trichothiodystrophy] Ann Dermatol Venereol. 1987;114:1529–36. [PubMed] [Google Scholar]
- 78.Milligan A, Fletcher A, Porter DI, Hutchinson PE. Trichothiodystrophy. Clin Exp Dermatol. 1991;16:264–7. doi: 10.1111/j.1365-2230.1991.tb00371.x. [DOI] [PubMed] [Google Scholar]
- 79.Motley RJ, Finlay AY. A patient with Tay’s syndrome. Pediatr Dermatol. 1989;6:202–5. doi: 10.1111/j.1525-1470.1989.tb00818.x. [DOI] [PubMed] [Google Scholar]
- 80.Murphy LA, Atherton DJ. Collodion baby, failure to thrive and frequent fever due to trichothiodystrophy syndrome. Br J Dermatol. 2003;149(Suppl 64):71–86. [Google Scholar]
- 81.Murrin KL, Clarke DJ. Behavioural aspects of Pollitt syndrome: a 32-year follow-up of a case described by R. J. Pollitt and colleagues in 1968. J Intellect Disabil Res. 2002;46(Pt 3):273–8. doi: 10.1046/j.1365-2788.2002.00369.x. [DOI] [PubMed] [Google Scholar]
- 82.Ostergaard JR, Christensen T. The central nervous system in Tay syndrome. Neuropediatrics. 1996;27:326–30. doi: 10.1055/s-2007-973803. [DOI] [PubMed] [Google Scholar]
- 83.Peserico A, Battistella PA, Bertoli P. MRI of a very rare hereditary ectodermal dysplasia: PIBI(D)S. Neuroradiology. 1992;34:316–7. doi: 10.1007/BF00588190. [DOI] [PubMed] [Google Scholar]
- 84.Peter C, Tomczok J, Hoting E, Behrendt H. Trichothiodystrophy without associated neuroectodermal defects. Br J Dermatol. 1998;139:137–40. doi: 10.1046/j.1365-2133.1998.02331.x. [DOI] [PubMed] [Google Scholar]
- 85.Petrin JH, Meckler KA, Sybert VP. A new variant of trichothiodystrophy with recurrent infections, failure to thrive, and death. Pediatr Dermatol. 1998;15:31–4. doi: 10.1046/j.1525-1470.1998.1998015031.x. [DOI] [PubMed] [Google Scholar]
- 86.Poissonnier M, Blanc A, Bat P. [Genetic counseling in a case of neuroectodermosis: Vera Price trichothiodystrophy. Brittle hair with reduced sulfur content] J Genet Hum. 1988;36:361–5. [PubMed] [Google Scholar]
- 87.Porto L, Weis R, Schulz C, Reichel P, Lanfermann H, Zanella FE. Tay’s syndrome: MRI. Neuroradiology. 2000;42:849–51. doi: 10.1007/s002340000443. [DOI] [PubMed] [Google Scholar]
- 88.Price Veal . Trichothiodystrophy: sulfur-deficient brittle hair. In: Brown ACCR, editor. Hair, trace elements and human illness. Praeger Publishers, Inc; New York: 1980. [Google Scholar]
- 89.Przedborski S, Ferster A, Goldman S, Wolter R, Song M, Tonnesen T, Pollitt RJ, Vamos E. Trichothiodystrophy, mental retardation, short stature, ataxia, and gonadal dysfunction in three Moroccan siblings. Am J Med Genet. 1990;35:566–73. doi: 10.1002/ajmg.1320350424. [DOI] [PubMed] [Google Scholar]
- 90.Quintero RA, Morales WJ, Gilbert-Barness E, Claus J, Bornick PW, Allen MH, Ackerman J, Koussef B. In utero diagnosis of richothiodystrophy by endoscopicallyguided fetal eyebrow biopsy. Fetal Diagn Ther. 2000;15:152–5. doi: 10.1159/000020995. [DOI] [PubMed] [Google Scholar]
- 91.Racioppi L, Cancrini C, Romiti ML, Angelini F, Di Cesare S, Bertini E, Livadiotti S, Gambarara MG, Matarese G, Lago PF, Stefanini M, Rossi P. Defective dendritic cell maturation in a child with nucleotide excision repair deficiency and CD4 lymphopenia. Clin Exp Immunol. 2001;126:511–8. doi: 10.1046/j.1365-2249.2001.01625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rebora A, Crovato F. The Tay syndrome. A reply to the author’s reply. Eur J Pediatr. 1984;143:76. doi: 10.1007/BF00442760. [DOI] [PubMed] [Google Scholar]
- 93.Rebora A, Guarrera M, Crovato F. Amino acid analysis in hair from PIBI(D)S syndrome. J Am Acad Dermatol. 1986;15:109–11. doi: 10.1016/s0190-9622(86)80141-4. [DOI] [PubMed] [Google Scholar]
- 94.Rebora A, Crovato F. PIBI(D)S syndrome—trichothiodystrophy with xeroderma pigmentosum (group D) mutation. J Am Acad Dermatol. 1987;16(5 Pt 1):940–7. doi: 10.1016/s0190-9622(87)70118-2. [DOI] [PubMed] [Google Scholar]
- 95.Rebora A, Crovato F. Trichothiodystrophy, xeroderma pigmentosum and PIBI(D)S syndrome. Hum Genet. 1988;78:106–8. doi: 10.1007/BF00291250. [DOI] [PubMed] [Google Scholar]
- 96.Riou L, Zeng L, Chevallier-Lagente O, Stary A, Nikaido O, Taieb A, Weeda G, Mezzina M, Sarasin A. The relative expression of mutated XPB genes results in xeroderma pigmentosum/Cockayne’s syndrome or trichothiodystrophy cellular phenotypes. Hum Mol Genet. 1999;8:1125–33. doi: 10.1093/hmg/8.6.1125. [DOI] [PubMed] [Google Scholar]
- 97.Rizzo R, Pavone L, Micali G, Calvieri S, Di Gregorio L. Trichothiodystrophy: report of a new case with severe nervous system impairment. J Child Neurol. 1992;7:300–3. doi: 10.1177/088307389200700311. [DOI] [PubMed] [Google Scholar]
- 98.Sarasin A, Blanchet-Bardon C, Renault G, Lehmann A, Arlett C, Dumez Y. Prenatal diagnosis in a subset of trichothiodystrophy patients defective in DNA repair. Br J Dermatol. 1992;127:485–91. doi: 10.1111/j.1365-2133.1992.tb14845.x. [DOI] [PubMed] [Google Scholar]
- 99.Sass JO, Skladal D, Zelger B, Romani N, Utermann B. Trichothiodystrophy: quantification of cysteine in human hair and nails by application of sodium azide-dependent oxidation to cysteic acid. Arch Dermatol Res. 2004;296:188–91. doi: 10.1007/s00403-004-0483-2. [DOI] [PubMed] [Google Scholar]
- 100.Savary JB, Vasseur F, Vinatier D, Manouvrier S, Thomas P, Deminatti MM. Prenatal diagnosis of PIBIDS. Prenat Diagn. 1991;11:859–66. doi: 10.1002/pd.1970111107. [DOI] [PubMed] [Google Scholar]
- 101.Schepis C, Elia M, Siragusa M, Barbareschi M. A new case of trichothiodystrophy associated with autism, seizures, and mental retardation. Pediatr Dermatol. 1997;14:125–8. doi: 10.1111/j.1525-1470.1997.tb00219.x. [DOI] [PubMed] [Google Scholar]
- 102.Stefanini M, Giliani S, Nardo T, Marinoni S, Nazzaro V, Rizzo R, Trevisan G. DNA repair investigations in nine Italian patients affected by trichothiodystrophy. Mutat Res. 1992;273:119–25. doi: 10.1016/0921-8777(92)90073-c. [DOI] [PubMed] [Google Scholar]
- 103.Stefanini M, Lagomarsini P, Giliani S, Nardo T, Botta E, Peserico A, Kleijer WJ, Lehmann AR, Sarasin A. Genetic heterogeneity of the excision repair defect associated with trichothiodystrophy. Carcinogenesis. 1993;14:1101–5. doi: 10.1093/carcin/14.6.1101. [DOI] [PubMed] [Google Scholar]
- 104.Takayama K, Salazar EP, Broughton BC, Lehmann AR, Sarasin A, Thompson LH, Weber CA. Defects in the DNA repair and transcription gene ERCC2(XPD) in trichothiodystrophy. Am J Hum Genet. 1996;58:263–70. [PMC free article] [PubMed] [Google Scholar]
- 105.Takayama K, Danks DM, Salazar EP, Cleaver JE, Weber CA. DNA repair characteristics and mutations in the ERCC2 DNA repair and transcription gene in a trichothiodystrophy patient. Hum Mutat. 1997;9:519–25. doi: 10.1002/(SICI)1098-1004(1997)9:6<519::AID-HUMU4>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 106.Toelle SP, Valsangiacomo E, Boltshauser E. Trichothiodystrophy with severe cardiac and neurological involvement in two sisters. Eur J Pediatr. 2001;160:728–31. doi: 10.1007/s004310100845. [DOI] [PubMed] [Google Scholar]
- 107.Tolmie JL, de Berker D, Dawber R, Galloway C, Gregory DW, Lehmann AR, McClure J, Pollitt RJ, Stephenson JB. Syndromes associated with trichothiodystrophy. Clin Dysmorphol. 1994;3:1–14. [PubMed] [Google Scholar]
- 108.van Neste D, Bore P. [Trichothiodystrophy: a morphological and biochemical study] Ann Dermatol Venereol. 1983;110:409–17. [PubMed] [Google Scholar]
- 109.van Neste D, de Greef H, van Haute N, van Hee J, van der Maesen J, Taieb A, Maleville J, Fontan D, Bakry N, Gillespie JM, Marshall RC. High-sulfur protein deficient human hair: clinical aspects and biochemical study of two unreported cases of a variant of trichothiodystrophy. In: Van Neste, editor. Trends in human hair and alopecia research. Klaver Academic; Dordrecht: 1989. pp. 195–206. [Google Scholar]
- 110.Vermeulen W, Rademakers S, Jaspers NG, Appeldoorn E, Raams A, Klein B, Kleijer WJ, Hansen LK, Hoeijmakers JH. A temperature-sensitive disorder in basal transcription and DNA repair in humans. Nat Genet. 2001;27:299–303. doi: 10.1038/85864. [DOI] [PubMed] [Google Scholar]
- 111.Viprakasit V, Gibbons RJ, Broughton BC, Tolmie JL, Brown D, Lunt P, Winter RM, Marinoni S, Stefanini M, Brueton L, Lehmann AR, Higgs DR. Mutations in the general transcription factor TFIIH result in beta-thalassaemia in individuals with trichothiodystrophy. Hum Mol Genet. 2001;10:2797–802. doi: 10.1093/hmg/10.24.2797. [DOI] [PubMed] [Google Scholar]
- 112.Wakeling EL, Cruwys M, Suri M, Brady AF, Aylett SE, Hall C. Central osteosclerosis with trichothiodystrophy. Pediatr Radiol. 2004;34:541–6. doi: 10.1007/s00247-004-1207-7. [DOI] [PubMed] [Google Scholar]
- 113.Wetzburger CL, Van Regemorter N, Szliwowski HB, Abramowicz MJ, Van Bogaert P. Gray matter heterotopia and acute necrotizing encephalopathy in trichothiodystrophy. Pediatr Neurol. 1998;19:392–4. doi: 10.1016/s0887-8994(98)00085-x. [DOI] [PubMed] [Google Scholar]
- 114.Yoon HK, Sargent MA, Prendiville JS, Poskitt KJ. Cerebellar and cerebral atrophy in trichothiodystrophy. Pediatr Radiol. 2005;35:1019–23. doi: 10.1007/s00247-005-1495-6. [DOI] [PubMed] [Google Scholar]
- 115.van Neste D, Miller X, Bohnert E. Clinical symptoms associated with trichothiodystrophy: a review of the literature with special emphasis on light sensitivity and the association with xeroderma pigmentosum (complementation group D) In: Van Neste, editor. Trends in human hair and alopecia research. Klaver Academic; Dordrecht: 1989. pp. 183–93. [Google Scholar]
- 116.Martin JA, Hamilton BE, Sutton PD, Ventura SJ, Menacker F, Kirmeyer S, Munson ML. Births: Final Data for 2005. National Vital Statistics Reports. 2007;56:1–104. [PubMed] [Google Scholar]
- 117.Levin AV. Congenital eye anomalies. Pediatr Clin North Am. 2003;50:55–76. doi: 10.1016/s0031-3955(02)00113-x. [DOI] [PubMed] [Google Scholar]
- 118.Alapetite C, Benoit A, Moustacchi E, Sarasin A. The comet assay as a repair test for prenatal diagnosis of Xeroderma pigmentosum and trichothiodystrophy. J Invest Dermatol. 1997;108:154–9. doi: 10.1111/1523-1747.ep12332692. [DOI] [PubMed] [Google Scholar]
- 119.Savary JB, Vasseur F, Vinatier D, Manouvrier S, Deminatti MM. First-trimester prenatal exclusion of PIBIDS syndrome with normal DNA excision repair on chorionic villus cells. Prenat Diagn. 1992;12:969–71. doi: 10.1002/pd.1970121119. [DOI] [PubMed] [Google Scholar]
- 120.Dubaele S, Proietti De SL, Bienstock RJ, Keriel A, Stefanini M, Van HB, Egly JM. Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Mol Cell. 2003;11:1635–46. doi: 10.1016/s1097-2765(03)00182-5. [DOI] [PubMed] [Google Scholar]
- 121.Lehmann AR. The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev. 2001;15:15–23. doi: 10.1101/gad.859501. [DOI] [PubMed] [Google Scholar]
- 122.Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH. Cockayne syndrome and xeroderma pigmentosum. Neurology. 2000;55:1442–9. doi: 10.1212/wnl.55.10.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Itin PH, Pittelkow MR. Trichothiodystrophy: review of sulfur-deficient brittle hair syndromes and association with the ectodermal dysplasias. J Am Acad Dermatol. 1990;22:705–17. doi: 10.1016/0190-9622(90)70096-z. [DOI] [PubMed] [Google Scholar]
- 124.Faghri S, DiGiovanna JJ, Tamura D, Kraemer KH. Trichothiodystrophy includes a broad spectrum of multisystem abnormalities and may have a high mortality at a young age. J Invest Dermatol April. 2007;127(Suppl 1):S106. [Google Scholar]