

Abstract
Introduction
Anesthetic preconditioning protects cardiomyocytes from oxidative stress-induced injury, but it is ineffective in patients with diabetes mellitus. To address the role of hyperglycemia in the inability of diabetic individuals to be preconditioned, we used human cardiomyocytes differentiated from induced pluripotent stem cells generated from patients with or without type 2 diabetes mellitus (DM-iPSC- and N-iPSC-CMs, respectively) to investigate the efficacy of preconditioning in varying glucose conditions (5, 11, and 25 mM).
Methods
Induced pluripotent stem cells were induced to generate cardiomyocytes by directed differentiation. For subsequent studies, cardiomyocytes were identified by genetic labeling with enhanced green fluorescent protein driven by a cardiac-specific promoter. Cell viability was analyzed by lactate dehydrogenase assay. Confocal microscopy was utilized to measure opening of the mitochondrial permeability transition pore and the mitochondrial adenosine-5′-triphosphate-sensitive potassium channels.
Results
Isoflurane (0.5 mM) preconditioning protected N-iPSC- and DM-iPSC-CMs from oxidative stress-induced lactate dehydrogenase release and mitochondrial permeability transition pore opening in 5 mM and 11 mM glucose. Isoflurane triggered mitochondrial adenosine-5′-triphosphate-sensitive potassium channel opening in N-iPSC-CMs in 5 mM and 11 mM glucose and in DM-iPSC-CMs in 5 mM glucose. 25 mM glucose disrupts anesthetic preconditioning-mediated protection in DM-iPSC- and N-iPSC-CMs.
Conclusions
The opening of mitochondrial adenosine-5′-triphosphate-sensitive potassium channels are disrupted in DM-iPSC-CMs in 11 mM and 25 mM glucose and in N-iPSC-CMs in 25 mM glucose. Cardiomyocytes derived from healthy donors and patients with a specific disease, such as diabetes in this study, open possibilities in studying genotype and phenotype related pathologies in a human relevant model.
Introduction
The World Health Organization estimates that diabetes mellitus affects nearly 200 million people worldwide with 90% of them having type 2. The risk of cardiovascular disease increases dramatically for those diagnosed with diabetes.1 Anesthetic preconditioning (APC) can reduce myocardial injury following an ischemia and reperfusion injury.2-4 However, diabetes or hyperglycemia in the non-diabetic myocardium have been shown to abolish APC in animal models.5-6
Tanaka et al. discovered that streptozotocin-induced diabetic canines could not be efficiently protected from myocardial infarction with isoflurane preconditioning.5 They also observed that infarct size was directly related to blood glucose levels. Kehl et al. observed that moderate hyperglycemia attenuated the protective effects of 0.5 minimum alveolar concentration isoflurane, but not 1.0 minimum alveolar concentration isoflurane in canines. However, severe hyperglycemia attenuated the protective effects of 0.5 and 1.0 minimum alveolar concentration isoflurane.6 These animal studies provided great insight into APC in diabetes mellitus; however, they could not address a genetic component of this phenomenon due to the nature of inducing diabetes in this experimental model. Moreover, it is unclear whether we could extend these mechanisms delineated in animal models to human myocardium.
Induced pluripotent stem cells (iPSCs) used in this study were generated from human skin fibroblasts by introducing pluripotency factors. Reprogramming the cells allows them to obtain characteristics of pluripotent stem cells.7 As such, iPSCs have the capability to differentiate into cardiomyocytes8-9 while maintaining their genotype. Our laboratory has previously shown that cardiomyocytes derived from human embryonic stem cells (hESC-CMs) exhibit competent responses to APC compared to various animal models and human myocardium.10 The generation of iPSCs from patient-specific-derived fibroblasts enables us to study human disease mechanisms that were nearly impossible to be investigated in the past. In this study, iPSCs derived from non-diabetic and type 2 diabetic individuals were used to generate cardiomyocytes (DM-iPSC-CMs, N-iPSC-CMs, respectively). Others have shown that differentiated cardiomyocytes behave very similar to adult cardiomyocytes on multiple levels, including exhibition of spontaneous and rhythmic contractions, and expression of cardiac specific markers.9-11 Thus, DM-iPSC-CMs and N-iPSC-CMs provide us with a unique in vitro model to investigate environmental factors, such as high glucose, that affect APC in diabetic myocardium. Due to the lack of experimentally available human myocardium, reliable type 2 diabetic animal models, and the disconnect between animal and human studies, our model has the unique ability to investigate a human disease and its ability to attenuate APC-induced cardioprotection for the first time. Our study investigates the detrimental effects of high glucose on APC-mediated cardiac protection in cardiomyocytes derived from a non-diabetic and a type 2 diabetic individual.
Materials and Methods
Human induced pluripotent stem cell culture
Dermal fibroblasts were isolated from one type 2 diabetic and one non-diabetic patient and these patient-specific fibroblasts were reprogrammed into iPSCs12-13 (DM-iPSCs, N-iPSCs, respectively); both lines were generous gifts from Dr. George Daley, M.D., Ph.D. (Professor, Director, Stem Cell Transplantation Program, Howard Hughes Medical Institute and Children's Hospital, Boston, MA) and Dr. Stephen Duncan, Ph.D. (Professor, Vice-Chairman Cell Biology, Neurobiology, and Anatomy, Director MCW Program in Regenerative Medicine and Stem Cell Biology, Medical College of Wisconsin, Milwaukee, WI), respectively. iPSC lines were cultured according to our previously described protocol.10 Mouse embryonic fibroblasts were inactivated with mitomycin C (Sigma-Aldrich, St. Louis, MO) and plated in Dulbecco's Modified Eagle Medium (Millipore Bioscience Research Reagents, Temecula, CA) complemented with 10% Fetal Bovine Serum (Invitrogen, Carlsbad, CA) and 1% nonessential amino acids (Millipore Bioscience Research Reagents). iPSCs were maintained on inactivated mouse embryonic fibroblasts in hypoxic conditions (4% O2, 5% CO2) with iPSC culture medium consisting of Dulbecco's Modified Eagle Medium/Ham F12 medium (Invitrogen) supplemented with 20% knockout serum (Invitrogen), 1% nonessential amino acids (Invitrogen), 1% penicillin-streptomycin (Invitrogen), L-glutamine (Millipore), β-mercaptoethanol (Sigma-Aldrich), and 4 ng/ml human recombinant basic fibroblast growth factor (Invitrogen). iPSC colonies were passaged every six to eight days. iPSC passages 45-65 were used in the subsequent study.
Cardiac differentiation of N-iPSCs and DM-iPSCs
iPSC colonies were mechanically dissociated into small aggregates and plated onto dishes precoated with Reduced Growth Factor Matrigel (BD Biosciences, San Jose, CA), and cultured under hypoxic (4% O2, 5% CO2) conditions. Cells were supplemented with iPSC medium conditioned by inactivated mouse embryonic fibroblasts for seven days. Next, cells were grown in Roswell Park Memorial Institute (RPMI)/B27 medium (Invitrogen) supplemented with growth factors activin A (50 ng/ml; R&D Systems, Minneapolis, MN) and bone morphogenetic protein-4 (10 ng/ml; R&D) for five days. Finally, cells were plated in normoxic conditions (20% O2) with RPMI/B27 and matured for three months prior to use in subsequent experiments.
Microdissection and single cell dissociation
Three months following differentiation, cell clusters were mechanically dissociated under a dissecting microscope (SMZ1000; Nikon, Tokyo, Japan) using a 27-gauge needle and enzymatically dispersed in 0.05% trypsin-EDTA (Invitrogen) for 5 min. Trypsin was inactivated by Dulbecco's Modified Eagle Medium containing 20% Fetal Bovine Serum and individual cells were plated onto matrigel-coated coverslips at low density.
Immunofluorescence
Cells cultured on matrigel-coated glass coverslips were fixed with 1% paraformaldehyde for 30 minutes and then were washed three times with phosphate-buffered saline. Cells were permeabilized with 0.5% Triton × -100 (Sigma) and blocked with 10% donkey serum for 30 minutes. Cells were incubated with primary antibodies for sarcomeric α-actinin (1:100 dilution; Sigma-Aldrich) or anti-cardiac-specific troponin T (1:100; Thermo Fisher Scientific, Waltham, MA) for 1 h at 37°C. After three washes, cells were incubated with secondary antibody Alexa Fluor 594 (1:1000, Invitrogen) for 1 h at room temperature. Nuclei were stained with TOPRO-3 (1:1000; Invitrogen). Coverslips were then mounted onto slides and images were acquired with a laser-scanning confocal microscope (Nikon Eclipse TE2000-U).
Genetic marking of iPSC-derived cardiomyocytes with a lentiviral vector
Genetic marking of iPSC-CMs was conducted similarily in our previous study.10 In brief, 3 days after dissociation, cardiomyocytes were transduced with a lentiviral vector encoding human myosin light chain-2v (MLC-2v)-driven enhanced green fluorescent protein (eGFP: multiplicity of infection, 2.2 × 104). The MLC-2v promoter is specific for ventricular myocytes.11,14-15 The MLC-2v-eGFP cassette (kindly provided by Lior Gepstein, M.D., Ph.D., Professor, The Bruce Rappaport Institute in the Medical Sciences, Faculty of Medicine, Technion-Israel Institute of Technology, Haifa, Israel16) was subcloned into lentiviral transfer plasmid pHR(+)c.Ub.MCSoligo.R(−)W(+). Lentiviral vector assembly and titering were performed as previously described.17-18
Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) activity was assessed for determination of cell viability. LDH was measured using a colorimetric cytotoxicity assay kit according to the manufacturer's directions as an indicator of cell membrane damage (Roche Diagnostics Corporation, Indianapolis, IN). iPSC-CMs were plated at 1 × 104 cells/well in 96-well plates and cultured in 100 μl of RPMI/B27 medium. The medium was exchanged every two days for one week. Cardiomyocytes were treated with either a control (RPMI/B27) medium or preconditioned with 0.5 mM isoflurane for 10 minutes and then replaced with control medium. Both control and experimental groups had a 2 h exposure of 10 mM H2O2 (Calbiochem, LA Jolla, CA) to induce oxidative stress. Experiments were conducted in RPMI/B27 media containing either 5 mM glucose with 20 mM mannitol, an osmotic control, 11 mM glucose with 14 mM mannitol, or 25 mM glucose throughout the experiment. Absorbance data were read in the assay plate spectrophotometrically at 490 nm (reference wavelength at 600 nm) with a microplate reader (BioTek Instruments, Inc., Winooski, VT). Only control and experimental groups that had an oxidative stress-induced LDH release were analyzed. After subtracting background absorbance of cell-free medium, total LDH released into medium was calculated.
Laser-scanning confocal microscopy for mitochondria membrane potential and mitochondrial permeability transition pore (mPTP) opening assays
Five days following lentiviral vector transduction, imaging was performed using a confocal microscope (Eclipse TE2000-U; Nikon) and data was analyzed with Image J software (National Institutes of Health, Bethesda, MD). Living iPSC-CMs were identified by detecting green fluorescent cells, indicating MLC-2v-driven eGFP expression. Experiments were conducted in either 5 mM glucose, 11 mM glucose, or 25 mM glucose. Tetramethylrhodamine ethyl ester (TMRE; 30 nM; Invitrogen) was used to detect mitochondrial membrane potential (ΔΨm) in iPSC-CMs for the purpose of detecting opening of mitochondrial adenosine-5′-triphosphate-sensitive potassium (mitoKATP) channels. Opening of mitoKATP channels induces a depolarization, observed as a loss of TMRE fluorescence. iPSC-CMs were exposed to either isoflurane (0.5 mM), diazoxide (100 μM; Sigma), a mitoKATP activator, or isoflurane plus 5-hydroxydecanoate (5-HD; 500 μM; Sigma), a mitoKATP inhibitor, to determine the activity of mitoKATP channels. Data are normalized to baseline (100%). Baseline fluorescence intensity was recorded for five frames (1 framssse/min) and then treatment was added and fluorescence intensity was recorded for ten frames (1 frame/min). For statistical analysis, the average values of the baseline measurements were compared to the average values of the last five frames following treatment to determine changes in TMRE fluorescence from baseline. Rarely, cardiomyocytes would shift during the experimental protocol and were not included in the analysis. Opening of the mPTP was assessed as described previously in our laboratory,19 a method based on mPTP induction by photoexcitation-generated oxidative stress.20-23 The mPTP opening was detected by rapid dissipation of ΔΨm, observed as a loss of TMRE fluorescence, which is sensitive to mPTP opening inhibition.19
Application of Anesthetic Preconditioning
Appropriate volumes of isoflurane stock solution (Baxter, Deerfield, IL) were sonicated into media to achieve 0.5 mM (∼1 minimum alveolar concentration) concentration as done in previous studies.10,24 Cardiomyocytes were anesthetically preconditioned with culture media containing 0.5 mM isoflurane for 10 minutes followed by a wash out with culture media in LDH and mPTP experiments. Culture medium containing 0.5 mM isoflurane was placed onto cardiomyocytes to trigger mitoKATP opening. At the end of each experiment, isoflurane concentration was analyzed by gas chromatography.
Statistical analysis
Data are presented as mean ± SD. Each experimental group consists of iPSC-derived CMs from at least three separate differentiations. n indicates the number of independent experiments. For the statistical analyses, SigmaStat 3.0 software (Systat Software, Inc., San Jose, CA) was used. Statistical comparisons were performed using one-way analysis of variance with Holm-Sidak correction for multiple testing over all comparisons within each cell line or unpaired t-test where appropriate. P < 0.05 was considered significant.
Results
Differentiation and characterization of iPSC-derived cardiomyocytes
Cardiomyocytes differentiated from both N-iPSCs and DM-iPSCs were observed contracting in culture dishes approximately 14 days following treatment with activin-A and bone morphogenetic protein-4 (fig. 1). Contracting N-iPSC-CMs and DM-iPSC-CMs are shown in Supplemental Digital Content 1 and 2, respectively. Immunostaining verified that dissociated cardiomyocytes from both N-iPSC and DM-iPSC populations had striation like patterns confirmed by both cardiac-specific troponin T (fig. 2, A, B) and sarcomeric α-actinin staining (fig. 2, C, D).
Figure 1. Experimental scheme for cardiomyocyte differentiation of induced pluripotent stem cells (iPSCs).

Non-diabetic and type 2 diabetic patient-derived pluripotent stem cells (N-iPSCs, DM-iPSCs, respectively) were expanded for seven days with mouse fibroblast-conditioned medium on matrigel-coated dishes in hypoxic conditions (4%O2, 5% CO2). The addition of activin-a and bone morphogenic protein-4 (BMP 4) for five days induced cardiac differentiation. Cells were then placed into normoxic conditions and growth factors were withdrawn and supplemented with an Roswell Park Memorial Institute (RPMI)/B27 defined medium. Cardiomyocytes were allowed to mature to 3 months following differentiation before subsequent studies were conducted.
Figure 2. Immunolabeling of non-diabetic and type 2 diabetic induced pluripotent stem cell-derived cardiomyocytes (N-iPSC- and DM-iPSC-CMs, respectively).
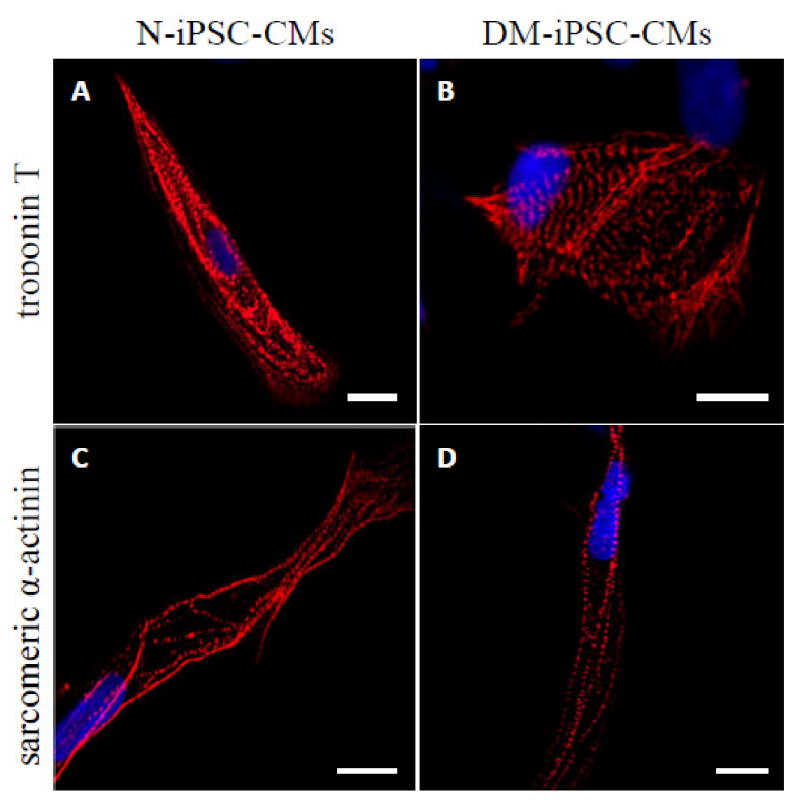
Confocal images of dissociated cells stained for sarcomeric proteins: cardiac-specific troponin T (A, B) and sarcomeric α-actinin (C, D). The majority of positively labeled cells contained striation-like patterns with no differences between N-iPSC- and DM-iPSC-CMs. Scale bar = 10 μm.
iPSC-derived cardiomyocytes express eGFP under the control of cardiac specific promoter MLC-2v
To properly identify living iPSC-CMs, determine differentiation efficiency, and ensure that subsequent confocal microscopy experiments were conducted on cardiomyocytes, we genetically labeled cells with a lentiviral vector that expressed eGFP under the transcriptional control of cardiac-specific promoter, MLC-2v (fig. 3). The differentiation efficiency was determined to be high in N-iPSC- and DM-iPSC-CMs, 67 ± 4%, 71 ± 4%, respectively (fig. 3, B-D; n = 6/group). eGFP-positive cardiomyocytes were selected for subsequent confocal microscopy experiments.
Figure 3. Labeling of non-diabetic and type 2 diabetic induced pluripotent stem cell-derived cardiomyocytes (N-iPSC- and DM-iPSC-CMs, respectively) using a lentiviral vector.
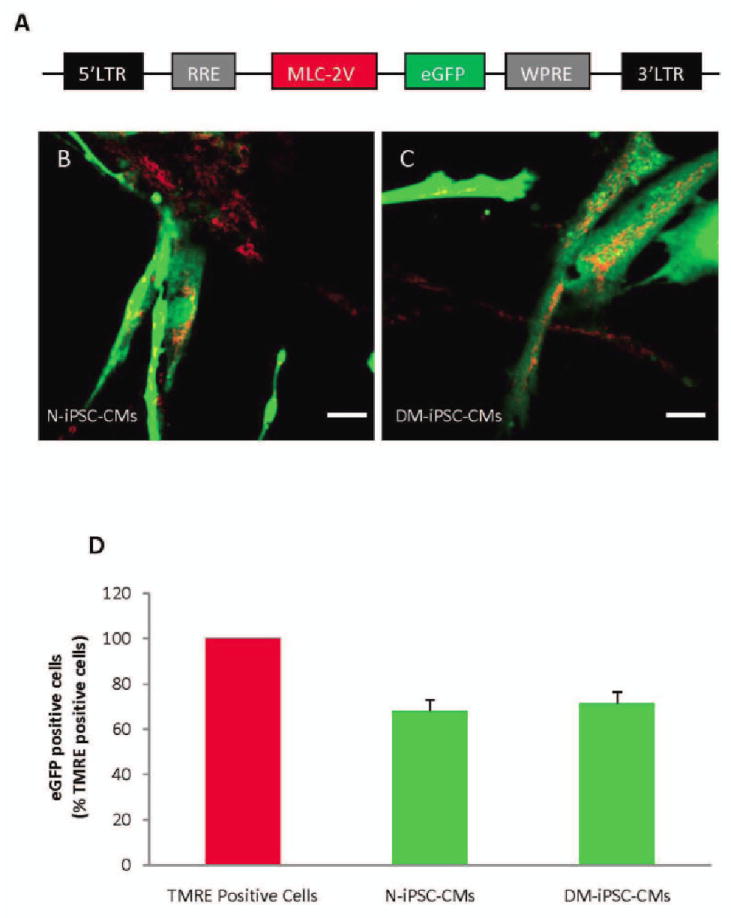
Schematic representation of lentiviral vector pHR(+)C.mlc-2V.egfp.r(−)w(+) used for labeling cardiomyocytes (A). To determine total cell number, cells were loaded with mitochondrial marker tetramethylrhodamine ethyl ester (TMRE, red); myosin light chain-2v (MLC-2v)- enhanced green fluorescent protein (eGFP)-positive cells were counted by detecting green fluorescence, giving the number of cardiomyocytes derived from N-iPSCs and DM-iPSCs (B, C, respectively). Summarized data from six separate differentiations, exhibiting a high percentage of induced pluripotent stem cell-derived cardiomyocytes (D). Scale bar = 40 um.
APC protects both N-iPSC- and DM- iPSC-derived cardiomyocytes in 5 mM and 11 mM but not 25 mM glucose against oxidative stress
To determine whether APC could protect cardiomyocytes derived from both N-iPSCs and DM-iPSCs from oxidative stress-induced damage, we exposed control and anesthetic preconditioned cardiomyocytes to 10 mM H2O2 for 2 h in 5 mM, 11 mM, or 25 mM glucose environment. N-iPSC-CMs that were preconditioned with isoflurane prior to H2O2 exposure had a significantly lower LDH release in 5 mM glucose compared to stress control, 128 ± 4% vs. 152 ± 4%, respectively (fig. 4A; n = 7/group) and in 11 mM glucose compared to stress control, 131 ± 6% vs. 151 ± 6%, respectively (fig. 4A; n = 8/group). APC did not reduce LDH release in N-iPSC-CMs in 25 mM glucose compared to the stress control (151 ± 9% vs. 150 ± 7%, respectively; fig. 4A; n = 7/group). DM-iPSC-CMs also had a reduction in LDH release when preconditioned with isoflurane compared to the stress control in 5 mM glucose (119 ± 3% vs. 148 ± 3%, respectively; fig. 4B; n = 7/group) and in 11 mM glucose (124 ± 4% vs. 146 ± 3%, respectively; fig. 4B; (n = 8/group). However, the APC-induced reduction in LDH release in DM-iPSC-CMs was lost in the presence of 25 mM glucose (fig. 4B; (n = 7/group).
Figure 4. Anesthetic preconditioning (APC) protects non-diabetic and type 2 diabetic induced pluripotent stem cell-derived cardiomyocytes (N-iPSC- and DM-iPSC-CMs, respectively) in 5 mM and 11 mM glucose from oxidative stress.
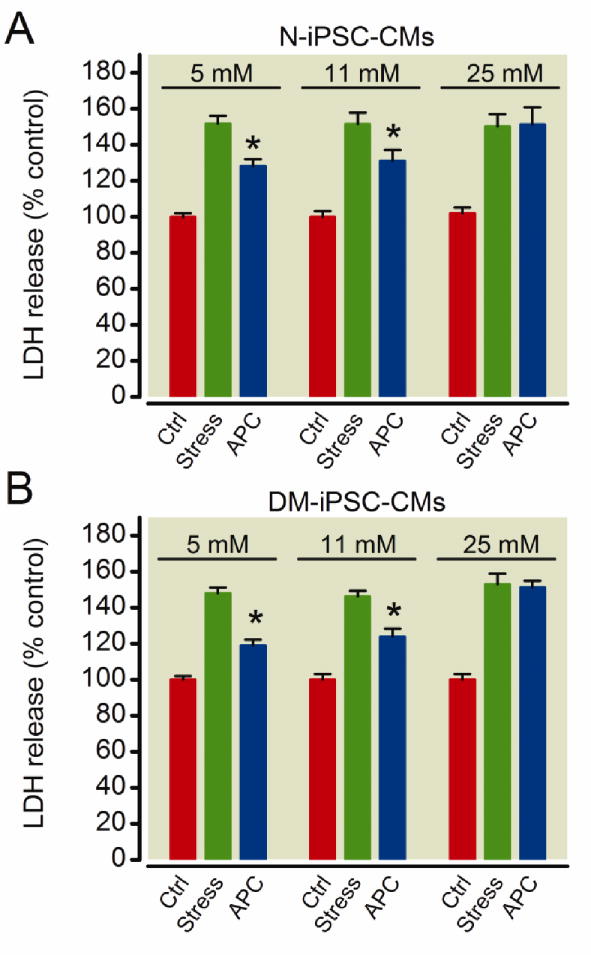
H2O2-induced oxidative stress increased lactate dehydrogenase (LDH) release from N-iPSC-(A) and DM-iPSC-CMs (B) in a 5 mM, 11 mM and 25 mM glucose environment. In both N-iPSC- and DM-iPSC-derived cardiomyocytes APC reduced LDH release in 5 mM and 11 mM glucose, but not in 25 mM glucose. *P < 0.05 versus stress. Ctrl = control.
Preconditioning with isoflurane delays opening of the mPTP in both N-iPSC- and DM-iPSC-derived cardiomyocytes in 5 mM and 11 mM but not 25 mM glucose
APC delays opening of the mPTP and thus reduces cell death in ischemia-reperfusion injury.10,19,25 Opening of the mPTP causes a rapid collapse in ΔΨm, which can be detected following rapid loss of TMRE fluorescence intensity in iPSC-CMs (fig. 5A). APC increased the mPTP opening time in N-iPSC-CMs in 5 mM glucose compared to control, 139 ± 6% vs. 100 ± 5% (fig. 5B; n = 22 cells/group). APC also increased mPTP opening time in DM-iPSC-CMs in 5 mM glucose compared to control, 128 ± 4% vs. 100 ± 5% (fig. 5C; n = 25 cells/group). Additionally, APC increased mPTP opening time in N-iPSC-CMs in 11 mM glucose, 131 ± 7% vs. 100 ± 5% (fig. 5B; n = 32) and in DM-iPSC-CMs in 11 mM glucose, 122 ± 9% vs. 100 ± 5%, respectively (fig. 5, B and C; n = 30 cells/group). The APC-induced delay in mPTP opening was lost in N-iPSC-CMS in 25 mM glucose, 100 ± 6% vs. 100 ± 5% (fig. 5B; n = 25 cells/group) and in DM-iPSC-CMs when the cells were exposed to 25 mM glucose, 102 ± 9% vs., 100 ± 6% (fig. 5C; n = 24 cells/group).
Figure 5. Preconditioning delays mitochondrial permeability transition pore (mPTP) opening in non-diabetic and type 2 diabetic induced pluripotent stem cell-derived cardiomyocytes (N-iPSC- and DM-iPSC-CMs, respectively) in a 5 mM and 11 mM glucose environment.

mPTP opening was induced by photoexcitation-generated oxidative stress and detected by rapid dissipation of tetramethylrhodamine ethyl ester (TMRE) fluorescence. Representative signal traces from control (Ctrl) and anesthetic preconditioning (APC) (A). Arbitrary mPTP opening time was determined as the time when TMRE fluorescence intensity decreased by half between initial and residual fluorescence intensity. APC delayed mPTP opening in N-iPSC-CMs in 5 mM and 11 mM glucose environments (B). APC was not able to delay mPTP opening in 25 mM glucose. APC delayed mPTP opening in DM-iPSC-CMs in 5 mM and 11 mM glucose, but not 25 mM glucose(C). *P< 0.05 versus control.
Isoflurane triggers opening of the mitoKATP channels and depolarizes mitochondria in iPSC-derived cardiomyocytes
Isoflurane induces opening of mitoKATP channels causing a mild mitochondrial depolarization which is observed as a partial loss of TMRE fluorescence intensity, the addition of 5-HD, a mitoKATP inhibitor, attenuates a portion of this depolarization (fig. 6A). Diazoxide, a mitoKATP opener, triggered opening of the mitoKATP channels and a subsequent loss of ΔΨm in N-iPSC-CMs in 5 mM (n = 56 cells/group), 11 mM (n = 60 cells/group), and 25 mM glucose (n = 63 cells/group) compared to baseline (fig. 6B). Diazoxide also opened mitoKATP channels in DM-iPSC-CMs in 5 mM (n = 62 cells/group), 11 mM (n = 65 cells/group), and 25 mM glucose (n = 68/cells group) compared to baseline (fig. 6C). The addition of isoflurane (0.5 mM) decreased TMRE fluorescence indicating a mitochondrial depolarization in N-iPSC-CMs in 5 mM glucose (decrease by 34 ± 5%), which was attenuated in the presence of 500 μM 5-HD (decrease by 18 ± 5%) (fig. 6B; n = 56 cells/group). Isoflurane induced a mitochondrial depolarization in DM-iPSC-CMs in 5 mM glucose (decrease by 21 ± 4%), which was attenuated in the presence of 500 μM 5-HD (decrease by 9 ± 4%) (fig. 6C; n = 62 cells/group). This indicated opening of mitoKATP channels by isoflurane. However, N-iPSC-CMs in 25 mM glucose had the same extent of decrease in TMRE fluorescence between isoflurane and isoflurane + 5-HD treatment, indicating the inability of isoflurane to open mitoKATP channels (fig 6B; n = 63 cells/group). Isoflurane did not open mitoKATP channels to a greater extent than when isoflurane + 5-HD was present in DM-iPSC-CMs in 11 mM and 25 mM glucose (fig 6C; n = 68 cells/group). The ability of isoflurane-induced mitochondrial depolarization in the presence of mitoKATP channel blocker, suggests that isoflurane decreases ΔΨm in part by other mechanisms.26
Figure 6. Isoflurane opens mitochondrial potassium adenosine-5′-triphosphate-sensitive (mitoKATP) channels to the greatest extent in non-diabetic induced pluripotent stem cell-derived cardiomyocytes (N-iPSC-CMs).

Mitochondrial membrane potential was monitored in dissociated iPSC-CMS. Representative signal traces from the addition (arrow) of isoflurane alone or in the presence of mitoKATP blocker 5-hydroxydecanoate (5-HD) in N-iPSC-CMs in 11 mM glucose (A). Isoflurane opens mitoKATP channels and causes a loss of tetramethylrhodamine ethyl ester (TMRE) fluorescence indicative of a mitochondrial depolarization. 5-HD partly blocks the isoflurane-induced mitochondrial depolarization. Diazoxide depolarized the mitochondria in both N-iPSC- and DM-iPSC-CMs in a 5 mM, 11 mM, and 25 mM glucose environment. Isoflurane induced the greatest mitochondrial depolarization in N-iPSC-CMs in a 5 mM glucose environment. 5-HD partly attenuated the isoflurane-induced mitochondrial depolarization (B). Isoflurane-induced mitochondrial depolarization was reduced in DM-iPSC-CMs compared to N-iPSC-CMs in their respective glucose conditions (C). *P < 0.05 versus baseline. †P < 0.05 versus Isoflurane treatment.
Discussion
In the present study, we investigated the effects of APC on iPSC-derived cardiomyocytes from a healthy individual and a type 2 diabetic patient in varying glucose conditions. Our major findings are summarized as follows: 1) iPSCs reprogrammed from non-diabetic and type 2 diabetic human fibroblasts can be differentiated into cardiomyocytes with directed differentiation, as supported by visualization of contracting cell regions; expression of cardiac-specific, highly organized contractile myofilaments; and positive genetic labeling with cardiac-specific marker (MLC 2v- eGFP). 2) Preconditioning with isoflurane reduced LDH release and delayed mPTP opening in both N-iPSC-CMs and unexpectedly in DM-iPSC-CMs in 5 mM and 11 mM, but not 25 mM glucose. 3) Isoflurane triggered the opening of mitoKATP channels in N-iPSC-CMs in 5 mM and 11 mM glucose and in DM-iPSC-CMs but only in 5 mM glucose. In summary, both N-iPSC- and DM-iPSC-CMs could be protected in 5 mM glucose, while 11 mM glucose attenuated isoflurane-induced mitoKATP opening in DM-iPSC-CMs, and 25 mM glucose abrogated all cardioprotective effects provided by APC in both DM-iPSC- and N-iPSC-CMs, indicating that 25 mM glucose was the predominant mechanism in the attenuation of APC in diabetic individuals.
We have shown that preconditioning with isoflurane attenuates cell damage and triggers cardioprotective mechanisms in iPSC-CMs against oxidative stress in 5 mM and 11 mM glucose. We recorded isoflurane-induced mitoKATP opening and a delay in oxidative stress-induced mPTP opening in N-iPSC-CMs in 5 mM and 11 mM glucose. iPSC-CMs responded similarly to APC as in studies of adult cardiomyocytes27 and hESC-CMs10, indicating that patient-specific iPSC-derived cardiomyocytes can be a vital tool in understanding human diseases not only at the genetic level, but also test how environmental conditions play a role in a disease phenotype.
We achieved a high rate of differentiation efficiency; approximately 70% of cells expressed eGFP driven by the cardiomyocyte specific promoter MLC-2v (fig. 3). N-iPSC-CMs and DM-iPSC-CMs both had striation-like patterns (fig. 2) indicating structural integrity similar to adult human cardiomyocytes and hESC-CMs.28 We utilized growth factors to differentiate our iPSCs into cardiomyocytes based on the presence of these factors in regulating heart development in the embryo.28 A previously published cardiac differentiation protocol for the induction of human embryonic stem cells differentiation into cardiomyocytes was used for iPSC differentiation.10 We are confident that cardiomyocytes derived from iPSCs can serve as a powerful tool to understand different human diseases and how environment plays a role in attenuating the cardioprotection provided by APC. Cardiomyocytes derived from iPSCs share many characteristics of human adult cardiomyocytes: contractions, cardio-specific immunostaining that illustrates the highly organized contractile sarcomeres, atrial and ventricular-like action potentials, and exhibits functioning APC mediators.
iPSC-derived cardiomyocytes have many unique advantages over models that are currently available to study human diseases. The first shortcoming of many disease animal models is that the model itself does not replicate the complexity of the disease in its entirety. Specifically, the streptozotocin-induced diabetic models do not alter the genome of the model. Many of the type 2 diabetic murine models29, although more relevant, do not exhibit all the components of type 2 diabetes in human. Additionally, there are many shortcomings of using an animal model compared to a human disease.30 As stem cell biology advances, pluripotent stem cell-derived cell types are being utilized in drug discovery and toxicity screening.31-32 The use of pluripotent stem cells to investigate disease models have become more prevalent as well.33-35 Clearly, a human-type model will have many advantages over animal models as we continue to study human diseases.
Our laboratory has extensively investigated APC in animal models,3,19,25 human myocardium,27 and in hESC-CMs.10 It was unclear whether iPSC-CMs could be protected from oxidative stress and what role glucose had in disabling the cardioprotection provided by preconditioning with isoflurane. In this study we showed that cardiomyocytes derived from both N-iPSCs and DM-iPSCs could be preconditioned with isoflurane; however, the presence of 25 mM glucose obliterated the APC-induced cardioprotective effects in each respective cell line. To further investigate the underlying mechanisms of the protection provided by APC in iPSC-CMs, we tested mitoKATP channel activity and mPTP opening. We showed that isoflurane partly depolarized mitochondria in a 5-HD-sensitive manner to the greatest extent in N-iPSC-CMs in 5 mM and 11 mM glucose and in DM-iPSC-CMs in 5 mM glucose. This isoflurane-induced depolarization suggests that opening of the mitoKATP channels plays a role in cardioprotection.3,36 N-iPSC-CMs in 25 mM glucose and DM-iPSC-CMs in 11 mM and 25 mM glucose did not show a significant difference in isoflurane-induced mitoKATP opening compared to groups treated with the mitoKATP channel blocker, 5-HD. This is not surprising as others have indicated that both diabetes and hyperglycemia have shown to attenuate isoflurane-induced mitoKATP opening.37-38
It is well known that isoflurane acts on the electron transport chain, specifically complex I26 and the isoflurane-induced inhibition of this complex could explain the mitochondrial depolarization that we observed in the 5-HD groups. However, APC protected DM-iPSC-CMs in 11 mM glucose from oxidative stress and caused an APC-induced delay in mPTP opening; suggesting that isoflurane may elicit cell protection via parallel pathways that do not fully depend on the opening of mitoKATP channels. Additionally, isoflurane may induce mitoKATP channel opening in a few channels or at levels that we could not accurately record with confocal microscopy. Further studies are needed to understand the role of mitoKATP channels in cardioprotective strategies in iPSC-CMs especially at moderately high glucose levels where APC provided protection without the measureable aid of mitoKATP channels opening.
Additionally, we investigated the effects of preconditioning on the delay of the mPTP opening. Typically, the mPTP opens during an ischemia-reperfusion injury resulting in an increase in the permeability of large solutes in the inner membrane of the mitochondria. mPTP opening dissipates ΔΨm, initiating cell death pathways.39 Preconditioning cardiomyocytes with volatile anesthetics has been shown to delay the opening of the mPTP.40-41 Delaying the mPTP opening preserves the mitochondrial bioenergetics and overall improves the cellular and mitochondrial function. Our results indicate that APC delays opening of the mPTP in N-iPSC- and DM-iPSC-CMs in 5 mM and 11 mM glucose, but not in 25 mM glucose conditions, which is in line with LDH experiments suggesting that iPSC-CMs derived from type 2 diabetic patients can be preconditioned and high glucose conditions may be playing a key role in attenuating cardioprotective effects provided by APC in these patients. The presence of 25 mM glucose alone attenuated APC-mediated delay in mPTP opening. Studies in rat cardiomyocytes have shown that pyruvate, a product of glycolysis, obliterated APC-induced delay in mPTP opening in ΔΨm-dependent manner.24 The delay in mPTP opening in N-iPSC-CMs in 5 mM and 11 mM glucose was similar to observations made in rat cardiomyocytes19, human adult cardiomyoyctes27, and hESC-CMs.10
A limitation of our study is the use of in vitro generated cardiomyocytes. Our iPSC-CMs may lack some of the characteristics of adult cardiomyocytes. However, with the parameters that we investigated, N-iPSC-CMs responded to APC similarly as other models: rat, rabbit, guinea pig, canine, human, and hESC-CMs that our department has previously investigated. To induce an injury, we generated oxidative stress with H2O2 or photoexcitation, which does not fully represent the conditions of an ischemia and reperfusion event. Specifically the use of 10 mM H2O2 to induce an oxidative stress environment in our iPSC-CMs was excessively higher than H2O2 levels used in animal studies. We have observed that our stem cell-derived cardiomyocytes are more resistant to oxidative stress-induced injury. Further studies are needed to investigate the mechanisms involved in the resistance of iPSC-CMs to oxidative stress. Never the less, H2O2-induced injury is widely accepted for studying reperfusion injury as it mimics the most important type of stress during this injury: oxidative stress. Finally, we solely investigated the effects of APC on cardiomyocytes. APC not only protects cardiomyocytes directly, but also indirectly through its action on other cell types in the heart, such as endothelial cells42, or by modulation of inflammatory response.43
In conclusion, for the first time our study shows that preconditioning with isoflurane protects cardiomyocytes derived from non-diabetic iPSCs from oxidative stress in 5 mM and 11 mM glucose. 25 mM glucose had detrimental effects on cardioprotection provided by APC in both non-diabetic and type 2 diabetic patient-derived cardiac lines. Unexpectedly, type 2 diabetic patient-specific iPSCs could be anesthetically preconditioned in 5 mM and 11 mM glucose. Additional studies and cell lines are needed to further understand the role of genetics in the disease phenotype, and identify potential components that may be contributing to the inability of human normoglycemic diabetic patients to be preconditioned.
The similarity between our study conducted on iPSC-CMs to our previous work in animal models, human cardiomyocytes, and hESC-CMs, proves that cardiomyocytes derived from patient-specific iPSCs are a suitable model to further understand preconditioning in a human model and underlying mechanisms by which disease phenotypes alter the protection provided by APC. Development of iPSC-derived ventricular cardiomyocytes from healthy individuals and patients with various diseases in conjunction with modulating external factors, such as varying glucose levels, provides a powerful tool for translational research.
Supplementary Material
Acknowledgments
The authors thank Katelyn Revak, B.S. (Research Technician, Department of Anesthesiology, Medical College of Wisconsin, Milwaukee, WI) for technical assistance and Aniko Szabo, Ph.D. (Associate Professor, Institute for Health and Society, Division of Biostatistics, Medical College of Wisconsin, Milwaukee, WI) for help with the statistical analysis.
Sources of Funding: This work was supported in part by P01GM066730 and R01HL034708 from the National Institutes of Health, Bethesda, MD, (to Z. J. Bosnjak).
Footnotes
Part of this work has been presented at the Experimental Biology meeting, Washington D.C, April 19, 2011, and at the American Heart Association Basic Cardiovascular Sciences meeting, New Orleans, Louisiana, July 19, 2011.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Solomon CG. Reducing cardiovascular risk in type 2 diabetes. N Engl J Med. 2003;348:457–9. doi: 10.1056/NEJMe020172. [DOI] [PubMed] [Google Scholar]
- 2.Stadnicka A, Marinovic J, Bienengraeber M, Bosnjak ZJ. Impact of in vivo preconditioning by isoflurane on adenosine triphosphate-sensitive potassium channels in the rat heart: Lasting modulation of nucleotide sensitivity during early memory period. Anesthesiology. 2006;104:503–10. doi: 10.1097/00000542-200603000-00018. [DOI] [PubMed] [Google Scholar]
- 3.Ljubkovic M, Mio Y, Marinovic J, Stadnicka A, Warltier DC, Bosnjak ZJ, Bienengraeber M. Isoflurane preconditioning uncouples mitochondria and protects against hypoxia-reoxygenation. Am J Physiol Cell Physiol. 2007;292:C1583–90. doi: 10.1152/ajpcell.00221.2006. [DOI] [PubMed] [Google Scholar]
- 4.Kersten JR, Schmeling TJ, Pagel PS, Gross GJ, Warltier DC. Isoflurane mimics ischemic preconditioning via activation of K(ATP) channels: Reduction of myocardial infarct size with an acute memory phase. Anesthesiology. 1997;87:361–70. doi: 10.1097/00000542-199708000-00024. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka K, Kehl F, Gu WD, Krolikowski JG, Pagel PS, Warltier DC, Kersten JR. Isoflurane-induced preconditioning is attenuated by diabetes. Am J Physiol-Heart C. 2002;282:H2018–H23. doi: 10.1152/ajpheart.01130.2001. [DOI] [PubMed] [Google Scholar]
- 6.Kehl F, Krolikowski JG, Mraovic B, Pagel PS, Warltier DC, Kersten JR. Hyperglycemia prevents isoflurane-induced preconditioning against myocardial infarction. Anesthesiology. 2002;96:183–8. doi: 10.1097/00000542-200201000-00032. [DOI] [PubMed] [Google Scholar]
- 7.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP, Thomson JA, Kamp TJ. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104:e30–41. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zwi L, Caspi O, Arbel G, Huber I, Gepstein A, Park IH, Gepstein L. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation. 2009;120:1513–23. doi: 10.1161/CIRCULATIONAHA.109.868885. [DOI] [PubMed] [Google Scholar]
- 10.Sepac A, Sedlic F, Si-Tayeb K, Lough J, Duncan SA, Bienengraeber M, Park F, Kim J, Bosnjak ZJ. Isoflurane preconditioning elicits competent endogenous mechanisms of protection from oxidative stress in cardiomyocytes derived from human embryonic stem cells. Anesthesiology. 2010;113:906–16. doi: 10.1097/ALN.0b013e3181eff6b7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huber I, Itzhaki I, Caspi O, Arbel G, Tzukerman M, Gepstein A, Habib M, Yankelson L, Kehat I, Gepstein L. Identification and selection of cardiomyocytes during human embryonic stem cell differentiation. Faseb J. 2007;21:2551–63. doi: 10.1096/fj.05-5711com. [DOI] [PubMed] [Google Scholar]
- 12.Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–86. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C, North PE, Dalton S, Duncan SA. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henderson SA, Spencer M, Sen A, Kumar C, Siddiqui MA, Chien KR. Structure, organization, and expression of the rat cardiac myosin light chain-2 gene Identification of a 250-base pair fragment which confers cardiac-specific expression. J Biol Chem. 1989;264:18142–8. [PubMed] [Google Scholar]
- 15.Zhu H, Garcia AV, Ross RS, Evans SM, Chien KR. A conserved 28-base-pair element (HF-1) in the rat cardiac myosin light-chain-2 gene confers cardiac-specific and alpha-adrenergic-inducible expression in cultured neonatal rat myocardial cells. Mol Cell Biol. 1991;11:2273–81. doi: 10.1128/mcb.11.4.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caspi O, Itzhaki I, Kehat I, Gepstein A, Arbel G, Huber I, Satin J, Gepstein L. In vitro electrophysiological drug testing using human embryonic stem cell derived cardiomyocytes. Stem Cells Dev. 2009;18:161–72. doi: 10.1089/scd.2007.0280. [DOI] [PubMed] [Google Scholar]
- 17.Park F, Sweeney WE, Jia G, Roman RJ, Avner ED. 20-HETE mediates proliferation of renal epithelial cells in polycystic kidney disease. J Am Soc Nephrol. 2008;19:1929–39. doi: 10.1681/ASN.2007070771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park F. Correction of bleeding diathesis without liver toxicity using arenaviral-pseudotyped HIV-1-based vectors in hemophilia A mice. Hum Gene Ther. 2003;14:1489–94. doi: 10.1089/104303403769211691. [DOI] [PubMed] [Google Scholar]
- 19.Pravdic D, Sedlic F, Mio Y, Vladic N, Bienengraeber M, Bosnjak ZJ. Anesthetic-induced preconditioning delays opening of mitochondrial permeability transition pore via protein Kinase C-epsilon-mediated pathway. Anesthesiology. 2009;111:267–74. doi: 10.1097/ALN.0b013e3181a91957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–14. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–49. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huser J, Rechenmacher CE, Blatter LA. Imaging the permeability pore transition in single mitochondria. Biophys J. 1998;74:2129–37. doi: 10.1016/S0006-3495(98)77920-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huser J, Blatter LA. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem J. 1999;343(Pt 2):311–7. [PMC free article] [PubMed] [Google Scholar]
- 24.Sedlic F, Sepac A, Pravdic D, Camara AK, Bienengraeber M, Brzezinska AK, Wakatsuki T, Bosnjak ZJ. Mitochondrial depolarization underlies delay in permeability transition by preconditioning with isoflurane: Roles of ROS and Ca2+ Am J Physiol Cell Physiol. 2010;299:C506–15. doi: 10.1152/ajpcell.00006.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sedlic F, Pravdic D, Ljubkovic M, Marinovic J, Stadnicka A, Bosnjak ZJ. Differences in production of reactive oxygen species and mitochondrial uncoupling as events in the preconditioning signaling cascade between desflurane and sevoflurane. Anesth Analg. 2009;109:405–11. doi: 10.1213/ane.0b013e3181a93ad9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanley PJ, Ray J, Brandt U, Daut J. Halothane, isoflurane and sevoflurane inhibit NADH:Ubiquinone oxidoreductase (complex I) of cardiac mitochondria. J Physiol. 2002;544:687–93. doi: 10.1113/jphysiol.2002.025015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mio Y, Bienengraeber MW, Marinovic J, Gutterman DD, Rakic M, Bosnjak ZJ, Stadnicka A. Age-related attenuation of isoflurane preconditioning in human atrial cardiomyocytes: Roles for mitochondrial respiration and sarcolemmal adenosine triphosphate-sensitive potassium channel activity. Anesthesiology. 2008;108:612–20. doi: 10.1097/ALN.0b013e318167af2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lough J, Sugi Y. Endoderm and heart development. Dev Dyn. 2000;217:327–42. doi: 10.1002/(SICI)1097-0177(200004)217:4<327::AID-DVDY1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 29.Postic C, Mauvais-Jarvis F, Girard J. Mouse models of insulin resistance and type 2 diabetes. Ann Endocrinol (Paris) 2004;65:51–9. doi: 10.1016/s0003-4266(04)95630-2. [DOI] [PubMed] [Google Scholar]
- 30.Collins FS. Reengineering translational science: the time is right. Sci Transl Med. 2011;3:90cm17. doi: 10.1126/scitranslmed.3002747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inoue H, Yamanaka S. The use of induced pluripotent stem cells in drug development. Clin Pharmacol Ther. 2011;89:655–61. doi: 10.1038/clpt.2011.38. [DOI] [PubMed] [Google Scholar]
- 32.Laustriat D, Gide J, Peschanski M. Human pluripotent stem cells in drug discovery and predictive toxicology. Biochem Soc Trans. 2010;38:1051–7. doi: 10.1042/BST0381051. [DOI] [PubMed] [Google Scholar]
- 33.Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flugel L, Dorn T, Goedel A, Hohnke C, Hofmann F, Seyfarth M, Sinnecker D, Schomig A, Laugwitz KL. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363:1397–409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 34.Marchetto MC, Brennand KJ, Boyer LF, Gage FH. Induced pluripotent stem cells (iPSCs) and neurological disease modeling: Progress and promises. Hum Mol Genet. 2011;20:R109–15. doi: 10.1093/hmg/ddr336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–21. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- 36.Marinovic J, Bosnjak ZJ, Stadnicka A. Distinct roles for sarcolemmal and mitochondrial adenosine triphosphate-sensitive potassium channels in isoflurane-induced protection against oxidative stress. Anesthesiology. 2006;105:98–104. doi: 10.1097/00000542-200607000-00018. [DOI] [PubMed] [Google Scholar]
- 37.Ghosh S, Standen NB, Galinianes M. Failure to precondition pathological human myocardium. J Am Coll Cardiol. 2001;37:711–8. doi: 10.1016/s0735-1097(00)01161-x. [DOI] [PubMed] [Google Scholar]
- 38.Kersten JR, Montgomery MW, Ghassemi T, Gross ER, Toller WG, Pagel PS, Warltier DC. Diabetes and hyperglycemia impair activation of mitochondrial K(ATP) channels. Am J Physiol Heart Circ Physiol. 2001;280:H1744–50. doi: 10.1152/ajpheart.2001.280.4.H1744. [DOI] [PubMed] [Google Scholar]
- 39.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yellon DM, Davidson SM, Hausenloy D, Duchen MR. Signalling via the reperfusion injury signalling kinase (RISK) pathway links closure of the mitochondrial permeability transition pore to cardioprotection. Int J Biochem Cell B. 2006;38:414–9. doi: 10.1016/j.biocel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 41.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341(Pt 2):233–49. [PMC free article] [PubMed] [Google Scholar]
- 42.Novalija E, Varadarajan SG, Camara AK, An J, Chen Q, Riess ML, Hogg N, Stowe DF. Anesthetic preconditioning: Triggering role of reactive oxygen and nitrogen species in isolated hearts. Am J Physiol Heart Circ Physiol. 2002;283:H44–52. doi: 10.1152/ajpheart.01056.2001. [DOI] [PubMed] [Google Scholar]
- 43.Lango R, Mrozinski P. Clinical importance of anaesthetic preconditioning. Anestezjol Intens Ter. 2010;42:206–12. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.