

Abstract
Sequencing technology enables the complete characterization of human genetic variation. Statistical genetics studies identify numerous loci linked to or associated with phenotypes of direct medical interest. The major remaining challenge is to characterize functionally significant alleles that are causally implicated in the genetic basis of human traits. Here, I review three sources of evidence for the functional significance of human DNA variants in protein-coding genes. These include (i) statistical genetics considerations such as co-segregation with the phenotype, allele frequency in unaffected controls and recurrence; (ii) in vitro functional assays and model organism experiments; and (iii) computational methods for predicting the functional effect of amino acid substitutions. In spite of many successes of recent studies, functional characterization of human allelic variants remains problematic.
INTRODUCTION
Large-scale sequencing projects have revealed the landscape of human genetic variation (1,2). Linkage and association studies identified a large number of loci involved in various human phenotypes. In spite of this spectacular progress, characterization of functionally significant human alleles causally involved in phenotypes (i.e. directly contributing to the biology of phenotypes) remains challenging.
The problem of establishing a causal relationship between a phenotype and a specific sequence variant arises at multiple levels (Table 1). It spans both Mendelian and complex trait genetics, even though many aspects of the problem and approaches to address them are different.
Table 1.
Importance of the functional analysis in various types of human genetics studies
| Analysis of Mendelian traits |
Analysis of rare variants in complex traits |
||||||
|---|---|---|---|---|---|---|---|
| Interpretation of variants in previously mapped genes |
Mapping genes by whole-genome/exome sequencing | Analysis of rare variants in candidate genes |
Mapping genes by whole-genome/exome sequencing | ||||
| Uncharacterized variants not known to be de novo | De novo mutations | Segregating variants | De novo mutations | Rare variants | De novo mutations | Rare variants | De novo mutations |
| Analysis of the functional effect and causality is essential | Usually regarded as sufficient evidence of functionality | Functional analysis is not essential to map genes but can potentially increase power | Functional analysis is essential for isolated mutations. Recurrence may provide a statistical argument in favor of functionality | Functional analysis was shown to increase power | Likely a sufficient evidence of functionality | Functional analysis was hypothesized to increase power | Functional analysis is essential for isolated mutations. Recurrence may provide a statistical argument in favor of functionality |
In the simplest case of a Mendelian monogenic trait unequivocally linked to a particular gene, the problem is in distinguishing between benign and pathogenic alleles in this gene. This creates a major bottleneck in clinical genetic diagnostics (3). Many allelic variants observed in genes of diagnostic importance remain classified as variants of unknown significance (VUSs).
For Mendelian phenotypes with unknown genetic background, sequencing studies now provide a powerful way to identify causal genes. Briefly, the strategy involves finding a gene where all or most patients carry functional variants that are not observed in multiple unaffected controls (4). Usually, all coding non-synonymous variants and variants disrupting canonic splice sites are considered functional and other variants are ignored. Although this strategy generated many successes, it lacks power if sample sizes are small (only two or three patients available) or in case of oligogenic phenotypes. Knowledge of functional significance of allelic variants would greatly empower sequencing studies aiming at mapping genes underlying Mendelian disorders.
Remarkable progress in sequencing technology now allows detecting de novo mutations using parent–child trio sequencing (5). This approach has been successfully applied to a number of Mendelian traits and to complex psychiatric phenotypes such as autism and schizophrenia (5–9). Relatively small number of de novo mutations facilitates the analysis. On average, humans carry of the order of 100 de novo mutations (10–13). However, these mutations are typically unique to individual patients. Therefore, it is impossible to use statistical approaches to infer their involvement in phenotypes in case of whole-exome or whole-genome sequencing experiments.
Naturally, the magnitude of the problem is amplified when considering variants involved in complex traits. Genome-wide association studies (GWAS) identified a multitude of common SNPs associated with human complex traits. However, most of these SNPs are not causal and simply tag causal alleles due to linkage disequilibrium (LD). LD greatly facilitates mapping but equally complicates pinpointing causal variants by statistical means because association signals of many variants are confounded. In many cases, even the identity of a causal gene, rather than a specific allele, is not known. The problem is exacerbated because most of GWAS peaks are in non-coding regions. Moreover, it is possible that multiple causal variants give rise to a single GWAS peak. A number of sequencing projects aiming at finding causal variants underlying GWAS peaks are ongoing. The dominant hypothesis is that the variants responsible for the observed associations are common. Scenarios where associations of common SNPs are caused by low-frequency variants or even by multiple rare variants have also been proposed (14), although subsequent work suggested that such scenarios do not explain many GWAS peaks (15).
Examples of the functional characterization of variants underlying GWAS signals are still rare. One early example includes demonstration that a common variant creating a transcription factor-binding site for the CCAAT/enhancer-binding protein alters the hepatic expression of the SORT1 gene. This variant explains the corresponding GWAS signal for association with LDL-cholesterol (16). Fine-mapping studies have been reported recently for strong association signals within the human leukocyte antigen region (17,18). For common non-coding variants, analysis of intermediate molecular phenotypes related to transcriptional regulation such as mRNA expression (19) and chromatin accessibility (20–22) offers a potential way forward. These early studies on functional effects of common non-coding variants are outside the scope of this review.
A number of successful candidate gene sequencing studies discovered associations of multiple rare coding variants with complex phenotypes (23–32). Ongoing whole-exome sequencing studies attempt an unbiased search for genes harboring multiple rare variants collectively associated with complex traits (33). In the simplest form, this analysis detects an excess of rare coding variants in cases versus controls. The association signal is provided by functional variants, whereas neutral alleles are a source of noise masking the association signal. Again, functional significance of individual rare variants cannot be inferred by statistical means. In contrast to common variants, LD does not confound the signal. However, the association test for individual rare variants lacks statistical power given that they are observed a handful of times (or even once) in the sample. The ability to discriminate between functional and neutral alleles would dramatically increase the potential of sequencing studies focusing on rare variants in complex traits. Several published studies demonstrated that highlighting functional variants using experimental (24,30) or computational approaches (25,27,34) increases the power of these studies.
Understanding the functional significance of human alleles is also of great importance for evolutionary and population genetics. Accurate inference of functional consequences of human DNA variants would help characterize the role of natural selection in shaping population genetic variation (2).
Overall, medical genetics is interested in finding ‘pathogenic’ mutations that causally influence traits of medical interest. Population genetics focuses on ‘deleterious’ alleles that evolve under purifying selection. In contrast, functional analysis is focused on the ‘damaging’ effect on molecular function. The rationale for this approach is that the effects on phenotypes and fitness must be mediated by the effects on molecular function, even though the converse is not necessarily true. Existence of many common loss-of-function variants in humans (34) and events of adaptive pseudogenization (35) clearly show that damaging alleles may be neutral or beneficial rather than deleterious. It is also feasible that most of human alleles that are subject to purifying selection have no detectable effects on medically relevant phenotypes in current environment. However, most studies implicitly assume the strong relationship between the effects on molecular function, fitness and phenotypes. For example, many computational methods for predicting the functional effects of human alleles are based on the inference of purifying selection from comparative genomics data.
Here, I review current strategies to infer causality and functional significance of human protein-coding DNA variants, including variants involved in Mendelian human traits and rare coding variants involved in complex phenotypes.
INFERRING THE FUNCTIONAL SIGNIFICANCE OF MISSENSE MUTATIONS INVOLVED IN MENDELIAN PHENOTYPES
As noted earlier, the problem of assigning functional significance to variants involved in Mendelian phenotypes arises both in the context of gene discovery and in the context of interpreting VUSs in known genes. Overwhelming majority of sequence variants causing Mendelian traits are coding. Among coding variants, ‘missense’ changes are the most difficult to interpret (most of synonymous changes are benign and most of nonsense or splice-site changes are damaging). Three potential strategies to infer causality and functional significance could be employed: (i) the strategy based on statistical genetics, (ii) in vitro or in vivo experimental analysis, and (iii) computational predictions based on evolutionary and structural considerations.
Statistical arguments
In some cases, purely statistical arguments can be employed in favor of the causal relationship between DNA variants and Mendelian traits. Importantly, the arguments discussed in what follows are specific to Mendelian genetics and, in most part, cannot be applied to variants underlying complex phenotypes.
Analysis of co-segregation of the DNA variant with the phenotype is probably the most accurate method to establish causality by statistical means. However, at least five informative meioses are needed to support causality (36), and sufficiently large pedigrees are usually unavailable. In addition, segregation analysis may be misleading if more than one rare variant is present in the locus and co-segregates with the phenotype.
Another important consideration is the analysis of allele frequency in unaffected controls. This analysis has been dramatically facilitated by large-scale sequencing efforts such as 1000 Genomes Project (1) and Exome Sequencing Project (ESP) (2). Presence in healthy controls at appreciable frequency may reveal whether the allelic variant is a benign polymorphism segregating in the population, which will exclude the possibility that this variant is involved in the disease phenotype with high penetrance (this approach is obviously non-informative for variance of incompletely penetrance unless larger case–control study is pursued). Although it is easy to infer that the variant is benign (or, at least, not of high penetrance) if it is seen in a number of unaffected individuals, it is much less clear if its absence in multiple controls may serve as a strong support for the pathogenicity. Most importantly, for some genes such as BRCA1 and BRCA2, the number of sequenced cases vastly exceeds the number of sequenced controls, making the analysis of allele frequency in unaffected controls non-informative. Next, differences in global and even local ancestry may complicate conclusions because many rare variants are specific to individual human populations. Also, ESP contains data on individuals with various diseases, so not all sequenced individuals should be automatically assumed to be unaffected.
Even in the simplest possible case of a variant observed in a single patient with a dominant phenotype absent in a panel of ideally ancestry matched control subjects, the number of control subjects should be very large.
At the first glance, population genetics supports the use of moderate numbers of controls. Under the standard model of a constant size population with no natural selection, the chance that a variant observed in a patient will not be seen in n normal controls is 1/(n + 1) (37). This suggests that if the variant is not found in a 100 controls, then the chance for the mutation to have no phenotypic effect is <1%. Therefore, absence in a moderate number of controls would support pathogenicity. The following factors suggest that this is a stark under-estimate for human populations: (i) human population growth which has resulted in an excess of rare alleles, (ii) selection against moderately deleterious alleles, and (iii) human migrations which have resulted in rare alleles not seen in multiple controls (38–44). As seen from Figure 1, a more complex population genetics model incorporating population growth and natural selection (40) but not migration predicts that there is >1% chance that a benign variant observed in a single patient would not be detected in as many as 10 000 controls. Taking into account the effects of migration would likely make this number even higher. Therefore, the sole observation of the absence in multiple unaffected controls is insufficient to convincingly imply functional significance of a sequence variant.
Figure 1.
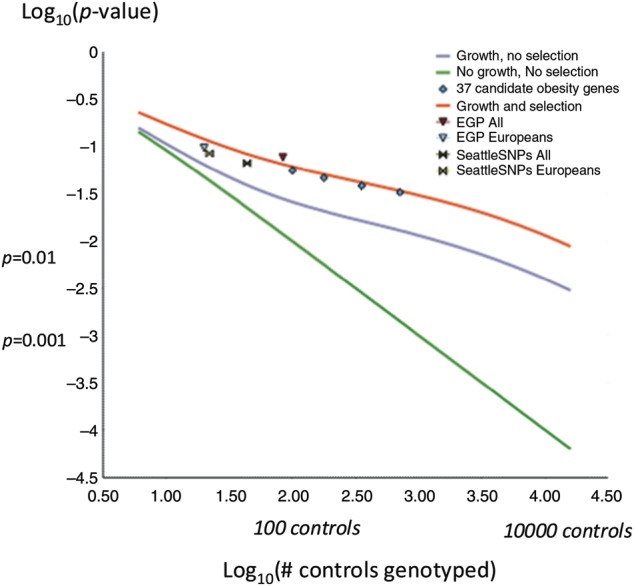
The Probability that a non-pathogenic variant observed in a single patient would not be observed in multiple controls. Log–log scale plot is shown for theoretical model assuming constant population and no natural selection (27) (green line); population genetic model assuming recent population growth and no natural selection (40) (blue line); population genetic model that incorporates both population growth and natural selection (40) (red line). Results of theoretical models are shown together with estimates based on real data obtained by re-sampling from three available systematic re-sequencing data sets [Environmental Genome Project data set (78), Seattle SNP data set (78) and Obesity Sequencing Study data set (27)].
In some cases, the evidence for pathogenicity of specific mutations can be provided by the observation of recurrence. For example, independent occurrence of two exactly same mutations has been observed in the Baraitser–Winter syndrome (45). Three different mutations in the same codon have been reported in the analysis of the Myhre syndrome (46), strongly suggesting the functional importance of this particular amino acid position.
A growing number of publications (examples include references 45,47–49) report de novo mutations as evident from parent–child trio sequencing. The observation of de novo mutation in a gene known to be involved in the phenotype (i.e. a gene under an independently reported linkage peak or a gene with multiple de novo mutations in other families) is highly informative about the functional significance of the mutation. Indeed, the rate of point mutations in humans is of the order of 10−8 per nucleotide per generation and about 10−5 per a protein-coding gene per generation (5,10–13). Therefore, it is unlikely that a de novo mutation unrelated to the phenotype is observed in a known gene. The situation is different, however, in the analysis of whole-exome or whole-genome sequencing without the knowledge of causal genes. Although de novo mutations can be considered excellent candidates, especially for dominantly inherited traits, independent functional validation is usually required.
Experimental evidence
Direct experimental functional analysis is a highly laborious but a highly convincing method to study the effect of human allelic variants. Experimental approaches include the analysis of protein expression and localization, in vitro functional assays and genetic manipulation on model organisms. The enthusiasm for direct experimental methods should be accompanied by a cautionary note that specific aspects of molecular function analyzed using in vitro assays in some cases may be unrelated to the phenotype and effects of mutations on model organisms may be sometimes uninformative about the human condition.
In many cases, missense mutations result in changes of protein expression and localization. Some recent studies relied on immunostaining to assess effects of individual human alleles (50,51). Testing other aspects of protein function requires development of specific functional assays. Phosphorylation assays can be applied for proteins involved in signaling. A recent study of implicated mutations in tyrosine kinase domain of the colony-stimulating factor 1 receptor (CSF1R) in hereditary diffuse leukoencephalopathy serves as an example (52). Autophosphorylation of CSF1R after stimulation with the colony-stimulating factor 1 (CSF1) was used to assay the function of human mutations. Phosphorylation of downstream targets was also examined in the study that identified mutations in AKT3, PIK3R2 and PIK3CA causing a spectrum of related megalencephaly syndromes (49).
Changes in protein–protein interactions can be used to detect the effect of mutations on proteins involved in complexes. In vitro protein aggregation assay was used to test for the function of the co-chaperone DNAJB6 that was shown to cause limb-girdle muscular dystrophy (53).
Functional assay to test lipid metabolism in incubated keranocytes was used in a recent study that linked PNPLA1 to congenital ichthyosis (54). The same study used differentiation assay.
In some cases, mapping mutations on protein 3D structure may provide a key insight into the functional mechanisms. For example, structural localization of KLHL3 mutations causing familial hyperkalemic hypertension shows spatial clustering that helped generating a biological hypothesis (55).
Model organisms amenable to genetic manipulation provide a possibility to test the phenotypic rather than molecular consequences of human allelic variants. The mammalian mouse model has been a model of choice for years to test the phenotypic effect of human genes. However, testing allelic series in the mouse is highly laborious. Zebrafish is being increasingly used to test the effect of human mutations because this vertebrate species is a powerful genetic model and a convenient system to screen for phenotypes. The approach involves knocking down the fish ortholog of the human gene and assaying the phenotypic effect. Next, if injecting human wild-type mRNA results in a phenotypic rescue, individual alleles can be tested for the potential to rescue the phenotype. Last, co-injection of wild type and mutant mRNAs provides a test for dominant negative effects. Recent examples of the successful application of this approach include the analysis of mutations in co-chaperone DNAJB6 (53) and mutations in the RNA exosome component causing pontocerebellar hypoplasia and spinal motor neuron degeneration (56). Zebrafish model was employed with great success in characterizing multiple variants in several genes involved in ciliopathies (57).
In some cases, much more distant model organisms appear helpful in interpreting human mutations. For example, a yeast system was successfully used to functionally characterize 84 human variants observed in patients with cystathionine-β-synthase deficiency (58).
Interestingly, dog is another species that helps establish the relationship between human mutations and phenotypes (54).
Computational predictions
Additional supporting evidence for the functional significance of missense mutations can be provided by computational prediction algorithms. At this time, the accuracy of computational predictions is ∼75–80% (59), with the accuracy estimates dependent on data sets or databases that are used to define pathogenic and benign variants. Thus, the computational analysis is less informative than direct experimental evidence. However, given that the computational methods do not involve any additional labor and cost and can be applied to any gene, many studies rely, at least in part, on the application of computational methods. The accuracy of the methods can be higher for highly confident predictions (60). If accompanied with rigorous accuracy estimation on a disease-specific data set, bioinformatically derived prediction information may assist clinical decision-making. Current ACMG and IARC recommendations endorse the application of computational methods in genetic diagnostics but only in combination with other criteria (3,61). As more protein sequences and structures accompanied by training data (known disease-causing mutations and neutral polymorphisms) are available, the classification accuracy will improve. At the same time, principle difficulties in improving the accuracy of prediction methods remain and are discussed at the end of this section.
Despite the variety of approaches that exist (Table 2), these methods all rely heavily on the two fundamental observations. First, the regions of proteins which are critical to function evolve under long-term negative selection; thus when the sequence of a human protein is aligned for comparison with its homologs from other species, these sites will display only specific patterns of amino acid residue variation or complete conservation. The analysis of phylogenetic information in the form of multiple sequence alignment is a powerful source of information about the spectrum of residues allowed at a particular position of the protein of interest (62–64). Second, most pathogenic mutations affect protein stability (65,66). In general, the prediction techniques based on protein spatial structure can be applied only if the structure has been resolved for the query protein or its close homolog, which is only true for a minor fraction of human proteins. However, even for the proteins with known spatial structure, structure-based methods work best only in addition to phylogeny-based approach and provide only a slight increase in the accuracy of the methods (67,68).
Table 2.
A selection of online tools for predicting the functional effect of protein-coding variants
| AlignGVGD | Conservation of physico-chemical properties | agvgd.iarc.fr |
| Condel | Prediction method based on combining other methods | bg.upf.edu/condel |
| MAPP | Conservation of physico-chemical properties | mendel.stanford.edu/sidowlab/downloads/MAPP |
| MutationTaster | Bayes classifier over multiple sequence features and conservation | mutationtaster.org |
| PMut | Evolutionary and structural features combined using a machine learning method | mmb2.pcb.ub.es:8080/PMut |
| PolyPhen-2 | Evolutionary and structural features combined using naïve Bayes classifier | genetics.bwh.harvard.edu/pph2 |
| SIFT | Evolutionary method based on position-specific scoring matrix | sift-dna.org |
| SNAP | Several evolutionary and structural features combined using a neural network | www.rostlab.org/services/snap |
| SNPs3D | Combination of a phylogenetic and a structural method. Uses support vector machine | www.snps3d.org |
Although existing methods all rely on evolutionary pattern and, sometimes, protein structure, they differ in algorithmic details. For example, SIFT (62) and PolyPhen-2 (67) estimate the probability that the mutant amino acid would fit the amino acid position given the observed substitution pattern. MAPP (69) analyzes the conservation of physico-chemical properties of amino acids, LRT (70) and GERP (71) estimate selective constraint. The methods based on multiple features also differ in machine learning algorithms they employ. For example, MutationTaster (72) and PolyPhen-2 (67) rely on the naïve Bayes classifier, and SNAP (73) utilizes a neural network. Although different methods use essentially the same information, the methods are surprisingly commonly discordant. This can be only in part explained by different threshold settings. This observation motivated the development of ‘umbrella’ methods that combine predictions made by different algorithms such as Condel (74).
The accuracy of the methods can be potentially improved if the scope of the methods would be narrower, so they would be specifically focused on a single phenotype and a group of genes involved in this phenotype. Such methods employ gene-specific training data sets, gene phylogeny, protein features and classification rules optimized for a particular set of genes involved in a specific disease. Recently developed methods include a method focused on the BRCA1 gene involved in risk of breast and ovarian cancer (75) and a method focused on genes encoding proteins of the heart sarcomere involved in hypertrophic cardiomyopathy (36).
Two important basic effects hamper further development of new prediction methods of higher accuracy. First, the existing approaches may have intrinsic difficulties differentiating between mutations of large effect, important for genetic diagnostics, and slightly deleterious sequence variants in phylogenetically conserved positions, whose existence in genomes of apparently healthy humans is confirmed by numerous resequencing studies. Second, it was shown that human disease mutations are occasionally observed as wild-type alleles in vertebrate orthologs (76). Most likely, this is due to epistatic interactions. Compensatory sequence changes enable amino acid changes corresponding to disease mutations in humans to be benign in a different genetic background. Current prediction methods analyze substitution patterns at individual positions and do not account for epistatic interactions. Compensatory changes should be taken into account to substantially increase the accuracy of computational approaches.
FUNCTIONAL ANALYSIS OF RARE NON-SYNONYMOUS VARIANTS INVOLVED IN COMPLEX PHENOTYPES
The analysis of complex traits presents a different set of issues. In this review, I limit the discussion to rare non-synonymous variants in complex traits and leave out the discussion of functional effects of common variants identified by GWAS.
There is a growing interest in the role of rare variants in human complex traits. This interest combined with the availability of next-generation sequencing technology that propels ongoing whole-exome sequencing studies [also discussed in the same journal issue (77)]. For individual very rare variants, the phenotypic effect cannot be identified by the association test. Co-segregation is non-informative about variants involved in complex traits. The existing statistical approaches analyze rare variants collectively, grouping them by gene or pathway (32). In this approach, the statistical signal provided by functionally significant variants is frequently masked by noise due to benign alleles included in the same statistical test. Candidate gene-based studies showed that focusing on functionally significant alleles can increase statistical signal and, hence, the power to detect association between presence of rare variants and complex traits. The signal of association of rare variants in melatonin receptor 1B (MTNR1B) with type 2 diabetes increases if only variants that affect melatonin binding are considered (30). In vitro experiments also helped to increase statistical signal of association in ANGPL genes with triglycerides (24).
Statistical power of exome sequencing studies is expected to be relatively low (40), so the knowledge of functional variants would potentially help identifying genes harboring rare variants associated with complex traits. Using experimental approaches at the whole-exome scale is not feasible. Some studies argued that computational methods for predicting the functional effect of human non-synonymous alleles might be used to increase the power of sequencing studies (27,33). Some statistical methods allow weighting alleles based on potential functional effects. Likely, most sequencing studies would employ both tests weighted with predicted functional significance and tests grouping all non-synonymous variants disregarding predicted effect on function.
CONCLUSION
Assigning functional significance to human alleles and inferring the causal relationship between DNA variants and phenotypes remains the central issue in human genetics. The most efficient way forward would combine statistical genetics considerations, in vivo and in vitro experimental studies and computational approaches. Low throughput of current experimental methods and insufficient accuracy of computational predictions should be addressed to confidently annotate massive data on human genetic variation from the functional perspective.
An additional issue raising the problem to even a greater level of complexity is that, in many cases, the same functional variant can have different phenotypic consequences varying in both expressivity and penetrance depending on other genetic and environmental factors.
FUNDING
I acknowledge the support of NIH grants R01MH084676 and R01GM078598.
ACKNOWLEDGEMENTS
I am grateful to Gregory Kryukov for the analysis presented in Figure 1.
Conflict of Interest statement. None declared.
REFERENCES
- 1.1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tennessen J.A., Bigham A.W., O'Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plon S.E., Eccles D.M., Easton D. IARC Unclassified Genetic Variants Working Group. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008;29:1282–1291. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A., et al. Exome sequencing identifies the cause of a Mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roach J.C., Glusman G., Smit A.F., Huff C.D., Hubley R., Shannon P.T., Rowen L., Pant K.P., Goodman N., Bamshad M., et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neale B.M., Kou Y., Liu L., Ma'ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V., et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D., et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L., et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu B., Roos J.L., Dexheimer P., Boone B., Plummer B., Levy S., Gogos J.A., Karayiorgou M. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat. Genet. 2011;43:864–868. doi: 10.1038/ng.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kondrashov A.S. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum. Mutat. 2003;21:12–27. doi: 10.1002/humu.10147. [DOI] [PubMed] [Google Scholar]
- 11.Nachman M.W., Crowell S.L. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kong A., Frigge M.L., Masson G., Besenbacher S., Sulem P., Magnusson G., Gudjonsson S.A., Sigurdsson A., Jonasdottir A., Jonasdottir A., et al. Rate of de novo mutations and the importance of father's age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun J.X., Helgason A., Masson G., Ebenesersdóttir S.S., Li H., Mallick S., Gnerre S., Patterson N., Kong A., Reich D., Stefansson K. A direct characterization of human mutation based on microsatellites. Nat. Genet. 2012 doi: 10.1038/ng.2398. doi, 10.1038/ng.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dickson S.P., Wang K., Krantz I., Hakonarson H., Goldstein D.B. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wray N.R., Purcell S.M., Visscher P.M. Synthetic associations created by rare variants do not explain most GWAS results. PLoS Biol. 2011;9:e1000579. doi: 10.1371/journal.pbio.1000579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musunuru K., Strong A., Frank-Kamenetsky M., Lee N.E., Ahfeldt T., Sachs K.V., Li X., Li H., Kuperwasser N., Ruda V.M., et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raychaudhuri S., Sandor C., Stahl E.A., Freudenberg J., Lee H.S., Jia X., Alfredsson L., Padyukov L., Klareskog L., Worthington J., et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 2012;44:291–296. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pereyra F., Jia X., McLaren P.J., Telenti A., de Bakker P.I., Walker B.D., Ripke S., Brumme C.J., Pulit S.L., et al. International HIV Controllers Study. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stranger B.E., Nica A.C., Forrest M.S., Dimas A., Bird C.P., Beazley C., Ingle C.E., Dunning M., Flicek P., Koller D., et al. Population genomics of human gene expression. Nat. Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McDaniell R., Lee B.K., Song L., Liu Z., Boyle A.P., Erdos M.R., Scott L.J., Morken M.A., Kucera K.S., Battenhouse A., et al. Heritable individual-specific and allele-specific chromatin signatures in humans. Science. 2010;328:235–239. doi: 10.1126/science.1184655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Degner J.F., Pai A.A., Pique-Regi R., Veyrieras J.B., Gaffney D.J., Pickrell J.K., De Leon S., Michelini K., Lewellen N., Crawford G.E., et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482:390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen J.C., Kiss R.S., Pertsemlidis A., Marcel Y.L., McPherson R., Hobbs H.H. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 24.Romeo S., Yin W., Kozlitina J., Pennacchio L.A., Boerwinkle E., Hobbs H.H., Cohen J.C. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J. Clin. Invest. 2009;119:70–79. doi: 10.1172/JCI37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji W., Foo J.N., O'Roak B.J., Zhao H., Larson M.G., Simon D.B., Newton-Cheh C., State M.W., Levy D., Lifton R.P. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008;40:592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johansen C.T., Wang J., Lanktree M.B., McIntyre A.D., Ban M.R., Martins R.A., Kennedy B.A., Hassell R.G., Visser M.E., Schwartz S.M., et al. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat. Genet. 2010;42:684–687. doi: 10.1038/ng.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahituv N., Kavaslar N., Schackwitz W., Ustaszewska A., Martin J., Hebert S., Doelle H., Ersoy B., Kryukov G., Schmidt S., et al. Medical sequencing at the extremes of human body mass. Am. J. Hum. Genet. 2007;80:779–791. doi: 10.1086/513471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rivas M.A., Beaudoin M., Gardet A., Stevens C., Sharma Y., Zhang C.K., Boucher G., Ripke S., Ellinghaus D., Burtt N., Fennell T., et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jordan C.T., Cao L., Roberson E.D., Duan S., Helms C.A., Nair R.P., Duffin K.C., Stuart P.E., Goldgar D., Hayashi G., et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am. J. Hum. Genet. 2012;90:796–808. doi: 10.1016/j.ajhg.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonnefond A., Clément N., Fawcett K., Yengo L., Vaillant E., Guillaume J.L., Dechaume A., Payne F., Roussel R., Czernichow S., et al. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat. Genet. 2012;44:297–301. doi: 10.1038/ng.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Momozawa Y., Mni M., Nakamura K., Coppieters W., Almer S., Amininejad L., Cleynen I., Colombel J.F., de Rijk P., Dewit O., et al. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat. Genet. 2011;43:43–47. doi: 10.1038/ng.733. [DOI] [PubMed] [Google Scholar]
- 32.Kiezun A., Garimella K., Do R., Stitziel N.O., Neale B.M., McLaren P.J., Gupta N., Sklar P., Sullivan P.F., Moran J.L. Exome sequencing and the genetic basis of complex traits. Nat. Genet. 2012;44:623–630. doi: 10.1038/ng.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Price A.L., Kryukov G.V., de Bakker P.I., Purcell S.M., Staples J., Wei L.J., Sunyaev S.R. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 2010;86:832–838. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacArthur D.G., Balasubramanian S., Frankish A., Huang N., Morris J., Walter K., Jostins L., Habegger L., Pickrell J.K., Montgomery S.B., et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X., Grus W.E., Zhang J. Gene losses during human origins. PLoS Biol. 2006;4:e52. doi: 10.1371/journal.pbio.0040052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jordan D.M., Kiezun A., Baxter S.M., Agarwala V., Green R.C., Murray M.F., Pugh T., Lebo M.S., Rehm H.L., Funke B.H., Sunyaev S.R. Development and validation of a computational method for assessment of missense variants in hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2011;88:183–192. doi: 10.1016/j.ajhg.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitchell A.A., Chakravarti A., Cutler D.J. On the probability that a novel variant is a disease-causing mutation. Genome Res. 2005;15:960–966. doi: 10.1101/gr.3761405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marth G.T., Czabarka E., Murvai J., Sherry S.T. The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics. 2004;166:351–372. doi: 10.1534/genetics.166.1.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williamson S.H., Hernandez R., Feldel-Alon A., Zhe L., Nielsen R., Bustamante C.D. Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc. Natl Acad. Sci. USA. 2005;102:7882–7887. doi: 10.1073/pnas.0502300102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kryukov G.V., Shpunt A., Stamatoyannopoulos J.A., Sunyaev S.R. Power of deep, all-exon resequencing for discovery of human trait genes. Proc. Natl Acad. Sci. USA. 2009;106:3871–3876. doi: 10.1073/pnas.0812824106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sunyaev S.R., Lathe W.C., III, Ramensky V.E., Bork P. SNP frequencies in human genes an excess of rare alleles and differing modes of selection. Trends Genet. 2000;16:335–337. doi: 10.1016/s0168-9525(00)02058-8. [DOI] [PubMed] [Google Scholar]
- 42.Kryukov G.V., Pennacchio L.A., Sunyaev S.R. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am. J. Hum. Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyko A.R., Williamson S.H., Indap A.R., Degenhardt J.D., Hernandez R.D., Lohmueller K.E., Adams M.D., Schmidt S., Sninsky J.J., Sunyaev S.R., et al. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet. 2008;4:e1000083. doi: 10.1371/journal.pgen.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y., Vinckenbosch N., Tian G., Huerta-Sanchez E., Jiang T., Jiang H., Albrechtsen A., Andersen G., Cao H., Korneliussen T., et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat. Genet. 2010;42:969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 45.Rivière J.B., van Bon B.W., Hoischen A., Kholmanskikh S.S., O'Roak B.J., Gilissen C., Gijsen S., Sullivan C.T., Christian S.L., Abdul-Rahman O.A., et al. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat. Genet. 2012;44:440–444. doi: 10.1038/ng.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le Goff C., Mahaut C., Abhyankar A., Le Goff W., Serre V., Afenjar A., Destrée A., di Rocco M., Héron D., Jacquemont S., et al. Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat. Genet. 2011;44:85–88. doi: 10.1038/ng.1016. [DOI] [PubMed] [Google Scholar]
- 47.Van Houdt J.K., Nowakowska B.A., Sousa S.B., van Schaik B.D., Seuntjens E., Avonce N., Sifrim A., Abdul-Rahman O.A., van den Boogaard M.J., Bottani A., et al. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat. Genet. 2012;44:445–449. doi: 10.1038/ng.1105. [DOI] [PubMed] [Google Scholar]
- 48.Heinzen E.L., Swoboda K.J., Hitomi Y., Gurrieri F., Nicole S., de Vries B., Tiziano F.D., Fontaine B., Walley N.M., Heavin S., et al. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat. Genet. 2012;44:1030–1034. doi: 10.1038/ng.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rivière J.B., Mirzaa G.M., O'Roak B.J., Beddaoui M., Alcantara D., Conway R.L., St-Onge J., Schwartzentruber J.A., Gripp K.W., Nikkel S.M., et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet. 2012;44:934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boileau C., Guo D.C., Hanna N., Regalado E.S., Detaint D., Gong L., Varret M., Prakash S.K., Li A.H., d'Indy H., et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012;44:916–921. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wortmann S.B., Vaz F.M., Gardeitchik T., Vissers L.E., Renkema G.H., Schuurs-Hoeijmakers J.H., Kulik W., Lammens M., Christin C., Kluijtmans L.A., et al. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat. Genet. 2012;44:797–802. doi: 10.1038/ng.2325. [DOI] [PubMed] [Google Scholar]
- 52.Rademakers R., Baker M., Nicholson A.M., Rutherford N.J., Finch N., Soto-Ortolaza A., Lash J., Wider C., Wojtas A., DeJesus-Hernandez M., et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 2011;44:200–205. doi: 10.1038/ng.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sarparanta J., Jonson P.H., Golzio C., Sandell S., Luque H., Screen M., McDonald K., Stajich J.M., Mahjneh I., Vihola A., et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 2012;44:450–455. doi: 10.1038/ng.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grall A., Guaguère E., Planchais S., Grond S., Bourrat E., Hausser I., Hitte C., Le Gallo M., Derbois C., Kim G.J., et al. PNPLA1 mutations cause autosomal recessive congenital ichthyosis in golden retriever 4s and humans. Nat. Genet. 2012;44:140–147. doi: 10.1038/ng.1056. [DOI] [PubMed] [Google Scholar]
- 55.Louis-Dit-Picard H., Barc J., Trujillano D., Miserey-Lenkei S., Bouatia-Naji N., Pylypenko O., Beaurain G., Bonnefond A., Sand O., Simian C., et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet. 2012;44:456–460. doi: 10.1038/ng.2218. [DOI] [PubMed] [Google Scholar]
- 56.Wan J., Yourshaw M., Mamsa H., Rudnik-Schöneborn S., Menezes M.P., Hong J.E., Leong D.W., Senderek J., Salman M.S., Chitayat D., et al. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat. Genet. 2012;44:704–708. doi: 10.1038/ng.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zaghloul N.A., Katsanis N. Zebrafish assays of ciliopathies. Methods Cell Biol. 2011;105:257–272. doi: 10.1016/B978-0-12-381320-6.00011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mayfield J.A., Davies M.W., Dimster-Denk D., Pleskac N., McCarthy S., Boydston E.A., Fink L., Lin X.X., Narain A.S., Meighan M., Rine J. Surrogate genetics and metabolic profiling for characterization of human disease alleles. Genetics. 2012;190:1309–1323. doi: 10.1534/genetics.111.137471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hicks S., Wheeler D.A., Plon S.E., Kimmel M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum. Mutat. 2011;32:661–668. doi: 10.1002/humu.21490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jordan D.M., Ramensky V.E., Sunyaev S.R. Human allelic variation: perspective from protein function, structure, and evolution. Curr. Opin. Struct. Biol. 2010;20:342–350. doi: 10.1016/j.sbi.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Richards C.S., Bale S., Bellissimo D.B., Das S., Grody W.W., Hegde M.R., Lyon E., Ward B.E. Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet. Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 62.Ng P.C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sunyaev S.R., Ramensky V.E., Koch I., Lathe W., III, Kondrashov A.S., Bork P. Prediction of deleterious human alleles. Hum. Mol. Genet. 2001;15:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 64.Chasman D., Adams R.M. Predicting the functional consequences of non-synonymous single nucleotide polymorphisms: structure-based assessment of amino acid variation. J. Mol. Biol. 2001;307:683–706. doi: 10.1006/jmbi.2001.4510. [DOI] [PubMed] [Google Scholar]
- 65.Yue P., Li Z., Moult J. Loss of protein structure stability as a major causative factor in monogenic disease. J. Mol. Biol. 2005;353:459–473. doi: 10.1016/j.jmb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 66.Potapov V., Cohen M., Schreiber G. Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Eng. Des. Sel. 2009;22:553–560. doi: 10.1093/protein/gzp030. [DOI] [PubMed] [Google Scholar]
- 67.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kumar S., Suleski M.P., Markov G.J., Lawrence S., Marco A., Filipski A.J. Positional conservation and amino acids shape the correct diagnosis and population frequencies of benign and damaging personal amino acid mutations. Genome Res. 2009;19:1562–1569. doi: 10.1101/gr.091991.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stone E.A., Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005;15:978–986. doi: 10.1101/gr.3804205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chun S., Fay J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cooper G.M., Goode D.L., Ng S.B., Sidow A., Bamshad M.J., Shendure J., Nickerson D.A. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat. Methods. 2010;7:250–251. doi: 10.1038/nmeth0410-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 73.Bromberg Y., Yachdav G., Rost B. SNAP predicts effect of mutations on protein function. Bioinformatics. 2008;24:2397–2398. doi: 10.1093/bioinformatics/btn435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.González-Pérez A., López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 2011;88:440–449. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Karchin R., Monteiro A.N., Tavtigian S.V., Carvalho M.A., Sali A. Functional impact of missense variants in BRCA1 predicted by supervised learning. PLoS Comput. Biol. 2007;3:e26. doi: 10.1371/journal.pcbi.0030026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kondrashov A.S., Sunyaev S., Kondrashov F.A. Dobzhansky-Muller incompatibilities in protein evolution. Proc. Natl Acad. Sci. USA. 2002;99:14878–14883. doi: 10.1073/pnas.232565499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Do R., Kathiresan S., Abecasis G. Exome sequencing and complex disease: practical aspects of rare variant association studies. Hum. Mol. Genet. doi: 10.1093/hmg/dds387. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Livingston R.J., von Niederhausern A., Jegga A.G., Crawford D.C., Carlson C.S., Rieder M.J., Gowrisankar S., Aronow B.J., Weiss R.B., Nickerson D.A. Pattern of sequence variation across 213 environmental response genes. Genome Res. 2004;14:1821–1831. doi: 10.1101/gr.2730004. [DOI] [PMC free article] [PubMed] [Google Scholar]