

Abstract
The major histocompatibility complex (MHC) region on the short arm of chromosome 6 harbors the largest number of replicated associations across the human genome for a wide range of diseases, but the functional basis for these associations is still poorly understood. One fundamental challenge in fine-mapping associations to functional alleles is the enormous sequence diversity and broad linkage disequilibrium of the MHC, both of which hamper the cost-effective interrogation in large patient samples and the identification of causal variants. In this review, we argue that there is now a valuable opportunity to leverage existing genome-wide association study (GWAS) datasets for in-depth investigation to identify independent effects in the MHC. Application of imputation to GWAS data facilitates comprehensive interrogation of the classical human leukocyte antigen (HLA) loci. These datasets are, in many cases, sufficiently large to give investigators the ability to disentangle effects at different loci. We also explain how querying variation at individual amino acid positions for association can be powerful and expand traditional analyses that focus only on the classical HLA types.
INTRODUCTION
Across the entire genome, the major histocompatibility complex (MHC) region stands out by virtue of harboring the largest number of bona fide genetic associations for a range of conditions. These not only include inflammatory, autoimmune and infectious diseases but also different forms of cancer, drug-induced hypersensitivity and, more recently, neuropsychiatric disease. The earliest associations within the MHC were identified in the 1970s, using serological reagents and cell-based assays that measure different human leukocyte antigens (HLAs) (1,2). These initial studies revealed large effects that could be detected with relatively small sample sizes. Decades later, despite the impressive cumulative list of MHC associations, there has been a surprising lack of progress in pinpointing causal variants underlying these disease associations.
The MHC is often regarded as one of the most complex regions of the genome because of its enormous sequence diversity, broad linkage disequilibrium (LD) and high gene density (3–5). This complexity has severely hampered efforts to study the region in detail. As a result of LD, multiple nearby variants may have equivalent statistical evidence of association, and thus it can be difficult to assign causality to a specific variant (6). Inability to pin down a causal variant limits the ability of investigators to elucidate the functional mechanisms of validated associations and to understand disease pathogenesis. Even larger sample sizes than were required for their initial discovery will likely be needed to reach sufficient power to tease apart the role of correlated variants. The question remains to what extent imputation with dense reference panels (like 1000 Genomes) or genotyping with dense SNP arrays (like Immunochip) might help to narrow associations with functional variation at known loci (7,8). The MHC region provides a ripe opportunity to investigate these specific issues.
In this review, we highlight the potential for fine-mapping associations within the MHC. For these studies to be done effectively large numbers of samples need to be typed for HLA alleles, which are potentially driving many of the observed associations. Imputation-based approaches, specifically developed for the HLA genes within the MHC, make it possible to type classical alleles in silico for large sample sets with available data from genome-wide association studies (GWASs) (9–13). We make the case that such methods will likely add substantial value to the existing GWAS datasets. Our aim is to demystify some of the perceived complexities in this subfield and to propose a systematic way to interrogate genetic variation in the MHC.
MHC IN A NUTSHELL
The MHC comprises a contiguous 4 Mb region on the short arm of chromosome 6. The extended MHC (xMHC), as its name suggests, spans an even larger 7.6 Mb region and comprises more than 400 annotated genes and pseudogenes (3). Figure 1 provides an overview of the region, highlighting key genes and known genetic associations. Of these genes, the classical HLA genes encode cell surface antigen-presenting proteins, and as key components of the immune system, they are the most intensely studied (in particular, HLA-A, -B and -C in the class I region and HLA-DQA1, -DQB1 and -DRB1 in the class II region). The class I molecules HLA-A, -B and -C present foreign peptides to CD8+ cytotoxic T cells, whereas the class II molecules HLA-DR, HLA-DQ and HLA-DP present peptides to CD4+ helper T cells. The class II molecules form heterodimers based on subunits encoded by HLA-DRA, -DQA1 and -DPA1 (for the α-subunit) and by HLA-DRB1 (and its homologs -DRB3, -DRB4 and -DRB5), -DQB1 and -DPB1 (for the β-subunit). Other genes that have been extensively studied within the xMHC region include tumor necrosis factor, and members of the complement and antigen-processing pathways. Other MHC genes with a less obvious link to immunity include the histones, tRNAs and olfactory receptors. The combination of the high gene density and extensive LD results in disease associations spanning many genes with diverse function, and this can challenge the interpretation of observed genetic associations. The so-called 8.1 ancestral haplotype is a case in point; this haplotype spans essentially the entire MHC from class I to II and has been implicated in multiple immune disorders (14). Of course, there is also the possibility that non-HLA genes play a role in complex diseases; for example, C2/CFB in age-related macular degeneration (15).
Figure 1.

Overview of the MHC region with validated GWAS associations from the NHGRI GWAS catalog (http://www.genome.gov/gwastudies/) plotted as a function of position along chromosome 6 (hg18). Strongest SNP associations are annotated by trait or disease. Classical HLA-A, -B, -C, -DP, -DQ and -DR genes are highlighted (red vertical ticks) among the many hundreds of genes across the region.
A COMPLEX NOMENCLATURE
Extreme sequence variation is one of the characteristic features of HLA genes. This is evidenced by sequence data from the International Immunogenetics database (IMGT/HLA v 3.9); more than 8000 unique alleles have been documented for the classical HLA genes (16). With >2600 reported haplotypes, HLA-B is probably the most polymorphic gene of the genome. Within the HLA coding regions, some codons contain multiple SNPs (sometimes with more than two alleles), creating a range of possible amino acids at certain sites in HLA proteins. Over the years, an elaborate nomenclature system has been developed to catalog allelic variation observed in each HLA gene (17), firmly rooted in the original antibody-based serotyping used to study HLA molecular isoforms. In this nomenclature, the two-digit type (e.g. HLA-DRB1*01) refers to groups of alleles defined based on serological reagents (‘serotyping’) that were widely used before the advent of polymerase chain reaction (PCR)-based reagents and DNA sequencing. Since antibodies bind to the exposed extra-cellular aspects of an HLA molecule, differences in two-digit alleles reflect mostly variation in the exposed parts of HLA protein structures. The four-digit type (e.g. HLA-DRB1*01:01) refers to a unique amino acid sequence of the gene product; that is, synonymous changes are ignored. Synonymous changes within exons and non-coding changes outside exons are defined by six- and eight-digit types, respectively. All these haplotype definitions are codified by the standard HLA dictionary and use naming conventions that may seem rather cryptic to a non-expert. Figure 2 shows HLA-DRB1*01 and *02 alleles in a multiple sequence alignment, highlighting relations between two-, four- and six-digit types.
Figure 2.

Illustration of HLA-DRB1 variation at two-, four- and six-digit resolution. Here we show a subset of the HLA-DRB1 cDNA sequences, using HLA-DRB1*01:01:01 as a reference. Yellow indicates reference alleles within a sequence, and black represents the presence of a non-reference allele. At the two-digit level, allelic differences cluster in specific regions that may influence binding of serological reagents. At the four-digit level, additional non-synonymous polymorphisms are present, so that each four-digit allele defines a unique amino acid sequence. At the six-digit level, synonymous polymorphisms are taken into account.
It is important to emphasize that the nomenclature does not naturally capture all SNPs, indels or polymorphic amino acid positions. Instead, it defines gene-wide haplotypes, where each unique haplotype gets its own unique number. Historically, the nomenclature has evolved from grouping HLA molecules according to serotype (at two-digit resolution) to a more complete classification on the basis of protein-changing and non-coding variation in DNA sequence (four-digit and beyond). The hierarchical structure of the HLA nomenclature reveals the implicit assumption about the relative functional impact of non-synonymous, synonymous and non-coding variation.
GENOTYPING HLA ALLELES
There is a long history in the immunogenetics community of sharing protocols, reagents and expertise, facilitated by the International HLA and Immunogenetics Workshop series (the 16th meeting took place in May/June 2012 in Liverpool, UK). The current standard for HLA typing involves the design of sequence-specific primers to isolate and amplify the desired HLA gene(s) of interest followed by Sanger sequencing. Most studies focus on capturing coding variations within the classical HLA genes (that is, to four-digit resolution), typically achieved by querying the exons coding for the peptide-binding grooves of the HLA proteins where most of the inter-individual diversity is concentrated. Indeed, a commonly used protocol for typing class I genes involves sequencing exons 2 and 3, whereas typing of class II genes is often based on sequencing only exon 2. More comprehensive (unbiased) protocols exist, but are more costly due to the additional PCR steps and greater sequencing burden to cover the remaining exons.
One limitation of Sanger sequencing is that it is often difficult to resolve closely related alleles (referred to as genotyping ambiguity), because Sanger sequencing assays a diploid mixture with no straightforward way to establish the haplotype phase between nearby sites. Novel protocols based on next-generation sequencing may offer an attractive alternative (18–20).
We note that it is not feasible to use high-throughput microarray-based SNP typing (which has propelled GWAS in the last few years) to query HLA genes directly, because the sequence diversity and polymorphism density makes it virtually impossible to design a robust set of oligonucleotide probes. As a result, most SNPs within the MHC on genotyping microarrays are located away from the polymorphic hotspots in the classical HLA genes. Their corresponding oligonucleotide probes for these SNPs can robustly differentiate between the known alleles and do not suffer from interference from neighboring SNPs within the probe sequence. In terms of SNP density, the earliest microarrays contain a few hundred SNPs across the MHC, whereas the more recent Immunochip includes thousands of SNPs.
IMPUTATION OF CLASSICAL HAPLOTYPES
Instead of genotyping, it is possible to impute classical HLA alleles by inferring them from surrounding SNP markers across the MHC (4). In the simplest case, an HLA allele might be ‘tagged’ by an SNP nearby that is in perfect LD. Alternatively, an HLA allele might be tagged by a set (haplotype) of multiple markers. More complex haplotype patterns can be exploited using sophisticated imputation algorithms, which are now well established in the context of GWAS and meta-analysis (7).
Admittedly, SNP-based imputation is unlikely to replace classical typing for clinical applications, such as organ matching for transplantation, where accuracy in a single individual is critical. Imputation might however be an attractive alternative for large-scale association studies, considering that SNPs are not only easy and cheap to genotype but are also widely available due to the recent investment into GWAS. If errors are randomly distributed, and not systematic in nature, imputation might result in a loss in power without an increased false-positive rate. This makes imputation ideal for the re-analysis of the existing GWAS datasets.
Since the MHC is characterized by extensive LD, it is highly amenable to effective imputation, despite its high overall haplotype diversity. This LD is the consequence of a relatively low recombination rate (almost 3-fold lower compared with the genome-wide average of 1.2 cM/Mb) (4). The first high-resolution genetic map of the MHC was constructed by genotyping >6000 SNPs and by collecting classical four-digit HLA types in four HapMap populations (CEU for Northern and Western European, YRI for African, and CHB and JPT for East-Asian populations) (4). This public resource provided for the first time a detailed picture of the LD structure between SNPs and classical HLA alleles, and demonstrated that specific SNPs (often at considerable distance) were informative for classical HLA alleles. This led to the important realization that classical HLA alleles could be tagged and imputed through LD to surrounding SNPs.
Prior to application of HLA imputation within the MHC, several studies have leveraged tight LD between SNPs and HLA alleles to interpret MHC associations. For example, rs2395029 emerged as the top hit for two unrelated GWAS of HIV-1 virus load set point (21) and drug-induced liver injury due to flucloxacillin (22). This SNP lies about 100 kb upstream of HLA-B, but it was already known to be in near-perfect LD with HLA-B*57:01 in Europeans (4). This same SNP was also successfully evaluated as a screening tool for HLA-B*57:01-associated abacavir hypersensitivity (23). In another example, six tag SNPs were identified (24) and independently validated in another population to predict classical HLA-DQ alleles associated with celiac disease (DQA1*05/DQB1*02 (DQ2.5), DQA1*0201/DQB1*0202 (DQ2.2), DQA1*0505/DQB1*0301 (DQ7) and DQA1*03/DQB1*0302 (DQ8)) (25).
Investigators have since devised more sophisticated imputation methods ((9,11–13)) and with large reference panels these tools have since been applied to association studies. Most of these methods leverage the observation that chromosomes with high identity by descent within the MHC region are likely to share the same HLA alleles. Then for a given individual with SNP genotypes within the MHC, statistical methods can identify MHC segments with high identity-by-descent, and use that information to infer HLA alleles. We expect that the major determinant of imputation quality will be the sample size and representativeness of the reference panel. How ancestry differences between the reference panel and GWAS dataset affect imputation quality must be evaluated empirically, but the MHC may be more sensitive to this given the high degree of population differentiation of HLA alleles across populations.
Multiple sclerosis
Multiple sclerosis (MS) is an inflammatory disease of the central nervous system characterized by neurodegeneration and progressive disability. It has been well known that MHC variation has the largest genetic influence on disease risk (26), but attempts to fine-map the MHC association signal beyond the primary DRB1*15:01 association has proved challenging with conflicting results (27,28). Recently, a large-scale GWAS meta-analysis of 10 000 MS cases and 17 000 controls, drawn from multiple European populations, had adequate power for the discovery of 29 novel MS loci outside MHC (29). Investigators imputed classical HLA alleles with the HLA*IMP tool (12). As expected, the primary signal mapped to HLA-DRB1*15:01 (P < 10−320). Subsequent conditional analysis identified additional classical allele associations with consistent effects across countries/cohorts for HLA-A*02:01, HLA-DRB1*03:01 (which was indistinguishable from HLA-DQB1*02:01), HLA-DRB1*13:03 and possibly for HLA-DPB1*03:01. These results suggest that the MHC association cannot be mapped to a single allele at a single locus—but rather constitutes independent effects at multiple loci. Further fine-mapping analysis proposed a possible model where three mutations on specific branches of the estimated genealogical tree of HLA-DRB1 were shared by the associated classical alleles at this locus. This model implicated an additional (risk) association for DRB1*08:01 by virtue of sharing the same putative mutation as DRB1*13:03; this hypothesis will need to be tested.
TESTING FUNCTIONAL VARIATION
We argue that HLA genes should be investigated much in the same way that we test variations outside of the MHC. That is, in addition to haplotype-based testing (tantamount to testing classical alleles for association), we assert that testing individual polymorphic sites within a gene might be informative, and may produce a more parsimonious model to explain an observed association. Since classical HLA alleles are defined by unique nucleotide sequences, variations at individual coding and non-coding sites at the nucleotide level can easily be inferred by simple lookup. Because the classical alleles also define unique amino acid sequences, it is equally straightforward to infer variations at polymorphic amino acid positions in the corresponding HLA proteins. Thus, it is possible to impute not only classical HLA alleles but also individual nucleotide variants and amino acid variants.
Our working hypothesis is that some amino acid positions in a protein are structurally or functionally more important than others, and can therefore be tested (through imputation) for association. This is particularly appropriate if disease susceptibility is modulated by specific amino acid sites (or multiple sites); in that case, a testing approach based solely on classical HLA alleles may not be as powerful. If a model based on specific amino acids can explain the data better than a model based on classical haplotypes, this should provide an additional biological insight as the association signal is much more localized compared with a four-digit allele, which by definition spans the entire gene/protein.
We note that the goal of explaining genetic associations in terms of protein structure and function is not a novel idea. Previously, amino acid associations have been described for HLA-DQβ1 position 57 in type 1 diabetes (30), HLA-DRβ1 position 74 in Graves' disease (31,32), HLA-DRβ1 position 11 in sarcoidosis (33), HLA-DRβ1 positions 67, 71 and 74 in autoimmune polyglandular syndrome (34), HLA-DRβ1 position 13 in MS (35), and HLA-DRβ1 positions 37 and 86 in primary sclerosing cholangitis (36). Comparative analyses of a growing number of three-dimensional structures from the Protein Data Bank have focused mostly on canonical ‘pockets’ within the peptide-binding groove to rationalize classical HLA allele associations (37–39). An illustrative example comes from the crystal structure of HLA-DQ bound to an immunogenic epitope from dietary gluten (the known antigen in celiac disease), which provides structural clues as to how DQ2 molecules may preferentially bind proline-rich gluten peptides (40). For most autoimmune diseases, however, we still do not know the (auto)antigens involved. Fine-mapping functional variants may produce useful information to narrow the search for possible peptides presented by HLA molecules associated with disease risk.
By coupling HLA imputation in large GWAS datasets to association testing at amino acid resolution, we anticipate that the potential gains can be greater than that has been achieved so far. The combination of large sample sizes and the expected effect sizes in the HLA is likely to yield much better power for stepwise conditional analysis allowing more precise ranking of models by their goodness-of-fit. In addition, GWAS data also provide for excellent control against population stratification, a key confounding variable for HLA association studies (41). HLA alleles are strongly differentiated across global populations and therefore highly sensitive to type 1 error when slight differences in the ancestry of cases and controls are not adequately controlled for.
Host control of HIV replication
In the International HIV Controllers Study, we performed a GWAS in about 1000 HIV controllers and 2700 HIV progressors. HIV controllers constitute a relatively small subgroup of HIV-positive individuals able to suppress viral replication down to levels that often go below detection in the absence of treatment. We observed 313 SNPs at genome-wide significance, concentrated in the class I region. One of those SNPs (rs2395029, mentioned above) had already been described as an effective tag SNP for classical HLA allele B*57:01. To assess whether there were other independent associations, we imputed classical alleles and corresponding amino acid polymorphisms at the HLA-A, -B and -C loci, allowing us to test individual classical alleles as well as polymorphic amino acid positions. Three HLA-B amino acid sites (97, 67 and 70) clearly stood out and explained the MHC association significantly more effectively than the well-known classical alleles such as B*57(01) and B*27(05). These sites are located in the peptide-binding groove, highlighting the importance of the peptide–MHC interaction in the control of HIV. Position 97 (P = 10−45) has as many as six common alleles and explained most of the observed MHC association (Figure 3). Three amino acids at this position (valine, asparagine and tryptophan) are carried exclusively by protective haplotypes (B*57:01, B*27:05 and B*14, respectively). Threonine is carried by protective haplotype B*52 (and others), while serine and arginine are carried by a variety of risk haplotypes including B*07 and B*35.
Figure 3.
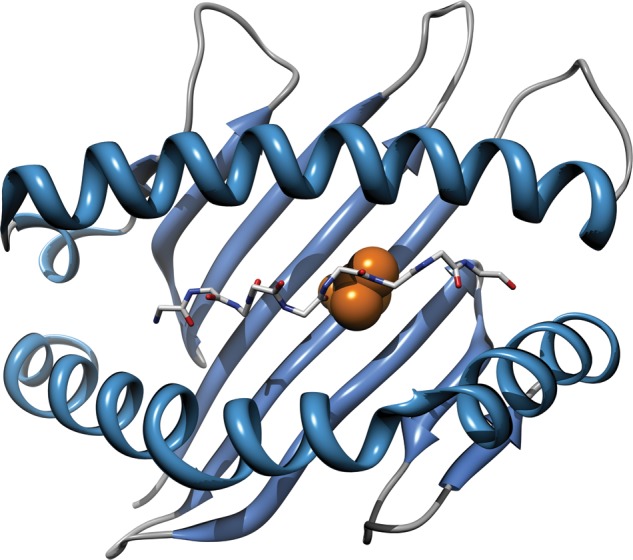
Three-dimensional model of the HLA-B protein based on the Protein Data Bank entry 2bvp. Amino acid position 97 is highlighted in the peptide-binding groove, as is the backbone of a peptide bound to the HLA molecule. Variations at position 97 are strongly associated with durable host control of HIV-1 replication, and can explain the observed effects of classical HLA-B alleles, including B*57 and B*27.
We subsequently performed a similar amino acid-based analysis of HIV control in African Americans and were able to replicate position 97 as the most important polymorphism in HLA-B (42). In African Americans, position 97 explains the association of not only the B*57:01 allele (the most prevalent B*57 allele in Northern–Western European populations) but also the B*57:03 allele (the most prevalent B*57 allele in African Bantu populations). Interestingly, we also detected an effect at position 245 located far from the peptide-binding groove. Threonine at this position is uniquely carried by HLA-B*81:01, an allele that is essentially absent from European populations. It has been suggested by others that this position may influence the binding of the CD8 co-receptor to the HLA-B molecule. These findings underscore the value of fine-mapping in multiple ethnicities.
Rheumatoid arthritis
Separately, our group has applied a similar imputation and fine-mapping approach to understand the basis of the MHC association with rheumatoid arthritis (43). For more than 20 years, the widely accepted ‘shared epitope’ model postulates that MHC risk to RA is conferred by a short stretch of amino acids (positions 70–74) in one of the two alpha-helices that enclose the peptide-binding groove of the HLA–DR molecule (44). Taking advantage of available GWAS data in 5000 cases and 15 000 controls from six different cohorts, we imputed classical alleles and polymorphic amino acids and tested them for association. Surprisingly, the strongest association signal did not map to any of the shared epitope positions, but mapped to position 11 (P < 10−581) located on the beta-sheet floor of the peptide-binding groove. Once position 11 was corrected for, we observed independent, but more modest effects, at the shared epitope positions 71 and 74. Adjusting for these DRB1 effects, additional amino acid associations were found in HLA-B and HLA-DPB1. Interestingly, the HLA-B effect is due to B*08, which is often described as part of the long 8.1 ancestral haplotype across the class I and II regions. The presence of both class I and II involvement suggests a role for both CD8+ cytotoxic T cells and CD4 helper T cells. The biological connection to other autoimmune diseases that share these same allelic associations is an interesting observation.
SEQUENCING THE MHC
With the primary focus of most HLA studies on four-digit coding variation, functional non-coding alleles are usually not directly ascertained. To what extent MHC association signals are (in part) caused by non-coding variation remains therefore an outstanding question. Recent studies however suggest a prominent role for the regulatory variation in the MHC including eQTLs (45–48). This signals the urgent need for a more comprehensive approach to study all variation across the MHC, not limited to the coding regions of classical HLA genes. This will likely happen in two consecutive steps: first through imputation using an MHC-wide reference panel and second by complete sequencing.
There have been a few parallel attempts to characterize long-range haplotype sequences across the MHC. For example, the MHC Haplotype Project determined the full-length sequences of eight common MHC haplotypes derived from consanguineous cell lines, selected on the basis of high frequency in European populations and their putative role in type 1 diabetes and MS (49–51). These DNA sequences have provided a valuable framework for polymorphism discovery and gene annotation. The extensive variation observed between these MHC haplotypes illustrates the limitations of relying on a single reference sequence (the sequence from the PGF cell line is currently the de facto human genome reference sequence for the MHC).
Whole-genome sequencing is still too expensive on a large scale, but can generate comprehensive coverage across the MHC. We are hopeful that soon this will become routine, but better approaches are needed for aligning divergent sequence reads to the reference sequence (or all eight reference MHC sequences). De novo assembly of short reads (without aligning to a reference) looks promising, but these approaches are still in their infancy (52). Some progress has been made with flow cytometric sorting of chromosome 6 for more cost-effective sequencing (as it accounts for ∼6% of the whole genome), but this approach would also demand better alignment or assembly strategies. Once these analytical issues are resolved and large-scale sequencing becomes more affordable, it will be possible to obtain complete sequencing information. Whole-exome sequencing datasets have demonstrated a relative excess of rare, functional variants, reflecting the effects of negative selection, with the implication that sequencing in larger samples will continue to discover novel variants (53). The abundance of rare alleles may be even more pronounced in the HLA according to a recent analysis (54), underscoring the potential value of MHC sequencing when this can be done efficiently.
CONCLUSION
With HLA imputation, it will be possible to extract much more information from existing GWAS datasets with an initial MHC association signal. Large-scale reference panels and effective imputation methods are currently available to type HLA genes in silico and fine-map associations within classical HLA genes. Unfortunately, adequately sized reference panels are not available still for many ethnic groups. Another major limitation is that these reference panels lack MHC-wide ascertainment of variation. We are hopeful that the onslaught of whole-genome sequencing efforts will allow the construction of large reference panels for multiple global populations. With those emerging tools and resources, it will become possible to study the relative importance of coding and non-coding variation, and of rare and common variation, and the role of non-HLA genes. We hope that detailed insights into functional variations in the MHC will generate opportunities to develop novel therapies for a wide range of diseases.
Conflict of Interest statement. None declared.
FUNDING
We acknowledge financial support from the National Institutes of Health (1 R01 AR062886-01, 5K08AR055688), the Arthritis Foundation, and the Collaboration for AIDS Vaccine Discovery of the Bill and Melinda Gates Foundation. P.I.W.d.B. is a recipient of a VIDI Award from the Netherlands Organization for Scientific Research (NWO).
REFERENCES
- 1.Bach F.H., van Rood J.J. The major histocompatibility complex—genetics and biology. (First of three parts) N. Engl. J. Med. 1976;295:806–813. doi: 10.1056/NEJM197610072951504. [DOI] [PubMed] [Google Scholar]
- 2.Ryder L.P., Svejgaard A., Dausset J. Genetics of HLA disease association. Annu. Rev. Genetics. 1981;15:169–187. doi: 10.1146/annurev.ge.15.120181.001125. [DOI] [PubMed] [Google Scholar]
- 3.Horton R., Wilming L., Rand V., Lovering R.C., Bruford E.A., Khodiyar V.K., Lush M.J., Povey S., Talbot C.C., Jr., Wright M.W., et al. Gene map of the extended human MHC. Nat. Rev. Genet. 2004;5:889–899. doi: 10.1038/nrg1489. [DOI] [PubMed] [Google Scholar]
- 4.de Bakker P.I.W., McVean G., Sabeti P.C., Miretti M.M., Green T., Marchini J., Ke X., Monsuur A.J., Whittaker P., Delgado M., et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat. Genet. 2006;38:1166–1172. doi: 10.1038/ng1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Traherne J.A. Human MHC architecture and evolution: implications for disease association studies. Int. J. Immunogenet. 2008;35:179–192. doi: 10.1111/j.1744-313X.2008.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ioannidis J.P., Thomas G., Daly M.J. Validating, augmenting and refining genome-wide association signals. Nat. Rev. Genet. 2009;10:318–329. doi: 10.1038/nrg2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marchini J., Howie B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 8.Shea J., Agarwala V., Philippakis A.A., Maguire J., Banks E., Depristo M., Thomson B., Guiducci C., Onofrio R.C., Kathiresan S., et al. Comparing strategies to fine-map the association of common SNPs at chromosome 9p21 with type 2 diabetes and myocardial infarction. Nat. Genet. 2011;43:801–805. doi: 10.1038/ng.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leslie S., Donnelly P., McVean G. A statistical method for predicting classical HLA alleles from SNP data. Am. J. Hum. Genet. 2008;82:48–56. doi: 10.1016/j.ajhg.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pereyra F., Jia X., McLaren P.J., Telenti A., de Bakker P.I.W., Walker B.D., Ripke S., Brumme C.J., Pulit S.L., Carrington M., et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie M., Li J., Jiang T. Accurate HLA type inference using a weighted similarity graph. BMC Bioinformatics. 2010;11(Suppl 11):S10. doi: 10.1186/1471-2105-11-S11-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dilthey A.T., Moutsianas L., Leslie S., McVean G. HLA*IMP—an integrated framework for imputing classical HLA alleles from SNP genotypes. Bioinformatics. 2011;27:968–972. doi: 10.1093/bioinformatics/btr061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Setty M.N., Gusev A., Pe'er I. HLA type inference via haplotypes identical by descent. J. Comput. Biol. 2011;18:483–493. doi: 10.1089/cmb.2010.0258. [DOI] [PubMed] [Google Scholar]
- 14.Price P., Witt C., Allcock R., Sayer D., Garlepp M., Kok C.C., French M., Mallal S., Christiansen F. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol. Rev. 1999;167:257–274. doi: 10.1111/j.1600-065x.1999.tb01398.x. [DOI] [PubMed] [Google Scholar]
- 15.Thakkinstian A., McEvoy M., Chakravarthy U., Chakrabarti S., McKay G.J., Ryu E., Silvestri G., Kaur I., Francis P., Iwata T., et al. The Association Between Complement Component 2/Complement Factor B Polymorphisms and Age-related Macular Degeneration: A HuGE Review and Meta-Analysis. Am. J. Epidemiol. 2012;176:361–372. doi: 10.1093/aje/kws031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson J., Mistry K., McWilliam H., Lopez R., Parham P., Marsh S.G. The IMGT/HLA database. Nucleic Acids Res. 2011;39:D1171–D1176. doi: 10.1093/nar/gkq998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marsh S.G., Albert E.D., Bodmer W.F., Bontrop R.E., Dupont B., Erlich H.A., Fernandez-Vina M., Geraghty D.E., Holdsworth R., Hurley C.K., et al. Nomenclature for factors of the HLA system, 2010. Tissue Antigens. 2010;75:291–455. doi: 10.1111/j.1399-0039.2010.01466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erlich R.L., Jia X., Anderson S., Banks E., Gao X., Carrington M., Gupta N., DePristo M.A., Henn M.R., Lennon N.J., et al. Next-generation sequencing for HLA typing of class I loci. BMC Genomics. 2011;12:42. doi: 10.1186/1471-2164-12-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holcomb C.L., Hoglund B., Anderson M.W., Blake L.A., Bohme I., Egholm M., Ferriola D., Gabriel C., Gelber S.E., Goodridge D., et al. A multi-site study using high-resolution HLA genotyping by next generation sequencing. Tissue Antigens. 2011;77:206–217. doi: 10.1111/j.1399-0039.2010.01606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C., Krishnakumar S., Wilhelmy J., Babrzadeh F., Stepanyan L., Su L.F., Levinson D., Fernandez-Vina M.A., Davis R.W., Davis M.M., et al. High-throughput, high-fidelity HLA genotyping with deep sequencing. Proc. Natl Acad. Sci. USA. 2012;109:8676–8681. doi: 10.1073/pnas.1206614109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fellay J., Shianna K.V., Ge D., Colombo S., Ledergerber B., Weale M., Zhang K., Gumbs C., Castagna A., Cossarizza A., et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daly A.K., Donaldson P.T., Bhatnagar P., Shen Y., Pe'er I., Floratos A., Daly M.J., Goldstein D.B., John S., Nelson M.R., et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 2009;41:816–819. doi: 10.1038/ng.379. [DOI] [PubMed] [Google Scholar]
- 23.Colombo S., Rauch A., Rotger M., Fellay J., Martinez R., Fux C., Thurnheer C., Gunthard H.F., Goldstein D.B., Furrer H., et al. The HCP5 single-nucleotide polymorphism: a simple screening tool for prediction of hypersensitivity reaction to abacavir. J. Infect. Dis. 2008;198:864–867. doi: 10.1086/591184. [DOI] [PubMed] [Google Scholar]
- 24.Monsuur A.J., de Bakker P.I.W., Zhernakova A., Pinto D., Verduijn W., Romanos J., Auricchio R., Lopez A., van Heel D.A., Crusius J.B., et al. Effective detection of human leukocyte antigen risk alleles in celiac disease using tag single nucleotide polymorphisms. PLoS One. 2008;3:e2270. doi: 10.1371/journal.pone.0002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koskinen L., Romanos J., Kaukinen K., Mustalahti K., Korponay-Szabo I., Barisani D., Bardella M.T., Ziberna F., Vatta S., Szeles G., et al. Cost-effective HLA typing with tagging SNPs predicts celiac disease risk haplotypes in the Finnish, Hungarian, and Italian populations. Immunogenetics. 2009;61:247–256. doi: 10.1007/s00251-009-0361-3. [DOI] [PubMed] [Google Scholar]
- 26.Sawcer S., Ban M., Maranian M., Yeo T.W., Compston A., Kirby A., Daly M.J., De Jager P.L., Walsh E., Lander E.S., et al. A high-density screen for linkage in multiple sclerosis. Am. J. Hum. Genet. 2005;77:454–467. doi: 10.1086/444547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernando M.M., Stevens C.R., Walsh E.C., De Jager P.L., Goyette P., Plenge R.M., Vyse T.J., Rioux J.D. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4:e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rioux J.D., Goyette P., Vyse T.J., Hammarstrom L., Fernando M.M., Green T., De Jager P.L., Foisy S., Wang J., de Bakker P.I., et al. Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc. Natl Acad. Sci. USA. 2009;106:18680–18685. doi: 10.1073/pnas.0909307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawcer S., Hellenthal G., Pirinen M., Spencer C.C., Patsopoulos N.A., Moutsianas L., Dilthey A., Su Z., Freeman C., Hunt S.E., et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Todd J.A., Bell J.I., McDevitt H.O. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature. 1987;329:599–604. doi: 10.1038/329599a0. [DOI] [PubMed] [Google Scholar]
- 31.Ban Y., Davies T.F., Greenberg D.A., Concepcion E.S., Osman R., Oashi T., Tomer Y. Arginine at position 74 of the HLA-DR beta1 chain is associated with Graves’ disease. Genes Immun. 2004;5:203–208. doi: 10.1038/sj.gene.6364059. [DOI] [PubMed] [Google Scholar]
- 32.Simmonds M.J., Howson J.M., Heward J.M., Cordell H.J., Foxall H., Carr-Smith J., Gibson S.M., Walker N., Tomer Y., Franklyn J.A., et al. Regression mapping of association between the human leukocyte antigen region and Graves disease. Am. J. Hum. Genet. 2005;76:157–163. doi: 10.1086/426947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foley P.J., McGrath D.S., Puscinska E., Petrek M., Kolek V., Drabek J., Lympany P.A., Pantelidis P., Welsh K.I., Zielinski J., et al. Human leukocyte antigen-DRB1 position 11 residues are a common protective marker for sarcoidosis. Am. J. Respir. Cell Mol. Biol. 2001;25:272–277. doi: 10.1165/ajrcmb.25.3.4261. [DOI] [PubMed] [Google Scholar]
- 34.Menconi F., Osman R., Monti M.C., Greenberg D.A., Concepcion E.S., Tomer Y. Shared molecular amino acid signature in the HLA-DR peptide binding pocket predisposes to both autoimmune diabetes and thyroiditis. Proc. Natl Acad. Sci. USA. 2010;107:16899–16903. doi: 10.1073/pnas.1009511107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramagopalan S.V., McMahon R., Dyment D.A., Sadovnick A.D., Ebers G.C., Wittkowski K.M. An extension to a statistical approach for family based association studies provides insights into genetic risk factors for multiple sclerosis in the HLA-DRB1 gene. BMC Med. Genet. 2009;10:10. doi: 10.1186/1471-2350-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hov J.R., Kosmoliaptsis V., Traherne J.A., Olsson M., Boberg K.M., Bergquist A., Schrumpf E., Bradley J.A., Taylor C.J., Lie B.A., et al. Electrostatic modifications of the human leukocyte antigen-DR P9 peptide-binding pocket and susceptibility to primary sclerosing cholangitis. Hepatology. 2011;53:1967–1976. doi: 10.1002/hep.24299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cucca F., Lampis R., Congia M., Angius E., Nutland S., Bain S.C., Barnett A.H., Todd J.A. A correlation between the relative predisposition of MHC class II alleles to type 1 diabetes and the structure of their proteins. Hum. Mol. Genet. 2001;10:2025–2037. doi: 10.1093/hmg/10.19.2025. [DOI] [PubMed] [Google Scholar]
- 38.Jones E.Y., Fugger L., Strominger J.L., Siebold C. MHC class II proteins and disease: a structural perspective. Nat. Rev. Immunol. 2006;6:271–282. doi: 10.1038/nri1805. [DOI] [PubMed] [Google Scholar]
- 39.Karp D.R., Marthandan N., Marsh S.G., Ahn C., Arnett F.C., Deluca D.S., Diehl A.D., Dunivin R., Eilbeck K., Feolo M., et al. Novel sequence feature variant type analysis of the HLA genetic association in systemic sclerosis. Hum. Mol. Genet. 2010;19:707–719. doi: 10.1093/hmg/ddp521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim C.Y., Quarsten H., Bergseng E., Khosla C., Sollid L.M. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc. Natl Acad. Sci. USA. 2004;101:4175–4179. doi: 10.1073/pnas.0306885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 42.McLaren P.J., Ripke S., Pelak K., Weintrob A.C., Patsopoulos N.A., Jia X., Erlich R.L., Lennon N.J., Kadie C.M., Heckerman D., et al. Fine-mapping classical HLA variation associated with durable host control of HIV-1 infection in African Americans. Hum. Mol. Genet. 2012 doi: 10.1093/hmg/dds226. doi:10.1093/hmg/dds226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raychaudhuri S., Sandor C., Stahl E.A., Freudenberg J., Lee H.S., Jia X., Alfredsson L., Padyukov L., Klareskog L., Worthington J., et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 2012;44:291–296. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gregersen P.K., Silver J., Winchester R.J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 45.Fairfax B.P., Makino S., Radhakrishnan J., Plant K., Leslie S., Dilthey A., Ellis P., Langford C., Vannberg F.O., Knight J.C. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat. Genet. 2012;44:502–510. doi: 10.1038/ng.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fehrmann R.S., Jansen R.C., Veldink J.H., Westra H.J., Arends D., Bonder M.J., Fu J., Deelen P., Groen H.J., Smolonska A., et al. Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet. 2011;7:e1002197. doi: 10.1371/journal.pgen.1002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas R., Apps R., Qi Y., Gao X., Male V., O'hUigin C., O'Connor G., Ge D., Fellay J., Martin J.N., et al. HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat. Genet. 2009;41:1290–1294. doi: 10.1038/ng.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kulkarni S., Savan R., Qi Y., Gao X., Yuki Y., Bass S.E., Martin M.P., Hunt P., Deeks S.G., Telenti A., et al. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature. 2011;472:495–498. doi: 10.1038/nature09914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stewart C.A., Horton R., Allcock R.J., Ashurst J.L., Atrazhev A.M., Coggill P., Dunham I., Forbes S., Halls K., Howson J.M., et al. Complete MHC haplotype sequencing for common disease gene mapping. Genome Res. 2004;14:1176–1187. doi: 10.1101/gr.2188104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Traherne J.A., Horton R., Roberts A.N., Miretti M.M., Hurles M.E., Stewart C.A., Ashurst J.L., Atrazhev A.M., Coggill P., Palmer S., et al. Genetic analysis of completely sequenced disease-associated MHC haplotypes identifies shuffling of segments in recent human history. PLoS Genet. 2006;2:e9. doi: 10.1371/journal.pgen.0020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Horton R., Gibson R., Coggill P., Miretti M., Allcock R.J., Almeida J., Forbes S., Gilbert J.G., Halls K., Harrow J.L., et al. Variation analysis and gene annotation of eight MHC haplotypes: the MHC Haplotype Project. Immunogenetics. 2008;60:1–18. doi: 10.1007/s00251-007-0262-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iqbal Z., Caccamo M., Turner I., Flicek P., McVean G. De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat. Genet. 2012;44:226–232. doi: 10.1038/ng.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kiezun A., Garimella K., Do R., Stitziel N.O., Neale B.M., McLaren P.J., Gupta N., Sklar P., Sullivan P.F., Moran J.L., et al. Exome sequencing and the genetic basis of complex traits. Nat. Genet. 2012;44:623–630. doi: 10.1038/ng.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klitz W., Hedrick P., Louis E.J. New reservoirs of HLA alleles: pools of rare variants enhance immune defense. Trends. Genet. 2012 doi: 10.1016/j.tig.2012.06.007. http://dx.doi.org/10.1016/j.tig.2012.06.007. [DOI] [PubMed] [Google Scholar]