

Abstract

In this study we developed an efficient one-pot procedure for the preparation of 3-substituted and 3,4-disubstituted quinolines from stable starting materials (activated acetylenes reacting with o-tosylamidobenzaldehydes and o-tosylamidophenones, respectively) under mild conditions. The reaction appears to operate under a general base catalysis mechanism, instigated by the β-phosphonium enoate α-vinyl anion generated in situ through nucleophilic addition of PPh3 to the activated alkyne. Michael addition of the deprotonated tosylamides to the activated alkynes and subsequent rapid aldol cyclization led to the formation of labile N-tosyldihydroquinoline intermediates. Driven by aromatization, detosylation of the dihydroquinoline intermediates occurred readily in the presence of dilute aqueous HCl to give the final quinoline products.
Keywords: quinoline; 3-substituted quinoline; 3,4-disubstituted quinoline; phosphine catalysis; o-aminobenzaldehyde; o-aminophenone; activated alkyne; Michael addition; aldol addition; aromatization; detosylation
Introduction
The quinoline unit is found in a wide variety of pharmacologically and biologically active compounds.1 Not surprisingly, quinoline derivatives continue to attract the attention of medicinal chemists, and strategies for accessing new scaffolds of quinoline derivatives are of great interest to synthetic chemists. In recent years, many transition metal–catalyzed processes have been developed for mild and efficient syntheses of quinolines.2 Notably, the number of conventional metal-free paths for quinoline syntheses have also been growing.3 Several classical methods for targeting the quinoline core—including the Skraup, Doebner–von Miller, Friedländer, Pfitzinger, Conrad–Limpach, and Combes syntheses4a,b—remain relevant today for the preparation of quinoline-containing materials, ligands, and pharmaceutical agents.4a Nevertheless, such reactions are often performed under unfavorably harsh conditions, typically with either a strong acid or base and thermal assistance.4a,b For example, the Friedländer quinoline synthesis, a particularly powerful tool for generating quinoline core systems, is performed at high temperature in the presence of either a strong acid or base.4 In addition to unattractive reaction conditions, the instability of the coupling partners in the Friedländer quinoline synthesis further limits its synthetic potential.4,5 In particular, when the synthesis of 3-substituted quinolines is attempted using Friedländer methodology (R2 = R3 = H), self-condensation of both aldehyde coupling partners can lower the reaction efficiency and complicate the product’s purification (Scheme 1).11 To prevent self-condensation of aminobenzaldehydes in Friedländer methodology, several classical approaches, namely the Borsche, Pfitzinger, and Niemantowski quinoline syntheses, have been developed employing alternative starting materials.4 In a recent report, for example, aminobenzaldehydes were generated in situ from corresponding nitrobenzaldehydes and subsequently used in the Friedländer synthesis.5
Scheme 1.

Friedläder Quinoline Synthesis
For the synthesis of 3-substituted quinolines in particular, in 2009 Verpoort reported a one-pot methodology to avoid self-condensation of the aminobenzaldehyde by employing an aminobenzyl alcohol as a precursor; they also prevented self-condensation of the other coupling partner through late introduction of the strong base.6 This approach, however, requires an elevated temperature and a stoichiometric amount of the strong base. More recently, Li reported an alternative approach to 3-substituted quinolines by coupling alkynones with aminobenzaldehyde in the presence of a catalytic amount of Lewis acid as the activator.7 Despite the use of a mild Lewis acid, this approach requires thermal assistance, long reaction times, and the use of unstable aminobenzaldehydes. The synthesis of 3-substituted quinolines is also generally difficult when using other methods.4a,8 Herein, we report a simple and efficient one-pot phosphine-catalyzed procedure for the synthesis of 3-substituted quinolines under mild conditions from stable starting materials.
We developed the title quinoline synthesis during an expansion of our original double-Michael reaction.9,10 We attempted to develop alternative modes of this tandem reaction to generate a variety of heterocyclic scaffolds (Scheme 2). The double-Michael reaction requires two pronucleophilic groups in one of the starting materials to undergo the two successive Michael additions. Replacing one pronucleophilic group with an electrophilic group would alter the nature of the reaction to a cascade of nucleophilic additions (Michael addition followed by an aldol reaction, or vice versa). The presence of an N-tosyl group in the substrates not only activated the pronucleophile but also completely inhibited the self-condensation normally observed for Friedländer substrates. In fact, N-tosylated o-aminobenzaldehydes are bench-stable at room temperature for months of storage without any deterioration. The activated acetylenes are likewise very stable under storage and during the reaction.
Scheme 2.

3-Substituted Quinoline Synthesis Versus Double-Michael Reaction
Results and Discussion
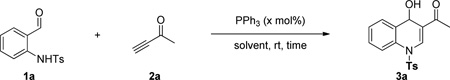
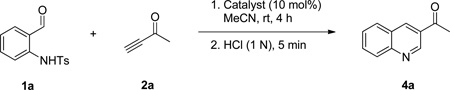
In a preliminary study of the reaction, we reacted 0.1 M of N-tosyl-2-aminobenzaldehyde (1a) with 2.0 equivalents of 3-butyn-2-one (2a) in THF in the presence of 20 mol% PPh3 as the catalyst. We stopped the reaction prior to completion, after 17 h, for a quick evaluation of its feasibility, identifying the dihydroquinoline 3a as the product in 28% yield (Table 1, entry 1). We suspected that the long reaction time might have caused 2a to oligomerize in the presence of the nucleophilic catalyst PPh3, thereby explaining the poor reaction yield. Therefore, we tested the slow addition (syringe pump, 2 h) of 2a to the reaction mixture, but the yield was unchanged (entry 2). We observed (1H NMR spectra) almost no oligomerization in a mixture of 2a and 20 mol% PPh3 after several days. Increasing the reaction time from 17 to 24 h barely increased the reaction yield (entry 3), implying that a significantly longer time might be required for the reaction to reach completion. Surprisingly, the addition of a stoichiometric amount of PPh3 did not accelerate the reaction; indeed, it completely shut down the reaction and yielded no product (entry 4). 1H NMR spectra revealed the complete destruction of 2a within 5 min in the presence of a stoichiometric amount of PPh3. When the concentration of 1a was increased to 0.2 M or greater, the reaction yield improved significantly (entries 1 and 5–7). Among the solvents tested for the reaction, MeCN provided the highest yield (entry 8); reactions performed in DMSO and DCM (data not shown) provided product yields lower than those obtained in THF and MeCN. The reactions of 1a in THF at concentrations of 0.4 and 0.2 M resulted in the same yields (entries 6 and 7). Because 1a is not soluble in MeCN at a concentration of 0.4 M, we considered the optimal concentration of 1a in MeCN to be 0.2 M. Decreasing the loading of the catalyst PPh3 to 10 mol% improved the reaction yield by 4% (entry 10). The reaction yield decreased, however, after decreasing the catalyst loading of 5 mol% (entry 11). Increasing the number of equivalents of the activated acetylene 2a did not improve the reaction performance (entry 12). Careful monitoring revealed that the reaction required only 4 h to reach completion in MeCN (entry 13). Finally, decreasing the amount of 2a to 1.5 equivalents did not change the reaction yield (entry 14), but it did drop dramatically to 14% when we used 1.2 equivalents of 2a (entry 15).
Table 1.
Optimization of Dihydroquinoline Synthesisa
 | ||||||
|---|---|---|---|---|---|---|
| entry | 2a (equiv) |
PPh3 (mol%) |
solvent | concentrationb (M) |
time (h) |
yield (%) |
| 1 | 2.0 | 20 | THF | 0.1 | 17 | 28 |
| 2 c | 2.0 | 20 | THF | 0.1 | 17 | 28 |
| 3 | 2.0 | 20 | THF | 0.1 | 24 | 32 |
| 4 | 2.0 | 100 | THF | 0.1 | 24 | 0 |
| 5 | 2.0 | 20 | THF | 0.05 | 17 | 25 |
| 6 | 2.0 | 20 | THF | 0.2 | 17 | 67 |
| 7 | 2.0 | 20 | THF | 0.4 | 17 | 68 |
| 8 | 2.0 | 20 | MeCN | 0.2 | 17 | 71 |
| 9 | 2.0 | 20 | MeCN | 0.4 | N/Ad | N/Ad |
| 10 | 2.0 | 10 | MeCN | 0.2 | 17 | 75 |
| 11 | 2.0 | 5 | MeCN | 0.2 | 17 | 49 |
| 12 | 2.5 | 5 | MeCN | 0.2 | 17 | 47 |
| 13 | 2.0 | 10 | MeCN | 0.2 | 4 | 76 |
| 14 | 1.5 | 10 | MeCN | 0.2 | 4 | 76 |
| 15 | 1.2 | 10 | MeCN | 0.2 | 4 | 14 |
Butynone 2a was added in one portion to a solution of 1a and PPh3 in the indicated solvent.
Concentration of the aldehyde 1a.
Addition of butynone 2a over 2 h via syringe pump.
Not available.
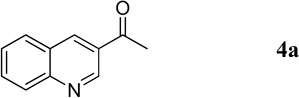
The isolated dihydroquinoline adduct 3a was unstable and readily decomposed at room temperature.5a Treating the vinylogous hemiaminal with acetyl chloride and pyridine resulted in the clean production of the stable quinoline 4a in excellent yield (Scheme 3a). In this two-pot procedure, decomposition of the unstable dihydroquinoline 3a during its isolation resulted in a lower yield of the quinoline product 4a. Such loss could be avoided through a one-pot procedure to directly convert the unstable dihydroquinoline intermediate into the corresponding quinoline. The ideal reagent for such a procedure would necessarily contain a “chloride” to trap the tosyl group in the form of the byproduct TsCl; in addition, it should transform the OH group into a good leaving group. Hydrogen chloride met these requirements; simply quenching the reaction with 1 M aqueous HCl provided the quinoline 4a in 88% yield together with TsCl as a byproduct (Scheme 3b). This higher yield for the isolated quinoline relative to that of the isolated dihydroquinoline confirmed the loss of the latter product through decomposition in the two-pot procedure.
Scheme 3.

(a) Quinoline Formation from Dihydroquinoline. (b) One-Pot Phosphine-Catalyzed Synthesis of Quinoline
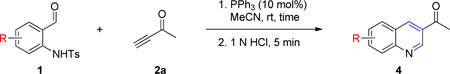
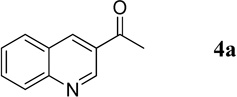
Having optimized the one-pot procedure for the synthesis of the 3-substituted quinoline, we further examined the scope of this reaction for the synthesis of 3-acetylquinolines (Table 2). Regardless of the electron-donating or -withdrawing ability of the substituents on the aminobenzaldehyde, the reactions were highly efficient, generating the desired quinolines in high yields. Nevertheless, the nature of the substituents had a significant impact on the rate of the reaction. The reaction of the non-substituted N-tosyl-2-aminobenzaldehyde (1a) reached completion within 4 h in 88% yield (entry 1). The electronically similar N-tosyl-3-amino-2-naphthaldehyde (1b) was converted into the quinoline 4b in a comparable yield of 89% after a similar reaction time of 5 h (entry 2). The presence of electron-withdrawing groups significantly slowed the rates of the reactions (entries 3–5), whereas electron-donating groups accelerated them (entries 6 and 7). In general, the presence of a substituent, regardless of its electronic nature, had a positive impact on the yield of the reaction (entry 1 vs. entries 2–7).
Table 2.
One-Pot Phosphine-Catalyzed Syntheses of Substituted 3-Acetylquinolinesa
 | ||||
|---|---|---|---|---|
| entry | substrate | product | time (h) | yield (%)b |
| 1 |  |
 |
4 | 88 |
| 2 | 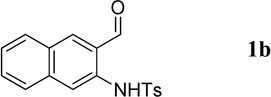 |
 |
5 | 89 |
| 3 | 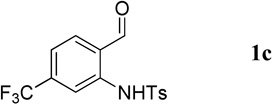 |
 |
19 | 94 |
| 4 | 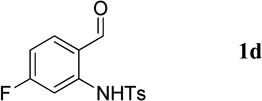 |
 |
19 | 96 |
| 5 | 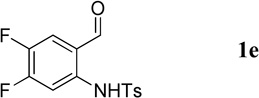 |
 |
11 | 95 |
| 6 | 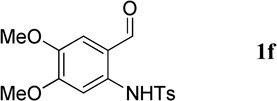 |
 |
1.5 | 99 |
| 7 | 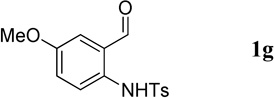 |
 |
1.5 | 92 |
2a (1.5 equiv.) was added in one portion to a solution of 1 (0.2 M) and PPh3 (10 mol%) in MeCN.
Isolated yield.
The successful syntheses of the 3-acetylquinolines in Table 2 encouraged us to expand the scope of the reaction to include other 3-substituted quinolines (Table 3). In general, the reaction was highly efficient for acetylenic ketones, but much less efficient for other activated alkynes (entries 1–10 vs. entries 12 and 13). We obtained lower yields for the reactions performed with methyl propiolate or an acetylenic sulfone, due to formation of the corresponding simple Michael adducts with the aldehyde functionality intact (entries 12 and 13). With regard to the acetylenic ketone, the reactions of aryl acetylenyl ketones were faster and provided higher yields than those of alkyl acetylenyl ketones (entry 1 vs. entries 2–9). The electronic nature of the aryl group of the acetylenic ketone greatly impacted the reaction rate and yield. With electron-deficient aryl groups, the reactions required only a few minutes to reach completion in excellent yields (entry 2 vs. entries 3–8 and 11). In contrast, electron-rich aryl groups prolonged the reaction and resulted in excellent but lower yields (entry 2 vs. entries 9 and 10). We also examined the versatility of the reaction when using a multifunctional acetylenic ketone, namely a bis(acetylenic ketone); here, the reaction proceeded smoothly to form the bisquinoline product in good yield (entry 11).
Table 3.
One-Pot Phosphine-Catalyzed Syntheses of 3-Substituted Quinolinesa
 | ||||
|---|---|---|---|---|
| entry | substrate | product | time (min) |
yield (%)b |
| 1 | 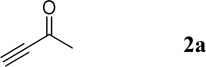 |
 |
240 | 88 |
| 2 | 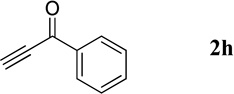 |
 |
30 | 95 |
| 3 | 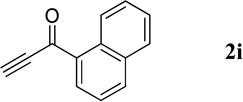 |
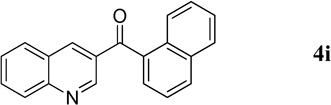 |
20 | 99 |
| 4 | 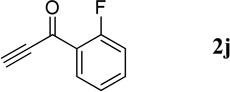 |
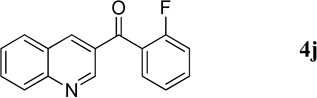 |
5 | 98 |
| 5 | 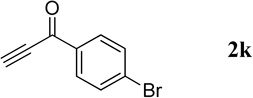 |
 |
5 | 98 |
| 6 | 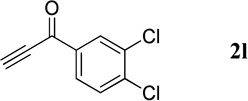 |
 |
5 | 97 |
| 7 | 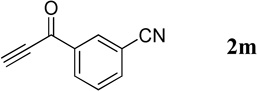 |
 |
5 | 96 |
| 8 | 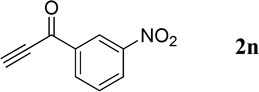 |
 |
5 | 98 |
| 9 | 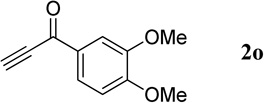 |
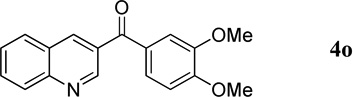 |
90 | 93 |
| 10 | 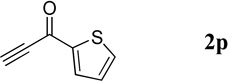 |
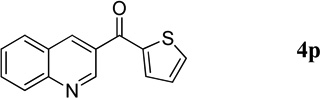 |
40 | 86 |
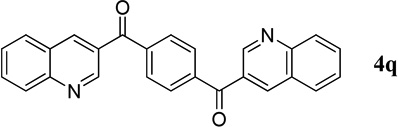
| 11c | 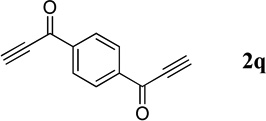 |
 |
5 | 63 |
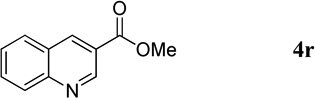
| 12 | 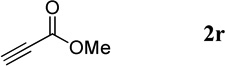 |
 |
600 | 61 |
| 13 | 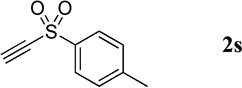 |
 |
5 | 30 |
2 (1.5 equiv.) was added in one portion to a solution of 1a (0.2 M) and PPh3 (10 mol%) in MeCN.
Isolated yield.
Diacetylene 2q (0.10 mmol) was added to a solution of 1a (0.21 mmol, 0.21 M) and PPh3 (20 mol%) in MeCN.
We further expanded the scope of the reaction to include less-reactive o-aminophenones as partners for the syntheses of 3,4-disubstituted quinolines. The reactions afforded the desired products, albeit in lower yields after longer reaction times (Table 3, entry 2 vs. Table 4). Longer reaction times were expected because of the lower reactivity of ketones relative to corresponding aldehydes. Among the selected o-aminophenones, those with larger R groups required the longest reaction times (entry 3 vs. entries 1, 2 and 4), although the size of the R group had only a minor impact on the reaction yield (entries 1–4).
Table 4.
One-Pot Phosphine-Catalyzed Syntheses of 3,4-Disubstituted Quinolinesa
 | ||||
|---|---|---|---|---|
| entry | substrate | product | time(h) | yield(%)b |
| 1 | 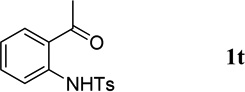 |
 |
3.5 | 79 |
| 2 | 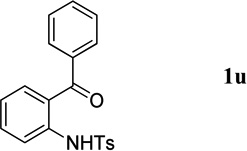 |
 |
3.5 | 72 |
| 3 | 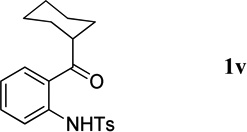 |
 |
6.5 | 75 |
| 4 | 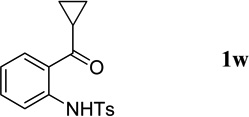 |
 |
2.5 | 67 |
2b (1.5 equiv.) was added in one portion to a solution of 1 (0.2 M) and PPh3 (10 mol%) in MeCN.
Isolated yield.
Mechanistic Discussion
Scheme 4 outlines three possible mechanistic scenarios. Mechanism 1 involves a general base catalysis. Nucleophilic addition of the free phosphine to the activated alkyne results in the phosphonium allenolate A, which acts as a base to activate the pronucleophile through deprotonation, resulting in a subsequent general base-catalyzed Michael/aldol reaction. Mechanism 2 is based on a nucleophilic phosphine catalysis, in which the phosphine is consumed and regenerated along the catalytic cycle; the ion pair B, which is also generated in mechanism 1, is presumably associated in a sufficiently tight manner to enforce the nucleophilic addition within the ion pair.9 Mechanism 3 is also based on a nucleophilic phosphine catalysis, where the aldol addition occurs immediately after the formation of the phosphonium allenolate A; hence, the ion pair B is not formed.
Scheme 4.


Possible Mechanistic Schemes for Phosphine-Catalyzed Dihydroquinoline Formation
Because we isolated the Michael adduct together with the quinoline product in the reaction of methyl propiolate (Table 3, entry 12),11 we speculate that mechanism 3 was most unlikely to operate. As indicated in Table 5, the reaction proceeded to form the quinoline product in the presence of a catalytic amount of a non-nucleophilic base. This result supports the operation of the general base catalysis mechanism in this reaction.
Table 5.
Non-Nucleophilic Base Catalysis for Dihydroquinoline Synthesis
Isolated yield.
Heterogeneous reaction mixture.
In the hopes of seeing reaction intermediates that could help to identify the true mechanistic pathway out of the three possibilities discussed above, we used NMR spectroscopy to monitor the reaction between 1a and 2a in the presence of 10 mol% PPh3 in CD3CN; unfortunately, no intermediates were evident at any time during the course of the reaction. The 1H NMR spectra of the reaction mixture revealed that the signal for the NH unit of reactant 1a disappeared within 10 min (Figure 1C). Apparently, the reaction was initiated rapidly in MeCN with partial deprotonation of the NH group; fast proton exchange made the NH proton undetectable, on the time scale of 1H NMR spectroscopy, at 10.83 ppm (the presence of N-tosylaminobenzaldehyde 1a was verified through TLC analysis during the reaction period). At the same time, two new signals, corresponding to protons Ha and Hb of the dihydroquinoline adduct, were growing (Figures 1C–F). Because of rapid proton exchange, the proton of the OH group of the dihydroquinoline product was also undetectable in the 1H NMR spectra recorded throughout the course of the reaction. In general, only the starting materials and the final dihydroquinoline products were evident (i.e., no intermediates were observed) at any time during the reactions monitored using 1H NMR spectroscopy.
Figure 1.


1H NMR spectra revealing the formation of the dihydroquinoline 3a. (A, B) Control experiments performed without (A) 3-butyn-2-one (2a) or (B) N-tosyl-2-aminobenzaldehyde (1a) in the reaction mixture; spectra recorded after 4 h. (C–F) Spectra of the reaction proper after (C) 10 min, (D) 1 h, (E) 3.5 h, and (F) 4 h.
Conclusion
We have developed an efficient one-pot procedure for the syntheses of 3-substituted and 3,4-disubstituted quinolines from the reactions of activated acetylenes with N-tosyl-2-aminobenzaldehydes and N-tosyl-2-aminobenzophenones, respectively. This approach provides a convenient and direct route toward 3-substituted quinolines, which are challenging to prepare using other methods. The reaction conditions are mild and many different substituents can be introduced without compromising yields.
Experimental Section
General Information
All reactions were performed in dry solvents under an Ar atmosphere and anhydrous conditions, unless otherwise indicated. DCM, THF, and MeCN were freshly distilled over CaH2 prior to use. Anhydrous DMSO was used as received from a commercial source. All other reagents were used as received from commercial sources. Reactions were monitored through thin layer chromatography (TLC) on 0.25-mm SiliCycle silica gel plates and visualized under UV light and with permanganate or 2,4-dinitrophenylhydrazine (DNP) staining. Flash column chromatography (FCC) was performed using SiliCycle Silica-P Flash silica gel (60-Å pore size, 40–63 µm). IR spectra were recorded using a Jasco FT-IR 4100 spectrometer. NMR spectra of the dihydroquinoline 3a were obtained using Bruker Avance-500 instruments, calibrated to residual THF-d8 as the internal reference (1.73 and 3.58 ppm for 1H NMR spectra; 25.4 and 67.6 ppm for 13C NMR spectra). NMR spectra of the quinolines 4 were recorded using Bruker Avance-500 instruments, calibrated to CD(H)Cl3 as the internal reference (7.26 and 77.0 ppm for 1H and 13C NMR spectra, respectively). 1H NMR spectral data are reported in terms of chemical shift (δ, ppm), multiplicity, coupling constant (Hz), and integration. 13C NMR spectral data are reported in terms of chemical shift (δ, ppm) and multiplicity, with the coupling constant (Hz) in the case of JCF coupling. The following abbreviations indicate the multiplicities: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet, br, broad. GC-MS data were recorded using an Agilent 6890-5975 GC mass spectrometer equipped with an autosampler and an HP5 column; samples were dissolved in DCM.
General Procedure for the Syntheses of Substrates 1
The N-tosylbenzaldehydes 1a–g were prepared from the corresponding anthranilic acids in three steps without purification of any intermediates.12 The N-tosyl-o-aminophenones 1t and 1u were prepared directly from the corresponding o-aminophenones through a single tosylation step.13 The N-tosyl-o-aminophenones 1v and 1w were prepared from the corresponding 2-aminobenzonitriles in two steps, without purification of any intermediates.14
Syntheses of Substrates 2
The activated acetylene 2 were prepared in two steps, without purification of any intermediates, according to reported procedures.15
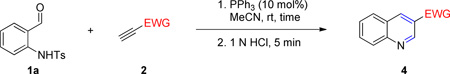
General Procedure for the Syntheses of Quinolines
o-Aminobenzaldehyde (1, 0.2 mmol), PPh3 (5.3 mg, 10 mol%), and MeCN (1 mL) were added sequentially to a flame-dried flask (10 mL); unless otherwise noted, the mixture was stirred until complete dissolution occurred. The activated acetylene 2 (0.3 mmol) was added in one portion and then the mixture was stirred under Ar at room temperature. Upon completion of the reaction, 1 M aqueous HCl (1 mL) was added and then the mixture was stirred for 5 min before saturated aqueous NaHCO3 (1 mL) was added to neutralize the mixture. The mixture was poured into a separatory funnel along with DCM (10 mL) and saturated aqueous NaHCO3 (10 mL). The aqueous phase was separated and extracted with DCM (2 mL). The combined organic phases were dried (Na2SO4) and concentrated in vacuo; the residue was purified through flash column chromatography [ethyl acetate (EtOAc)/hexanes (Hex), 3:7, unless specified otherwise] to afford the desired quinoline product.
1-(1-Tosyl-1,4-dihydroquinolin-3-yl)ethanone (3a)
To a flame-dried flask (10 mL) were sequentially added N-tosyl 2-aminobenzaldehyde (1a) (0.2 mmol), PPh3 (5.3 mg, 10 mol%), and MeCN (1 mL). The mixture was stirred until complete dissolution and then 3-butyn-2-one (2) (23.5 µL, 0.3 mmol) was added in one portion. The mixture was stirred under argon at room temperature; upon completion of the reaction (4.0–4.5 h), the mixture was concentrated in vacuo and purified through flash column chromatography (gradient EtOAc/Hex, 3:7 to 1L1) to furnish a slightly yellow solid (52.1 mg, 76% yield). IR (film) νmax 3391, 3088, 3050, 3006, 2923, 1645, 1632, 1597, 1566, 1485, 1456, 1343, 1210, 1160, 1088, 1017, 1009 cm−1; 1H NMR (500 MHz, THF-d8) δ 7.81 (d, J = 8.1 Hz, 1H), 7.41–7.37 (m, 4H), 7.32 (s, 1H), 7.21 (t, J = 7.7 Hz, 1H), 7.15 (d, J = 8.2 Hz, 2H), 6.81 (d, J = 6.1 Hz, 1H), 5.85 (d, J = 6.1 Hz, 1H), 2.32 (s, 3H), 2.26 (s, 3H); 13C NMR (125 MHz, THF-d8) δ 194.2, 143.3, 137.0, 135.0, 134.2, 132.2, 130.0, 128.9, 128.8, 126.5, 125.6, 124.8, 124.6, 73.3, 24.1, 20.2; HRMS (ESI-TOF) m/z: [M − OH]+ Calcd for C18H16NO3S 326.0851; Found 326.0847.
1-(Quinolin-3-yl)ethanone (4a).16
30.1 mg, 88% yield; slightly yellow solid. M.p. 98–99 °C. IR (film) νmax 3065, 3009, 2920, 1681, 1615, 1587, 1571, 1493, 1371 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.42 (s, 1H), 8.69 (s, 1H), 8.15 (d, J = 8.5 Hz, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.83 (t, J = 7.5 Hz, 1H), 7.62 (t, J = 7.5 Hz, 1H), 2.73 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 196.5, 149.5, 149.0, 137.3, 131.9, 129.22, 129.19, 129.1, 127.5, 126.7, 26.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C11H10NO 172.0762; Found 172.0762.
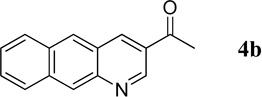
1-(Benzo[g]quinolin-3-yl)ethanone (4b)
39.5 mg, 89% yield; bright yellow solid. M.p. 150–152 °C. IR (film) νmax 3046, 2998, 2922, 1676, 1612, 1532, 1352, 1215 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.47 (s, 1H), 8.84 (s, 1H), 8.70 (s, 1H), 8.51 (s, 1H), 8.10 (d, J = 8.5 Hz, 1H), 8.06 (d, J = 8.5 Hz, 1H), 7.62–7.55 (m, 2H), 2.76 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 196.5, 149.5, 149.0, 137.3, 131.9, 129.22, 129.19, 129.1, 127.5, 126.7, 26.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C15H12NO 222.0919; Found 222.0921.
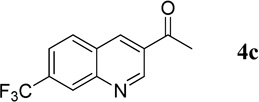
1-[7-(Trifluoromethyl)quinolin-3-yl]ethanone (4c)
44.8 mg, 94% yield; white crystalline solid. M.p. 126–127 °C. IR (film) νmax 3059, 3020, 2929, 1689, 1594, 1464, 1124 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.50 (s, 1H), 8.75 (s, 1H), 8.45 (s, 1H), 8.08 (d, J = 8.5 Hz, 1H), 7.80 (d, J = 8.5 Hz, 1H), 2.77 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 196.1, 150.4, 148.7, 136.8, 133.3 (q, J = 32.9 Hz), 130.5, 130.4, 128.3, 127.2 (q, J = 4.4 Hz), 123.5 (q, J = 271.0 Hz), 123.1 (q, J = 4.5 Hz), 26.8; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C12H9F3NO 240.0636; Found 240.0632.
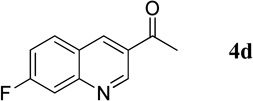
1-(7-Fluoroquinolin-3-yl)ethanone (4d)
36.4 mg, 96%; white solid. M.p. 116–118 °C. IR (film) νmax 3051, 2923, 1670, 1619, 1602, 1579, 1274, 1193 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.42 (s, 1H), 8.70 (s, 1H), 7.95 (dd, J = 8.9, 5.1 Hz, 1H), 7.77 (dd, J = 9.8, 2.3 Hz, 1H), 7.42 (dt, J = 8.7, 2.5, 1H), 2.73 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 196.2, 164.5 (d, J = 254.8 Hz), 150.9 (d, J = 12.7 Hz), 150.2, 140.0, 131.5 (d, J = 10.5 Hz), 128.8, 123.8, 118.2 (d, J = 25.7 Hz), 113.3 (d, J = 20.7 Hz), 26.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C11H9FNO 190.0668; Found 190.0670.
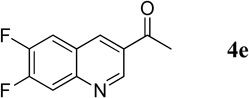
1-(6,7-Difluoroquinolin-3-yl)ethanone (4e)
39.5 mg, 95% yield; white solid. M.p. 151–153 °C. IR (film) νmax 3061, 2923, 1682, 1595, 1505, 1475, 1347, 1253, 1234 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.39 (s, 1H), 8.65 (s, 1H), 8.45 (s, 1H), 7.90 (dd, J = 7.7, 7.7 Hz, 1H), 7.68 (dd, J = 8.3, 8.3 Hz, 1H), 2.74 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 196.0, 153.7 (dd, J = 257.0, 17.8 Hz), 150.6 (dd, J = 257.0, 17.8 Hz), 147.1 (d, J = 11.2 Hz), 136.2, 129.3, 123.8 (d, J = 7.1 Hz), 115.9 (d, J = 17.2 Hz), 114.3 (d, J = 17.2 Hz), 26.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C11H8F2NO 208.0574; Found 208.0584.
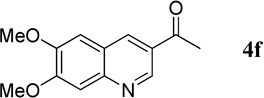
1-(6,7-Dimethoxyquinolin-3-yl)ethanone (4f).17
EtOAc/Hex, 1:1. 45.7 mg, 99% yield; white solid. M.p. 160–163 °C. IR (film) νmax 3008, 2960, 2925, 2831, 1667, 1597, 1504, 1438, 1427, 1228, 1144, 1003 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.24 (s, 1H), 8.56 (s, 1H), 7.46 (s, 1H), 7.14 (s, 1H), 4.06 (s, 3H), 4.03 (s, 3H), 2.71 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 196.6, 154.4, 150.4, 147.4, 147.3, 135.0, 127.9, 122.4, 107.8, 106.0, 56.2, 56.0, 26.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C13H14NO3 232.0974; Found 232.0964.
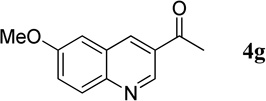
1-(6-Methoxyquinolin-3-yl)ethanone (4g)
37.1 mg, 92% yield; light-yellow solid. M.p. 122–124 °C. IR (film) νmax 2956, 2923, 2852, 1682, 1619, 1595, 1503, 1368, 1227, 1023 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.25 (s, 1H), 8.57 (s, 1H), 8.02 (d, J = 9.0 Hz, 1H), 7.45 (dd, J = 9.0, 2.5 Hz, 1H), 7.14 (d, J = 2.5 Hz, 1H), 3.93 (s, 3H), 2.71 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 196.8, 158.3, 146.7, 146.0, 135.8, 130.6, 129.4, 127.9, 127.8, 124.8, 55.5, 26.8; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C12H12NO2 202.0868; Found 202.0870.
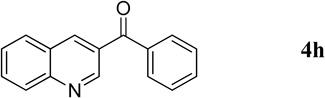
Phenyl(quinolin-3-yl)methanone (4h).7
44.2 mg, 95% yield; crystalline yellow solid. M.p. 73–75 °C. IR (film) νmax 3052, 1647, 1616, 1597, 1571, 1287, 1243 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.33 (s, 1H), 8.56 (s, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.88–7.86 (m, 3H), 7.69–7.63 (m, 2H), 7.58–7.48 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 194.8, 150.3, 149.4, 138.7, 136.9, 133.0, 131.8, 129.97, 129.95, 129.4, 129.1, 128.6, 127.5, 126.5; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H12NO 234.0919; Found 234.0915.
Naphth-1-yl(quinolin-3-yl)methanone (4i).7
56.3 mg, 99% yield; pale-yellow oil. IR (film) νmax 3051, 1652, 1616, 1589, 1568, 1493, 1286, 1237, 1186 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.43 (s, 1H), 8.54 (s, 1H), 8.21–8.19 (m, 2H), 8.08 (d, J = 8.2 Hz, 1H), 7.96 (d, J = 8.2 Hz, 1H), 7.87–7.84 (m, 2H), 7.66 (d, J = 7.1 Hz, 1H), 7.62–7.53 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 196.3, 150.4, 149.7, 139.5, 135.2, 133.8, 132.0, 130.8, 130.7, 129.4, 129.3, 128.5, 128.4, 127.6, 127.5, 126.7, 126.6, 125.4, 124.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C20H14NO 284.1075; Found 284.1072.
2-Fluorophenyl(quinolin-3-yl)methanone (4j)
49.2 mg, 98% yield; light-yellow solid. M.p. 84–86 °C. IR (film) νmax 3086, 1664, 1610, 1595, 1568, 1448, 1296, 1213 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.33 (s, 1H), 8.54 (s, 1H), 8.16 (d, J = 8.8 Hz, 1H), 7.90 (d, J = 7.9 Hz, 1H), 7.84 (t, J = 7.8 Hz, 1H), 7.66 (d, J = 14.1 Hz, 1H), 7.62–7.59 (m, 2H), 7.33 (t, J = 7.9 Hz, 1H), 7.20 (t, J = 8.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 191.7, 160.1 (d, J = 253.6 Hz), 149.7, 138.9, 133.9 (d, J = 8.4 Hz), 132.1, 130.9 (d, J = 2.7 Hz), 129.9, 129.4 (d, J = 10.1 Hz), 127.5, 126.6, 126.0 (d, J = 14.4 Hz), 124.6 (d, J = 3.8 Hz), 116.4 (d, J = 21.8 Hz); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H11FNO 252.0825; Found 252.0829.
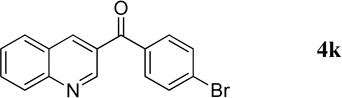
4-Bromophenyl(quinolin-3-yl)methanone (4k)
61.2 mg, 98% yield; white crystalline solid. M.p. 115–117 °C. IR (film) νmax 3090, 3056, 1645, 1618, 1584, 1566, 1491, 1366, 1231, 1168, 1069 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.30 (s, 1H), 8.53 (s, 1H), 8.20 (d, J = 8.1 Hz, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.87 (t, J = 8.1 Hz, 1H), 7.74 (d, J = 8.5 Hz, 2H), 7.69 (d, J = 8.5 Hz, 2H), 7.65 (t, J = 8.1 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 193.7, 150.0, 149.5, 138.6, 135.7, 131.94, 131.92, 131.4, 129.6, 129.5, 129.1, 128.2, 127.6, 126.4; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H11BrNO 312.0024; Found 312.0013.
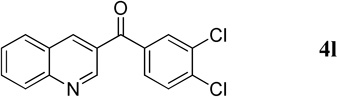
3,4-Dichlorophenyl(quinolin-3-yl)methanone (4l)
58.4 mg, 97% yield; light-yellow solid. M.p. 112–113 °C. IR (film) νmax 3076, 1637, 1617, 1577, 1385, 1293, 1238 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.28 (d, J = 1.9 Hz, 1H), 8.52 (d, J = 1.9 Hz, 1H), 8.19 (d, J = 7.9 Hz, 1H), 7.95–7.92 (m, 2H), 7.87 (ddd, J = 8.5, 7.0, 1.3 Hz, 1H), 7.69–7.61 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 192.4, 149.8, 149.6, 138.7, 136.5, 133.4, 132.1, 131.6, 130.7, 129.5, 129.12, 129.10, 128.9, 127.8, 126.4; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H10Cl2NO 302.0139; Found 302.0135.
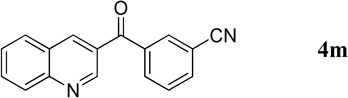
3-(Quinoline-3-carbonyl)benzonitrile (4m)
49.5 mg, 96% yield; off-white crystalline solid. M.p. 105–107 °C. IR (film) νmax 3067, 2227, 1653, 1616, 1596, 1567, 1291, 1179 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.30 (d, J = 2.7 Hz, 1H), 8.53 (d, J = 2.7 Hz, 1H), 8.21 (d, J = 10.6 Hz, 1H), 8.14 (s, 1H), 8.09 (dt, J = 9.8, 1.9 Hz, 1H), 7.95–7.92 (m, 2H), 7.89 (t, J = 9.7 Hz, 1H), 7.69 (q, J = 10.1 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 192.6, 149.74, 149.66, 138.9, 137.9, 135.8, 133.7, 133.2, 132.4, 129.6, 129.5, 129.2, 128.7, 127.9, 126.3, 117.6, 113.2; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H11N2O 259.0871; Found 259.0882.
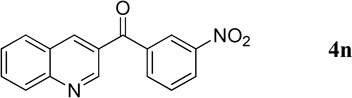
3-Nitrophenyl(quinolin-3-yl)methanone (4n)
54.6 mg, 98% yield; white solid. M.p 138–140 °C. IR (film) νmax 3086, 1642, 1612, 1527, 1491, 1346, 1285 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.31 (s, 1H), 8.69 (s, 1H), 8.56 (s, 1H), 8.50 (d, J = 8.5 Hz, 1H), 8.20 (t, J = 7.3 Hz, 2H), 7.94 (d, J = 8.5 Hz, 1H), 7.90 (t, J = 7.3 Hz, 1H), 7.77 (t, J = 7.3 Hz, 1H), 7.67 (t, J = 7.3 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 192.4, 149.71, 149.70, 148.2, 138.9, 138.3, 135.3, 132.4, 129.9, 129.5, 129.2, 128.7, 127.9, 127.2, 126.4, 124.5; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H11N2O3 279.0770; Found 279.0776.
3,4-Dimethoxyphenyl(quinolin-3-yl)methanone (4o)
54.6 mg, 93% yield; white solid. M.p. 93–95 °C. IR (film) νmax 3042, 2995, 2934, 1637, 1592, 1580, 1513, 1418, 1295, 1264, 1247, 1228, 1143, 1113, 1020 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.28 (d, J = 1.3 Hz, 1H), 8.54 (d, J = 1.3 Hz, 1H), 8.20 (d, J = 8.4 Hz, 1H), 7.93 (d, J = 8.1 Hz, 1H), 7.85 (t, J = 8.2 Hz, 1H), 7.65 (t, J = 8.2 Hz, 1H), 7.55 (s, 1H), 7.42 (d, J = 8.1 Hz, 1H), 6.94 (d, J = 8.4 Hz, 1H), 3.99 (s, 3H), 3.97 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 196.1, 150.4, 148.7, 136.8, 131.9, 129.22, 129.19, 129.1, 127.5, 126.7, 26.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H16NO3 294.1130; Found 294.1130.
Quinolin-3-yl(thien-2-yl)methanone (4p).7
41.1 mg, 86% yield; white solid. M.p. 89–91 °C. IR (film) νmax 3102, 3065, 1629, 1617, 1588, 1517, 1492, 1410, 1366, 1292, 1251, 1062 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.33 (s, 1H), 8.64 (s, 1H), 8.18 (d, J = 8.9 Hz, 1H), 7.93 (d, J = 8.9 Hz, 1H), 7.84 (t, J = 7.2 Hz, 1H), 7.79 (d, J = 5.0 Hz, 1H), 7.70 (d, J = 5.0 Hz, 1H), 7.64 (t, J = 7.5 Hz, 1H), 7.21 (t, J = 5.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 186.1, 149.5, 149.3, 143.1, 137.7, 135.01, 134.99, 131.7, 130.6, 129.4, 129.0, 128.3, 127.6, 126.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C14H10NOS 240.0483; Found 240.0494.
1,4-Phenylenebis(quinolin-3-ylmethanone) (4q)
Synthesized from o-aminobenzaldehyde 1 (57.8 mg, 0.21 mmol), PPh3 (5.3 mg, 20 mol%), and the activated bis-acetylene 2q (18.2 mg, 0.1 mmol) in MeCN (1 mL); gradient: EtOAc/Hex, from 3:7 to 1:1. 24.5 mg, 63% yield; pale-yellow powder. M.p. 237–239 °C. IR (film) νmax 3023, 1641, 1614, 1595, 1571, 1366, 1290, 1246, 1122 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.38 (s, 2H), 8.61 (s, 2H), 8.22 (d, J = 8.2 Hz, 2H), 8.03 (s, 4H), 7.96 (d, J = 8.2 Hz, 2H), 7.89 (t, J = 8.2 Hz, 2H), 7.68 (t, J = 8.2 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 194.0, 150.0, 149.6, 140.4, 139.1, 132.2, 130.0, 129.5, 129.22, 129.18, 127.8, 126.5; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C26H17N2O2 389.1290; Found 389.1291.
Methyl Quinoline-3-carboxylate (4r).18
22.7 mg, 61/% yield; white crystalline solid. M.p. 69–70 °C. IR (film) νmax 3056, 2948, 2924, 2849, 1714, 1618, 1572, 1433, 1367, 1290, 1240, 1193, 1100 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.44 (s, 1H), 8.84 (s, 1H), 8.16 (d, J = 7.9 Hz, 1H), 7.93 (d, J = 7.9 Hz, 1H), 7.83 (t, J = 7.9 Hz, 1H), 7.62 (t, J = 7.9 Hz, 1H), 4.01 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 165.7, 149.9, 149.7, 138.7, 131.8, 129.4, 129.0, 127.3, 126.7, 122.9, 52.4; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C11H10NO2 188.0712; Found 188.0709.
Quinolin-3-yl(tosyl)methanone (4s).19
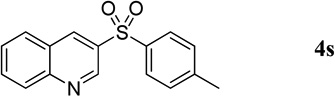
17.0 mg, 30% yield; yellow crystalline solid. M.p. 165–170 °C. IR (film) νmax 3068, 3056, 2925, 2851, 1616, 1593, 1585, 1497, 1312, 1304, 1290, 1153, 1140, 1091 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.25 (d, J = 1.7 Hz, 1H), 8.79 (d, J = 1.8 Hz, 1H), 8.14 (d, J = 8.5 Hz, 1H), 7.95 (d, J = 8.5 Hz, 1H), 7.90 (d, J = 8.2 Hz, 2H), 7.86 (t, J = 8.4 Hz, 3H), 7.67 (t, J = 8.4 Hz, 1H), 7.32 (d, J = 8.3 Hz, 2H), 2.39 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 149.2, 147.0, 144.8, 137.9, 136.5, 135.0, 132.5, 130.1, 129.5, 129.1, 128.2, 127.8, 126.3, 21.5; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H14NO2S 284.0745; Found 284.0753.
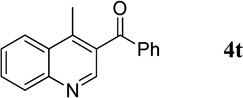
4-Methylquinolin-3-yl(phenyl)methanone (4t).7
EtOAc/Hex, 1:5. 40.3 mg, 82% yield; yellow crystalline solid. M.p. 82–86 °C. IR (film) νmax 3059, 3030, 2918, 1658, 1645, 1581, 1494, 1449, 1248, 1161 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.82 (s, 1H), 8.17 (d, J = 8.3 Hz, 1H), 8.12 (d, J = 8.5 Hz, 1H), 7.85–7.78 (m, 3H), 7.68–7.61 (m, 2H), 7.49 (t, J = 7.8 Hz, 2H), 2.67 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 196.9, 148.5, 148.0, 143.5, 137.4, 133.8, 131.8, 130.3, 130.1, 130.0, 128.7, 127.5, 127.2, 124.3, 15.8; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H14NO 248.1075; Found 248.1064.
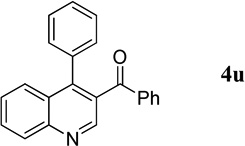
Phenyl(4-phenylquinolin-3-yl)methanone (4u).7
EtOAc/Hex, 1:5. 44.6 mg, 72% yield; yellow crystalline solid. M.p. 108–110 °C. IR (film) νmax 3062, 1654, 1570, 1486, 1324 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.00 (s, 1H), 8.24 (d, J = 8.5 Hz, 1H), 7.83–7.78 (m, 2H), 7.63–7.61 (m, 2H), 7.57–7.54 (m, 1H), 7.46–7.42 (m, 1H), 7.30–7.25 (m, 7H); 13C NMR (125 MHz, CDCl3) δ 196.7, 148.8, 148.4, 146.9, 137.3, 134.8, 133.1, 131.7, 130.4, 130.0, 129.7, 129.6, 128.4, 128.14, 128.12, 127.4, 126.7, 126.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C22H16NO 310.1232; Found 310.1230.
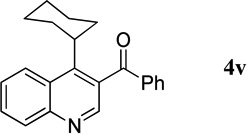
4-Cyclohexylquinolin-3-yl(phenyl)methanone (4v)
47.5 mg, 75% yield; pale-yellow oil. IR (film) νmax 3066, 2927, 2853, 1665, 1577, 1498, 1448, 1282, 1241 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.64 (s, 1H), 8.34 (br s, 1H), 8.16 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 6.3 Hz, 2H), 7.75 (t, J = 7.1 Hz, 1H), 7.61 (q, J = 7.3 Hz, 2H), 7.46 (t, J = 7.3 Hz, 2H), 3.25 (br s, 1H), 1.97 (br s, 2H), 1.85–1.77 (m, 4H), 1.26 (br s, 4H); 13C NMR (125 MHz, CDCl3) δ 198.2, 150.7, 148.7, 148.1, 137.6, 133.8, 132.0, 130.7, 130.1, 129.6, 128.9, 128.6, 126.7, 126.6, 32.0, 27.0, 25.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C22H22NO 316.1701; Found 316.1699.
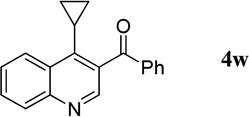
4-Cyclopropylquinolin-3-yl(phenyl)methanone (4w)
36.6 mg, 67% yield; pale-yellow oil. IR (film) νmax 3064, 3005, 2922, 1654, 1596, 1567, 1499, 1447, 1322, 1279, 1241, 1226, 1170 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.83 (s, 1H), 8.51 (d, J = 8.5 Hz, 1H), 8.17 (d, J = 8.5 Hz, 1H), 7.85–7.79 (m, 3H), 7.68–7.66 (m, 1H), 7.65–7.59 (m, 1H), 7.59–7.46 (m, 2H), 2.11–2.08 (m, 1H), 0.95–0.93 (m, 2H), 0.59–0.57 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 197.2, 148.9, 148.5, 147.4, 138.0, 133.4, 132.7, 130.3, 130.0, 129.6, 128.6, 128.3, 127.1, 125.5, 12.8, 8.2; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C19H16NO 274.1232; Found 274.1230.
Supplementary Material
Acknowledgment
Financial support was provided by the NIH (R01GM071779 and P41GM081282). This material is based upon work supported by the National Science Foundation under equipment grant no. CHE-1048804.
Footnotes
Supporting Information
Copies of 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Solomon VR, Lee H. Curr. Med. Chem. 2011;18:1488. doi: 10.2174/092986711795328382. [DOI] [PubMed] [Google Scholar]; (b) Kaur K, Jain M, Reddy RP, Jain R. Eur. J. Med. Chem. 2010;45:3245. doi: 10.1016/j.ejmech.2010.04.011. [DOI] [PubMed] [Google Scholar]; (c) Ahmed N, Brahmbhatt KG, Sabde S, Mitra D, Singh IP, Bhutani KK. Bioorg. Med. Chem. 2010;18:2872. doi: 10.1016/j.bmc.2010.03.015. [DOI] [PubMed] [Google Scholar]; (d) Musiol R, Serda M, Hensel-Bielowka S, Polanski J. Curr. Med. Chem. 2010;17:1960. doi: 10.2174/092986710791163966. [DOI] [PubMed] [Google Scholar]; (e) Bedoya LM, Abad MJ, Calonge E, Saavedra LA, Gutierrez CM, Kouznetsov VV, Alcami J, Bermejo P. Antiviral Res. 2010;87:338. doi: 10.1016/j.antiviral.2010.06.006. [DOI] [PubMed] [Google Scholar]; (f) Barluenga J, Rodriguez F, Fananas FJ. Chem. Asian J. 2009;4:1036. doi: 10.1002/asia.200900018. [DOI] [PubMed] [Google Scholar]; (g) Michael JP. Nat. Prod. Rep. 2008;25:166. doi: 10.1039/b612168n. [DOI] [PubMed] [Google Scholar]; (h) Sato M, Motomura T, Aramaki H, Matsuda T, Yamashita M, Ito Y, Kawakami H, Matsuzaki Y, Watanabe W, Yamataka K, Ikeda S, Kodama E, Matsuoka M, Shinkai H. J. Med. Chem. 2006;49:1506. doi: 10.1021/jm0600139. [DOI] [PubMed] [Google Scholar]
- 2.Metal-catalyzed quinoline syntheses. For Pd, see: Matsubara Y, Hirakawa S, Yamaguchi Y, Yoshida Z-I. Angew. Chem. Int. Ed. 2011;50:7670. doi: 10.1002/anie.201102076. Gabriele B, Mancuso R, Salerno G, Lupinacci E, Ruffolo G, Costa M. J. Org. Chem. 2008;73:4971. doi: 10.1021/jo8006495. Zhang Z, Tan J, Wang Z. Org. Lett. 2008;2:173. doi: 10.1021/ol702153x. Gabriele B, Mancuso R, Salerno G, Ruffolo G, Plastina P. J. Org. Chem. 2007;72:6873. doi: 10.1021/jo071094z. For Co, see: Li L, Jones WD. J. Am. Chem. Soc. 2007;129:10707. doi: 10.1021/ja070551q. For Rh, see: Horn J, Marsden SP, Nelson A, House D, Weingarten GG. Org. Lett. 2008;10:4117. doi: 10.1021/ol8016726. Beller M, Thiel OR, Trauthwein H, Hartung CG. Chem. Eur. J. 2000;6:2513. doi: 10.1002/1521-3765(20000717)6:14<2513::aid-chem2513>3.0.co;2-v. For Ti, see: Basuli F, Aneetha H, Huffman JC, Mindiola DJ. J. Am. Chem. Soc. 2005;127:17992. doi: 10.1021/ja0566026. For Au, see: Liu X-Y, Ding P, Huang J-S, Che C-M. Org. Lett. 2007;9:2645. doi: 10.1021/ol070814l. For Ni, see: Korivi RP, Cheng C-H. J. Org. Chem. 2006;71:7079. doi: 10.1021/jo060800d. For In, see: Sakai N, Annaka K, Fujita A, Sato A, Konakahara T. J. Org. Chem. 2008;73:4160. doi: 10.1021/jo800464u. Lekhok KC, Prajapati D, Boruah RC. Synlett. 2008:655. Sakai N, Annaka K, Konakahara T. J. Org. Chem. 2006;71:3653. doi: 10.1021/jo060245f. For Zn, see: Jiang B, Si Y-G. J. Org. Chem. 2002;67:9449. doi: 10.1021/jo0204606. For W, see: Sangu K, Fuchibe K, Akiyama T. Org. Lett. 2004;6:353. doi: 10.1021/ol036190a. For Fe, see: O’Dell DK, Nicholas KM. J. Org. Chem. 2003;68:6427. doi: 10.1021/jo034447c.
- 3.Metal-free quinoline syntheses. Ali S, Zhu H-T, Xia X-F, Ji K-G, Yang Y-F, Song X-R, Lian Y-M. Org. Lett. 2011;13:2598. doi: 10.1021/ol2007154. Peng C, Wang Y, Liu L, Wang H, Zhao J, Zhu Q. Eur. J. Org. Chem. 2010:818. doi: 10.1021/jo1017525. Majumder S, Gipson KR, Odom AL. Org. Lett. 2009;11:4720. doi: 10.1021/ol901855b. Wang Y, Xin X, Liang Y, Lin Y, Zhang R, Dong D. Eur. J. Org. Chem. 2009:4165. Sandelier MJ, DeShong P. Org. Lett. 2007;9:3209. doi: 10.1021/ol0710921. Zhao Y-L, Zhang W, Wang S, Liu Q. J. Org. Chem. 2007;72:4985. doi: 10.1021/jo070069q. Zhang Q, Zhang ZG, Yan ZH, Liu Q, Wang TY. Org. Lett. 2007;9:3651. doi: 10.1021/ol701536q. Wu Y-C, Liu L, Li H-J, Wang D, Chen Y-J. J. Org. Chem. 2006;71:6592. doi: 10.1021/jo060290n. Janza B, Studer A. Org. Lett. 2006;8:1875. doi: 10.1021/ol0604421. Tanaka S-y, Yasuda M, Baba A. J. Org. Chem. 2006;71:800. doi: 10.1021/jo052004y. Zhang XX, Campo MA, Yao TL, Larock RC. Org. Lett. 2005;7:763. doi: 10.1021/ol0476218. Hessian KO, Flynn BL. Org. Lett. 2006;8:243. doi: 10.1021/ol052518j.
- 4.Reviews on classical quinoline syntheses: Kouznetsov VV, Médez LYV, Góez CMM. Curr. Org. Chem. 2005;9:141. Madapa S, Tusi A, Batra S. Curr. Org. Chem. 2008;12:1116. Review on Friedläder quinoline synthesis: Marco-Contelles J, Perez-Mayoral E, Samadi A, Carreiras M. do C, Soriano E. Chem. Rev. 2009;109:2652. doi: 10.1021/cr800482c.
- 5.Li A-H, Beard DJ, Coate H, Honda A, Kadalbajoo M, Kleinberg A, Laufer R, Mulvihill KM, Nigro A, Rastogi P, Sherman D, Siu KW, Steinig AG, Wang T, Werner D, Crew AP, Mulvihill MJ. Synthesis. 2010;10:1678. and references therein. [Google Scholar]
- 6.Mierde HV, Voort PVD, Verpoort F. Tetrahedron Lett. 2009;50:201. [Google Scholar]
- 7.Li H, Xu X, Yang J, Xie X, Huang H, Li Y. Tetrahedron Lett. 2011;52:530. [Google Scholar]
- 8.(a) Uhle FC, Jacobs WA. J. Org. Chem. 1945;10:76. [Google Scholar]; (b) Monrad RN, Madsen R. Org. Biomol. Chem. 2011;9:610. doi: 10.1039/c0ob00676a. [DOI] [PubMed] [Google Scholar]
- 9.(a) Sriramurthy V, Barcan GA, Kwon O. J. Am. Chem. Soc. 2007;129:12928. doi: 10.1021/ja073754n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sriramurthy V, Kwon O. Org. Lett. 2010;12:1084. doi: 10.1021/ol100078w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reviews on phosphine catalysis: Lu X, Zhang C, Xu Z. Acc. Chem. Res. 2001;34:535. doi: 10.1021/ar000253x. Valentine DH, Jr, Hillhouse JH. Synthesis. 2003:317. Methot JL, Roush WR. Adv. Synth. Catal. 2004;346:1035. Lu X, Du Y, Lu C. Pure Appl. Chem. 2005;77:1985. Nair V, Menon RS, Sreekanth AR, Abhilash N, Biju AT. Acc. Chem. Res. 2006;39:520. doi: 10.1021/ar0502026. Ye L-W, Zhou J, Tang Y. Chem. Soc. Rev. 2008;37:1140. doi: 10.1039/b717758e. Kwong CK-W, Fu MY, Lam CS-K, Toy PH. Synthesis. 2008:2307. Denmark SE, Beutner GL. Angew. Chem. Int. Ed. 2008;47:1560. doi: 10.1002/anie.200604943. Aroyan CE, Dermenci A, Miller SJ. Tetrahedron. 2009;65:4069. Kumara Swamy KC, Bhuvan Kumar NN, Balaraman E, Pavan Kumar KVP. Chem. Rev. 2009;109:2551. doi: 10.1021/cr800278z. Cowen BJ, Miller SJ. Chem. Soc. Rev. 2009;38:3102. doi: 10.1039/b816700c. Marinetti A, Voituriez A. Synlett. 2010:174. Kolesinska B. Cent. Eur. J. Chem. 2010:1147. Wei Y, Shi M. Acc. Chem. Res. 2010;43:1005. doi: 10.1021/ar900271g. Pinho e Melo TMVD. Monatsh. Chem. 2011;142:681. Lalli C, Brioche J, Bernadat G, Masson G. Curr. Org. Chem. 2011;15:4108. Wang S-X, Han X, Zhong F, Wang Y, Lu Y. Synlett. 2011:2766. Lóez F, Mascareñs JL. Chem. Eur. J. 2011;17:418. Zhao Q-Y, Lian Z, Wei Y, Shi M. Chem. Commun. 2012;48:1724. doi: 10.1039/c1cc15793k. Fan YC, Kwon O. Phosphine Catalysis. In: List B, editor. Science of Synthesis. Vol 1. Stuttgart: Asymmetric Organocatalysis, Lewis Base and Acid Catalysts; Georg Thieme; 2012. pp. 723–782.
- 11.The Michael adduct was isolated in 30% yield as a mixture of cis and trans isomers.
- 12.(a) Fonseca MH, Eibler E, Zabelb M, Köig B. Tetrahedron: Asymmetry. 2003;14:1989. [Google Scholar]; (b) Theeraladanon C, Arisawa M, Nishida A, Nakagawa M. Tetrahedron. 2004;60:3017. [Google Scholar]; (c) Chernyak D, Chernyak N, Gevorgyan V. Adv. Synth. Catal. 2010;352:961. doi: 10.1002/adsc.201000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Carril M, SanMartin R, Churruca F, Tellitu I, Domíguez E. Org. Lett. 2005;7:4787. doi: 10.1021/ol051291p. [DOI] [PubMed] [Google Scholar]; (b) Dannhardt G, Fiebich BL, Schweppenhäser J. Eur. J. Med. Chem. 2002;37:147. doi: 10.1016/s0223-5234(01)01330-7. [DOI] [PubMed] [Google Scholar]
- 14.Bolli HM, Marfurt J, Grisostomi C, Boss C, Binkert C, Hess P, Treiber A, Thorin E, Morrison K, Buchmann S, Bur D, Ramuz H, Clozel M, Fischli W, Weller T. J. Med. Chem. 2004;47:2776. doi: 10.1021/jm031115r. [DOI] [PubMed] [Google Scholar]
- 15.(a) Husen M. Tetrahedron Lett. 2005;46:1651. [Google Scholar]; (b) Chen PC, Wharton RE, Patel PA, Oyelere AK. Bioorg. Med. Chem. 2007;15:7288. doi: 10.1016/j.bmc.2007.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fan YC, Kwon O. Molecules. 2011;16:3802. doi: 10.3390/molecules16053802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SW, Lee K, Seomoon D, Kim S, Kim H, Kim H, Shim E, Lee M, Lee S, Kim M, Lee PH. J. Org. Chem. 2004;69:4852. doi: 10.1021/jo0495790. [DOI] [PubMed] [Google Scholar]
- 17.Borsche W, Ried W. Liebigs Ann. Chem. 1943;554:269. [Google Scholar]
- 18.Cain M, Weber RW, Guzman F, Cook JM, Barker SA, Rice KC, Crawley JN, Paul SM, Skolnick P. J. Med. Chem. 1982;25:1081. doi: 10.1021/jm00351a015. [DOI] [PubMed] [Google Scholar]
- 19.Hayashi E, Shimada N. Yakugaku Zasshi. 1977;97:627. doi: 10.1248/yakushi1947.97.6_627. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.