

Abstract
Background
The commonest genetic form of juvenile or early adult onset macular degeneration is Stargardt Disease (STGD) caused by recessive mutations in the gene ABCA4. However, high phenotypic and allelic heterogeneity and a small but non-trivial amount of locus heterogeneity currently impede conclusive molecular diagnosis in a significant proportion of cases.
Methods
We performed whole exome sequencing (WES) of nine putative Stargardt Disease probands and searched for potentially disease-causing genetic variants in previously identified retinal or macular dystrophy genes. Follow-up dideoxy sequencing was performed for confirmation and to screen for mutations in an additional set of affected individuals lacking a definitive molecular diagnosis.
Results
Whole exome sequencing revealed seven likely disease-causing variants across four genes, providing a confident genetic diagnosis in six previously uncharacterized participants. We identified four previously missed mutations in ABCA4 across three individuals. Likely disease-causing mutations in RDS/PRPH2, ELOVL, and CRB1 were also identified.
Conclusions
Our findings highlight the enormous potential of whole exome sequencing in Stargardt Disease molecular diagnosis and research. WES adequately assayed all coding sequences and canonical splice sites of ABCA4 in this study. Additionally, WES enables the identification of disease-related alleles in other genes. This work highlights the importance of collecting parental genetic material for WES testing as the current knowledge of human genome variation limits the determination of causality between identified variants and disease. While larger sample sizes are required to establish the precision and accuracy of this type of testing, this study supports WES for inherited early onset macular degeneration disorders as an alternative to standard mutation screening techniques.
Keywords: Stargardt Disease, Macular Degeneration, Exome, Mutation Screening, Molecular Diagnostics, ABCA4, PRPH2
Background
Individuals with early-onset macular degeneration (MD) represent a diagnostic challenge. While observation of MD in a juvenile or young adult strongly implies an underlying genetic cause of disease, the spectrum of potential genes and mutations known to cause MD is extensive. Given the variability of clinical features that have been attributed to autosomal recessive mutations in the ABCA4 gene [1-3], the most common clinical diagnosis for MD is Stargardt Disease [OMIM: #248200/#600110] (STGD). Rare cases of STGD or “Stargardt-like” disease phenotypes have been reported with mutations in PRPH2[4,5], VMD2[6], ELOVL4[7-9] and PROM1[10]. These genes, as well as ABCA4, are also associated with clinically distinct phenotypes including retinitis pigmentosa, cone/rod dystrophy and pattern dystrophy. This complex arena of genes and clinical features complicates the nomenclature in this field; it is unclear how to classify individuals with classic Stargardt phenotype lacking recessive mutations in ABCA4. While we do not limit the term “retinitis pigmentosa” to a phenotype caused by mutations in a single gene, there are those who believe STGD should be restricted to only those cases caused by ABCA4 mutations and “Stargardt-like” or juvenile macular dystrophy should be used for other genetic etiologies. For the purposes of this study, we classify our participants with early-onset macular degeneration as “putative Stargardt Disease” cases.
Since the initial identification of recessive mutations in the ABCA4 gene in several large STGD pedigrees [3], a multitude of studies have attempted to assess the mutational spectrum of ABCA4 in various ethnically restricted populations (Italy [11,12]; Spain [13,14]; Japan [15]; Portugal [16]; Hungary [17]; Denmark [18]). While each population appears to carry some founder mutations accounting for a significant proportion of disease alleles, hundreds of extremely rare or private variants have also been identified [19]. This allelic heterogeneity hampers the viability of standard panel-based mutation screening techniques and has lead to the creation of an ABCA4 array designed to genotype over 600 individual DNA variants [19]. While this array is an effective screening tool, the high rate of private mutations in STGD prevents it from being a comprehensive solution.
Another factor making ABCA4 such a difficult diagnostic target is its genomic structure. The large number of coding exons in ABCA4 gene (50 coding exons resulting in a 6.8 kb mRNA [Refseq NM_000350]) makes PCR-based sequencing costly and time-consuming. Currently, the cost of PCR and dideoxy-based targeted ABCA4 exon sequencing is approximately half that of WES. The costs of data analysis are difficult to compare, but WES analysis is highly automatable and can be performed in parallel as opposed to the highly labor-intensive analysis required to identify rare variants from individual sequence traces. Approaches using targeted next-generation sequencing hold great potential for ABCA4 mutation screening [20,21], and may reduce the cost of single gene sequencing significantly.
Using a combination of methods to screen for ABCA4 mutations in 121 patients from a heterogeneous patient population in our natural history study of Stargardt Disease, only half (59/121, 49%) of the cases were found to have two known or putative disease-causing ABCA4 mutations on separate alleles. Nearly one third have a single mutation, and the remaining ~20% have no definite or probable disease-causing ABCA4 mutations (manuscript in preparation). Highly sensitive next-generation targeted re-sequencing of ABCA4 has been implemented on an independent sample set with similar results [20]. Whether individuals with putative Stargardt Disease with one or zero detected ABCA4 mutations have non-traditional variation (e.g. promoter variants or epigenetic changes) missed by current mutation screening techniques or mutations in other genes is unclear at this time.
Providing a genetic diagnosis for individuals with putative Stargardt Disease is thus an ongoing challenge. Here we apply whole exome sequencing to nine putative STGD cases to assess the efficacy of this approach for molecular genetic diagnosis of this condition. All patients were previously screened and found to have zero (eight samples) or one (one sample) disease-causing mutation in ABCA4. While some of these probands express ocular imaging phenotypes not typically observed in classic STGD, all were diagnosed clinically as having STGD due to the early age at which signs of macular degeneration were initially identified.
In this study, we use the same WES methods and analytical tools as those implemented by laboratories now offering CLIA-approved, clinical WES. As such, we report these research findings as a preliminary surrogate for expected results from clinical WES of patients with retinal dystrophy.
Methods
Study Participants
Participant samples were recruited into this study under approval of the UCLA Institutional Review Board and consented prior to participation. Each was diagnosed by a referring retinal specialist and confirmed by a retinal expert at the Jules Stein Eye Institute, University of California Los Angeles (Gorin M.B) [22]. All research was conducted in accordance with the Health Insurance Portability and Accountability Act of 1996. A basic ophthalmologic exam, electroretinogram (ERG), optical coherence tomography (OCT) and imaging of the fundus was performed for each individual. Clinical phenotyping summaries can be found in Table 1. Subjects were selected for WES if they had an initial clinical diagnosis of Stargardt Disease and lacked mutations in ABCA4 as screened by targeted dideoxy re-sequencing. One case, STGD-02, was included as a positive control having had one ABCA4 mutation detected previously by Sanger sequencing.
Table 1.
Clinical Information and Genetic Findings of Putative Stargardt Disease Cases
| Sample | Age Sex | Acuity OD Acuity OS | Clinical Notes | ERG Findings | Color vision | ABCA4variants | PRPH2variants | Other variants |
|---|---|---|---|---|---|---|---|---|
| STGD-01 |
19 Male |
20/400 20/160 |
Peripapillary sparing, discrete flecks; nummular atrophy |
Normal rod Abnormal cone |
No testing |
p.N965S p.R2038W |
. |
. |
| STGD-02 |
15 Female |
20/160 20/100 |
Few yellowish Flecks without autofluorescence |
Normal rod Normal cone |
Abnormal |
p.Q636X p.G1961E* |
. |
. |
| STGD-03 |
58 Female |
20/50 20/16 |
No significant atrophy |
Abnormal rod Abnormal cone |
No testing |
. |
p.W25C p.Q276X |
. |
| STGD-04 |
51 Male |
20/20 20/20 |
Peripapillary sparing both eyes; “stellate” pattern |
Abnormal rod Abnormal cone |
Abnormal |
. |
p.G167S |
. |
| STGD-05 |
55 Female |
20/25 20/20 |
Central atrophy with out-branching; "stellate" pattern |
Normal rod Normal cone |
Normal |
. |
p.T236X |
. |
| STGD-06 |
34 Female |
20/200 20/200 |
Classic Stargardt |
Normal rod Abnormal cone |
No testing |
p.V989A |
. |
. |
| STGD-07 |
41 Male |
20/40 20/640 |
Flecks, peripapillary sparing, central geographic atrophy |
Normal rod Abnormal cone |
No testing |
. |
. |
. |
| STGD-08 |
37 Male |
20/40 20/400 |
No peripapillary sparing/flecks; irregular geographic atrophy with RPE changes; thickening on OCT |
Normal rod Normal cone |
Abnormal |
. |
. |
CRB1 p.K801X |
| STGD-09 | 61 Male | 20/25 20/25 | "Horseshoe" pattern of atrophy | Normal rod Normal cone | No testing | . | . | ELOVL4 p.A311T |
“Age” is age at ascertainment. Abbreviations: ERG - Electroretinogram; RPE - Retinal pigment epithelium; OD - right eye; OS - left eye. * Variant previously observed by dideoxy sequencing. “Variants” indicates putatively causal variants and does not include predicted benign or common (polymorphic) variants.
Whole Exome Sequencing Data Generation
Randomly fragmented genomic DNA libraries were created following standard protocols for high-throughput paired-end sequencing on the Illumina GA2x or HiSeq2000 instruments (Illumina Inc., San Diego, CA). The Agilent SureSelect 50 Mb capture kit was used to enrich the libraries for known coding loci in the human genome (Agilent Technologies; Santa Clara, CA). This kit has been shown to effectively capture >90% of these loci when an adequate average read depth is reached [23].
Data Analysis
Nucleotide base calling and quality score assessment was performed using instrument-specific Real Time Analysis (RTA) software provided by Illumina. A combination of commercially or academically available tools and custom scripts were used to analyze the raw DNA sequence reads. Alignment to the human genome (hg19; NCBI build 37; Feb. 2009) was performed using Novoalign (Novocraft Technologies; Selangor, Malaysia). Merging, sorting, and other manipulation of aligned data was performed using SAMTools [24]. PCR clonal duplicate removal was performed using Picard http://picard.sourceforge.net. Quality score recalibration, genotyping, variant filtration, and coverage depth analysis were performed using the Genome Analysis Toolkit [25]. Variant consequence analysis was performed using SeattleSeq [26] which incorporates many databases including: “NCBI full genes”, dbSNP131, and the 1000 Genomes Project. Further analysis was performed using a combination of custom PERL scripts and Mathematica 7.0 (Wolfram Research; Champaign, IL).
Novel variants identified as putatively causal were screened for presence in the NHLBI Exome Sequencing Project database. Alleles present at ≥1% allele frequency in either sample population (European Caucasian or African American) were considered common alleles unlikely to contribute to rare disease, and thus removed from analysis.
Additional Sequencing
Targeted amplification of PRPH2 and ELOVL4 coding sequences was performed using PCR (primer sequences available in Additional file 1 Supplemental Materials). PCR products were then sent out for dideoxy sequencing (Beckman Coulter; Brea, CA) and analyzed using Sequencher software (Gene Codes Corp.; Ann Arbor, MI).
Results
Exome sequencing completeness and quality
All libraries were successfully sequenced with a minimum mean on-target coverage depth of 44x (Additional file 2 Table S1). Samples sequenced on the Illumina HiSeq2000 had significantly higher coverage (average of 103x-121x) due to the increased volume of sequence reads produced by this instrument compared to the GA2x. Samples contained on average approximately 156,000 DNA variants from hg19, the vast majority of which overlap a known polymorphism (Additional file 2 Table S1). The two individuals of African American descent had an increased proportion of novel variants, consistent with the higher levels of genetic heterogeneity in that sub-population. Both the total number of variants and proportion of which overlap a known polymorphism were tightly correlated with read depth and are within the expected limits based on other studies [23].
Whole exome sequencing using the Agilent SureSelect 50 Mb capture kit provided high quality base calls for a minimum of 97.5% of the 6,822 base pair positions corresponding the translated ABCA4 mRNA. For samples sequenced on the HiSeq2000 were covered, 100% of bases were covered ≥8x (Additional file 2 Table S1). Canonical splice acceptor and splice donor sites were captured at a similar rate.
Individual Case Descriptions and Identified Variants
Clinical summaries, including visual acuity, age of recruitment, gender, ethnicity, and relevant ophthalmological findings are found in Table 1.
STGD-01
This participant exhibited several classic features of STGD (Table 1, Additional file 3 Figure S1). Panretinal cone dysfunction with preserved rod function was documented by ERG. Exome sequencing identified two missense variants (p.N965S and p.R2038W) previously reported as disease-causing in STGD [19].
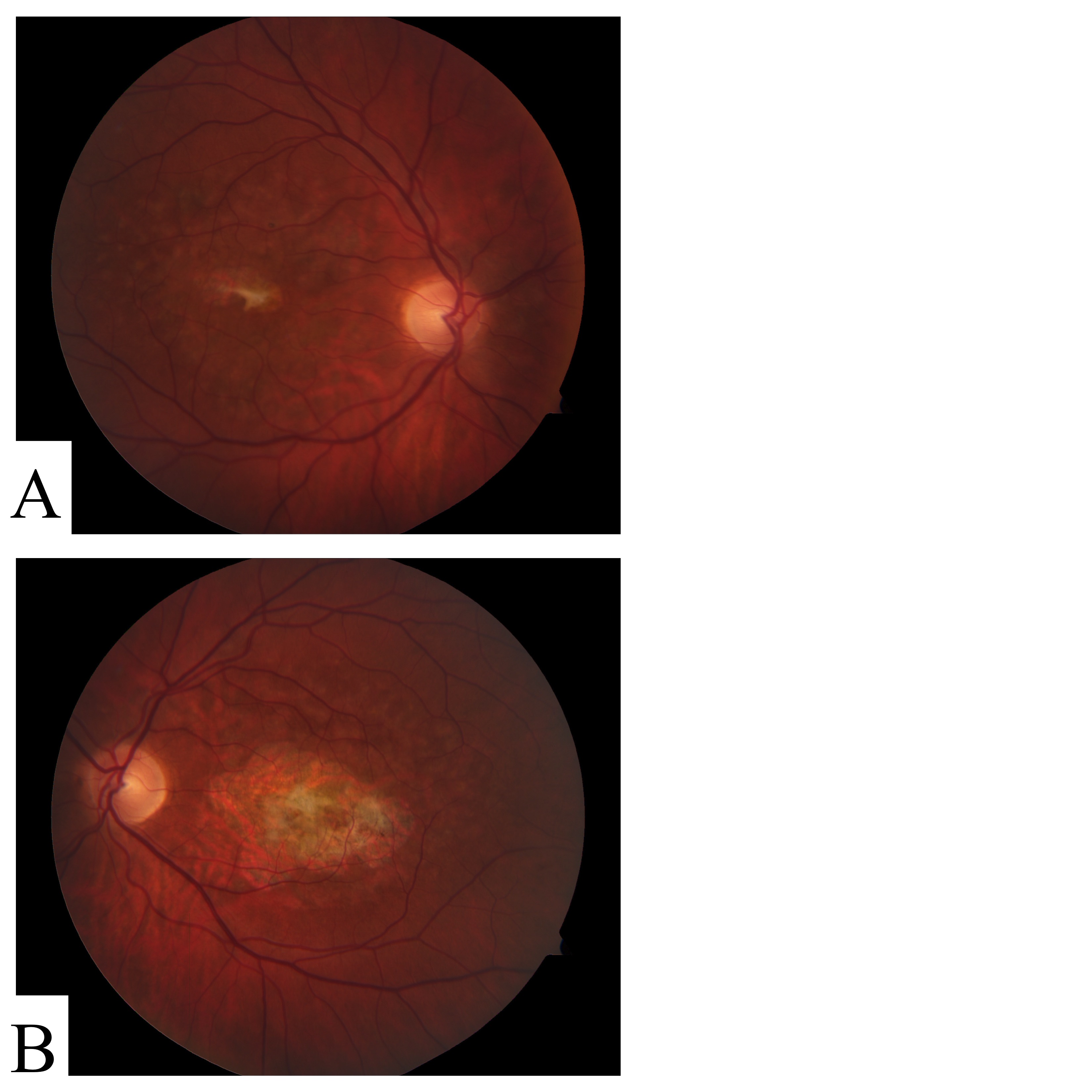
STGD-02
This participant presented with atypical macular degeneration (Table 1, Additional file 4 Figure S2). Two rare coding variants in ABCA4 were identified by exome sequencing in this participant. The p.G1961E missense variant is a known STGD mutation [19]. This variant was previously detected by PCR amplification and dideoxy sequencing and served as a positive control in this study. The second variant p.Q636X introduces a premature termination codon and is thus considered very likely deleterious.
STGD-03
This participant was documented to have central vision loss, more severe in the left eye (Table 1, Additional file 5 Figure S3a-S3b). Subretinal fluid was observed by OCT (Additional file 5 Figure S3b-S3c). Multifocal ERG showed near complete loss of the foveal peaks and uniformly reduced and delayed cone responses for the entire testing areas of both eyes. Two rare, novel coding variants in PRPH2 (p.W25C and p.Q276X) were identified in this participant. As parental genetic material is not available and these two variants are non-adjacent, it is not possible to assess phase. The missense variant is predicted to be deleterious by all three algorithms implemented by Condel [27]and the nonsense variant is assumed to be deleterious.
STGD-04
This participant presented with bilateral symptoms including central vision distortion, light/dark adaption difficulty, and intermittent photopsias. Clinical findings were consistent with a pattern dystrophy (Table 1, Additional file 6 Figure S4). Full field and multifocal ERGs detected abnormal cone and rod amplitudes and implicit times. Color vision (FM100 Hue) was bilaterally abnormal, with greater severity for the left eye. A single rare coding variant in PRPH2 was identified in this participant. This variant, p.G167S, has previously been linked to pattern dystrophy [28] and is predicted to be deleterious by Condel. The participant’s father’s vision became uncorrectable in the sixth decade of life, potentially consistent with a dominant mode of inheritance.
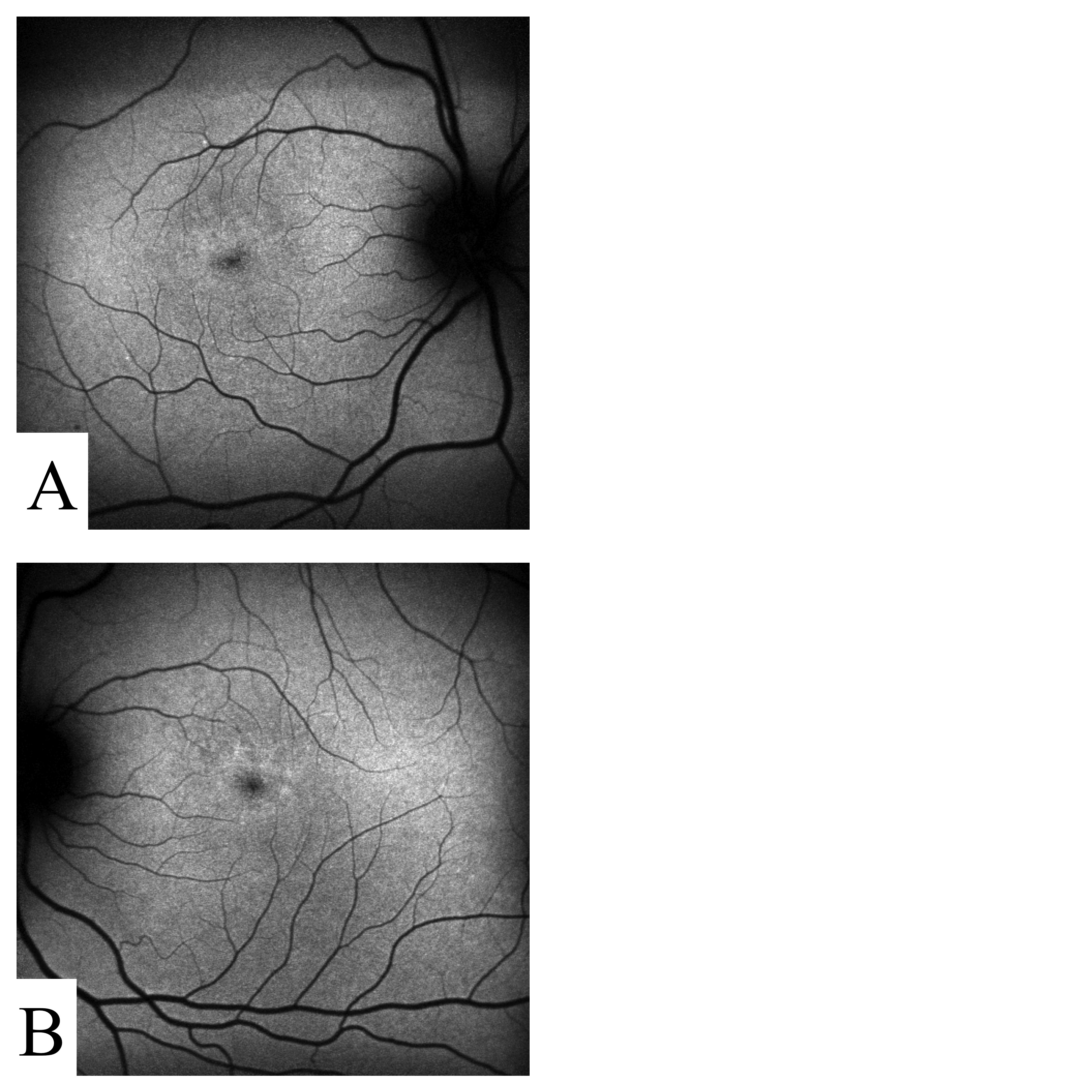
STGD-05
This participant showed signs of pattern dystrophy (Table 1, Additional file 7 Figure S5). A single rare nonsense variant (p.T236X) in PRPH2 was identified in this participant. This variant has not been previously described, but is likely deleterious as it introduces a premature termination codon. The proband’s father and paternal grandfather suffered from night blindness but, by history, the visual impairment was not as severe as that of the proband.
STGD-06
This participant had classic features of STGD (Table 1, Additional file 8 Figure S6). The full field ERG showed abnormal cone responses with preserved rod function. Exome sequencing identified a single, previously described rare missense disease-causing variant in ABCA4 (p.V989A). No other mutations were observed in any of the other genes known to cause retinal or macular dystrophies.
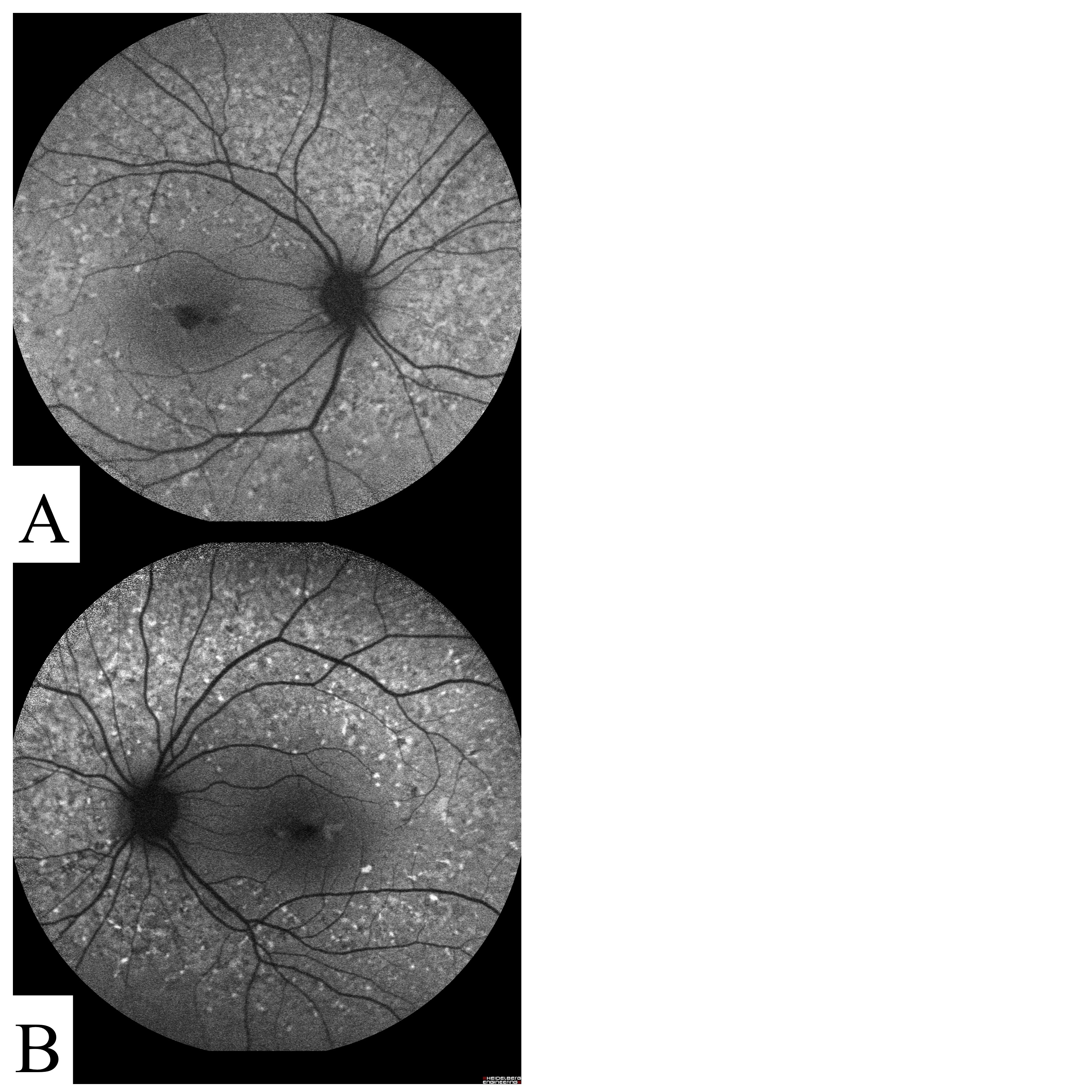
STGD-07
This participant had several classical STGD features including fundus flecks (Table 1, Additional file 9 Figure S7). The full field ERG showed abnormal cone responses with normal rod function. No clear candidate variants were found by exome sequencing in any of the known retinal or macular genes that might account for these clinical findings.
STGD-08
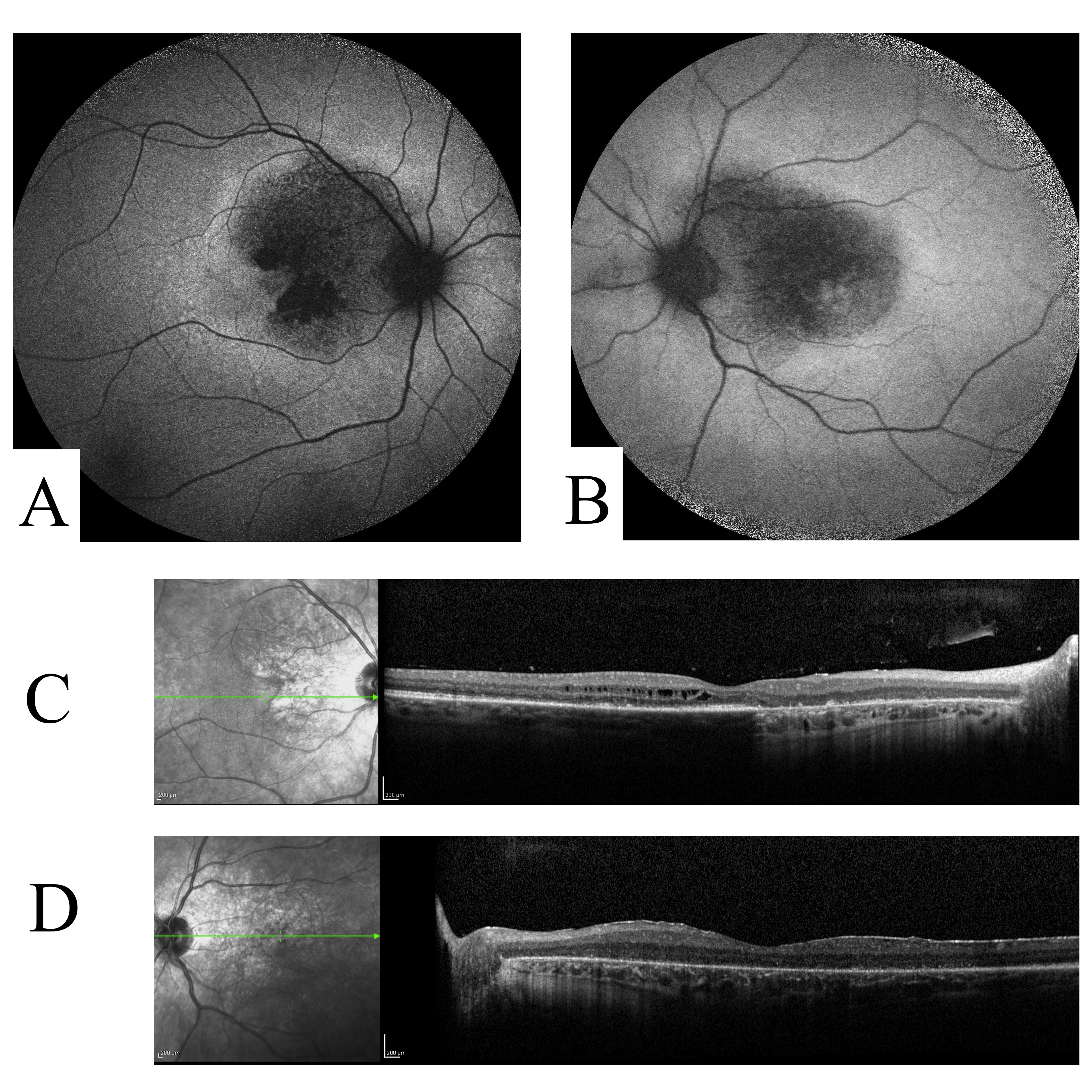
This participant was diagnosed with macular degeneration by age 16, but did not suffer from severe vision impairment until age 37. No fundus flecks were observable, though there was an irregular pattern of atrophy and RPE changes (Table 1, Additional file 10 Figure S8a-S8b). The central vision was impaired severely in the left eye but less so in the right. The optical coherence tomography study showed a diffuse thickening and loss of lamination of the central retina without the presence of cavitations or subretinal fluid (Additional file 10 Figure S8b-S8c). The retinal appearance, while atypical for patients with known Stargardt Disease, is similar to that seen in retinal dystrophies attributable to CRB1[29]. A novel nonsense mutation p.K801X in the developmental gene CRB1 was found by exome sequencing. There is no family history of visual problems suggestive of a dominant mode of inheritance.
STGD-09
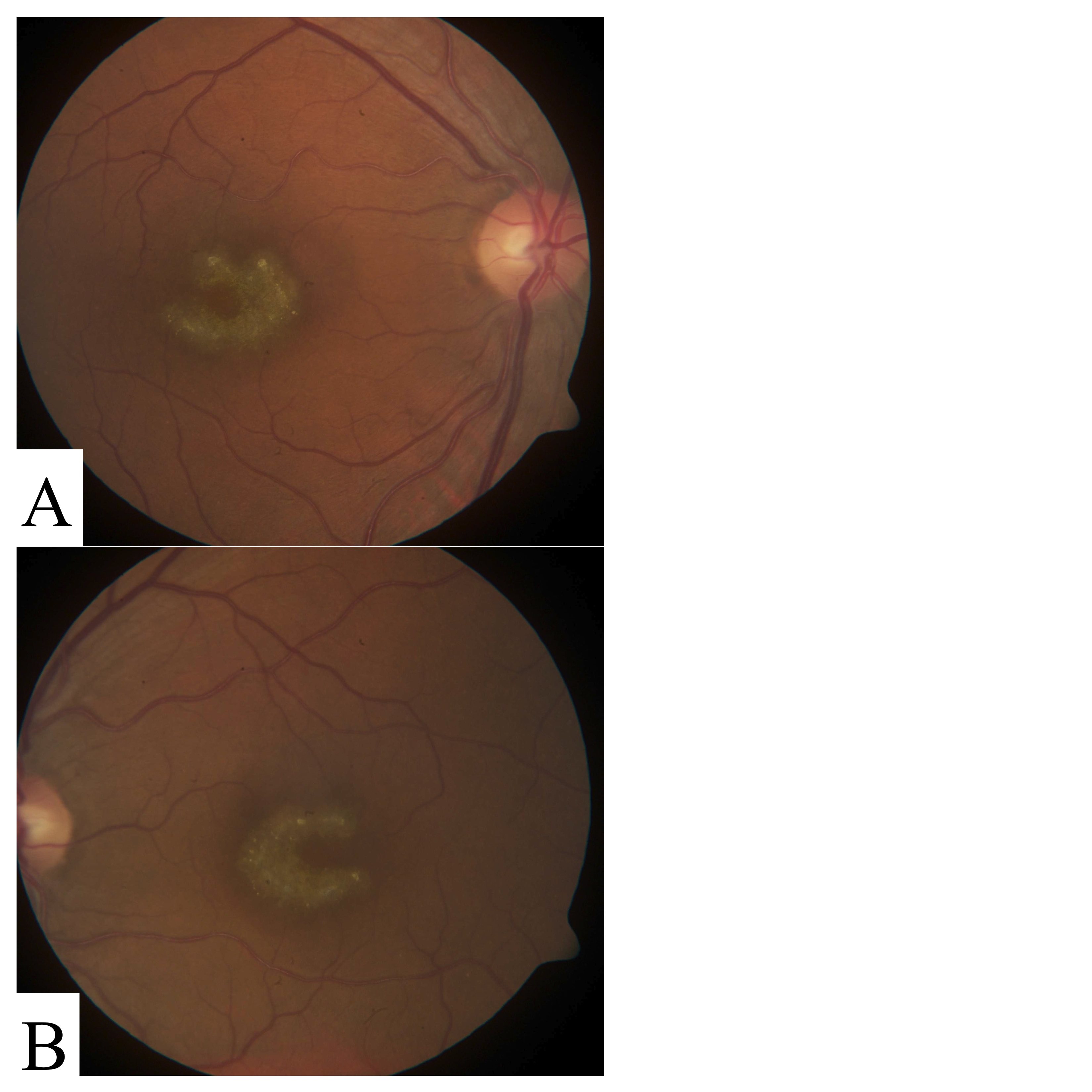
This participant presented with central vision loss and geographic atrophy in a perifoveal “horseshoe” pattern (Table 1, Additional file 11 Figure S9). Exome sequencing identified a novel missense variant in ELOVL4: p.A311T. An unconfirmed family history report indicated that his father may have had similar visual impairment.
Additional PHPR2 and ELOVL4 sequencing
Direct re-sequencing of 68 additional putative Stargardt Disease cases identified five individuals with either of two previously reported PRPH2 mutations (p.P210R [30] or p.172 W [5]). In total, rare coding variants in eight out of 77 (10.4%) putative STGD cases from six independent pedigrees were identified. This sample set is enriched for patients without two known ABCA4 mutations, suggesting this pickup rate is likely elevated compared to the general STGD patient population. No additional individuals were found to harbor putative disease-causing variants in ELOVL4.
All eight individuals with at least one putatively deleterious PRPH2 allele were over the age of 50 at ascertainment (age range 53–70 years). Two of these individuals also carry at least one disease mutation in ABCA4 identified by direct sequencing. Three had severe atrophy, precluding the clinical observation of pattern dystrophy that may or may not have been present earlier in life. Among the five remaining cases, a clear pattern dystrophy can be appreciated in only four out of a possible ten maculae.
Discussion
The most common cause of early-onset macular degeneration is Stargardt Disease (STGD). Even in the context of STGD - which is primarily caused by compound heterozygous missense, nonsense and/or splicing variants in ABCA4 - there are key complicating factors that lead to problems in precise molecular diagnosis. In this study we examine the feasibility of performing whole exome sequencing on putative STGD cases as either a supplement or replacement for ABCA4-specific targeted methodologies. To do this, we sequenced genomic DNA from nine individuals with negative or unclear molecular diagnosis following direct sequencing of the major STGD disease gene ABCA4. The sample size of this study limits the determination of the general validity of this approach, but our results strongly suggest that mutation detection of STGD patients by WES is highly effective as compared to other methods.
Four out of five ABCA4 mutations were not detected by PCR/dideoxy based exon sequencing and yet were detected in the context of whole exome sequencing in this study. Of the five total disease-causing ABCA4 alleles found by WES, four (all except p.Q636X) have previously been described and are genotyped by the current ABCR array available from Asper Biotech, supporting the use of the genotyping array as a first-pass screening tool due to its low cost.
Two of these mutations were clearly visible as heterozygous variants upon re-analysis of the original sequencing traces. One of the newly identified mutations is visible on the original sequence chromatogram but is located within the first five bases of the read and thus indistinguishable from normal background noise. The chromatogram corresponding to the remaining variant had a high level of noise throughout. Other laboratories have noted similarly missed variants [31]. While currently considered the “gold standard” for molecular genetic diagnosis of SNV-type mutations, PCR-based dideoxy sequencing relies heavily on manual sequence trace inspection and is thus subject to human error. This method should and will continue to be relied upon for many applications, including mutation validation and genotyping of known alleles. However, it is difficult for a molecular diagnostic laboratory to maintain a high level of surveillance quality when performing a large number of these assays, as is required for mutation screening in STGD and other macular dystrophies.
Variant detection by exome sequencing is by comparison far more automated and less susceptible to this type of error. The sources of error in WES are systematic and readily measured. The major sources of erroneous findings by WES are insufficient or uneven read depth (“coverage”), mismapping, and incorrect variant annotation. While these sources of error are complex to resolve, they are highly consistent which has allowed computational advancements to dramatically improve results. In contrast, the confounding issues facing capillary gel electrophoresis-based dideoxy sequencing have remained relatively stable since the inception of this method. Our data supports the contention that WES provides reliable and efficient mutation detection for SNVs and indels in ABCA4, and is an attractive alternative to other methods.
Surprisingly, RDS/PRPH2 mutations were identified by exome-seq in three individuals. This gene has been linked to several other retinopathies including pattern macular dystrophy [30,32-36] and retinitis pigmentosa [37], and it has been previously suggested that mutations in PRPH2 can mimic the STGD phenotype [38] or modify disease severity [5]. However, PRPH2 has not been considered a significant contributor to classic STGD. Indeed, two of the individuals carrying a heterozygous PRPH2 mutation show a “stellate” pattern dystrophy in one eye. While this is consistent with previous reports of PRPH2-related disease [4,28,37-40], it is too subtle a finding to confidently sub-classify patients. The lack of concordance between two eyes of the same individual in both cases (Additional file 6 Figure S4 and Additional file 7 Figure S5) suggests incomplete expressivity of this endophenotype, suggesting that some cases with PRPH2 related disease may show no identifiable signs of pattern dystrophy.
Additional mutations were detected by direct re-sequencing of PRPH2 in a broader cohort of STGD cases. Complicating matters, two out of the eight people with rare missense or nonsense mutations in PRPH2 also have mutations in ABCA4. One of these participants, a compound heterozygote for ABCA4 mutations has two affected relatives who share the PRPH2 mutations but neither of the ABCA4 mutations. For these relatives, the PRPH2 mutation appears to be disease-causing. In the proband with a single ABCA4 mutation and a PRPH2 mutation, the genetic etiology of the disease is less clear. At this time, it is not possible to assess the effect of this potentially digenic scenario, though it has been previously observed [5]. The presence of rare, coding PRPH2 variants in STGD cases with one or more ABCA4 mutations suggests that one must be cautious regarding attributing apparent cases of Stargardt Disease to ABCA4 mutations when only ABCA4 has been screened. Though rare, these cases support the implementation of exome sequencing to detect potentially confounding genetic mutations as well as enable the study of the effect of multi-gene mutation combinations on disease progression and severity.
Mutations in CRB1 have been associated with a wide array of retinal dystrophies, including retinitis pigmentosa and Leber congenital amaurosis [41]. Here we report the first finding of a heterozygous nonsense mutation in CRB1 in an individual with macular degeneration. Given that CRB1 is involved in normal retinal development, this variant is an intriguing functional candidate. A thickening of the fovea (without the cavitations associated with macular edema) observable by OCT in this participant is consistent with other cases of CRB1-related disease. In the absence of a functional validation of the p.K801X variant or analysis of additional cases with nonsense variants in CRB1, it is not possible at present to confidently assign a molecular diagnosis to this case and we thus categorize this variant as being of unknown significance. This inability to causally link a suspect variant with phenotype is a significant limitation of the technique, and will likely be typical of early clinical WES efforts.
While it is attractive to attempt to correlate clinical and phenotypic information with our newly acquired genetic data, sample size is a significant limitation. For example, one might hypothesize that individuals with PRPH2 dysfunction would have abnormal cone/rod responses due to its restricted expression to the outer disc segment of photoreceptors [42]. However, given the range of variability across Stargardt cases and the small number of PRPH2 positive cases, it is not possible to assess any such correlation at present. Collaboration and data sharing amongst multiple STGD research groups is needed to sufficiently increase the sample size to evaluate any genotype-phenotype correlations.
In this study, we suffer from the unavailability of complete family history information and genetic material from related individuals, particularly for participants with variants outside of ABCA4. These limitations are common in clinical practice. While both history and DNA would provide critical insight into mode of inheritance and shed light on the functional consequences of observed genetic variants, we identified a clear molecular basis of disease in six out of nine cases.
Despite near total coverage of most genes, including both ABCA4 and PRPH2 in all nine cases, several of our cases elude a definitive molecular diagnosis. One case has only a single ABCA4 mutation, one has a variant of unknown significance in CBR1 and one sample has no demonstrable mutations in any gene with known association to retinal or macular degeneration. Careful manual inspection of genes carrying potential compound heterozygous, homozygous, and protein-truncating variants in these individuals did not yield any strong functional candidates for novel STGD disease genes. It is now generally accepted that every individual carries a significant burden of potentially pathogenic DNA variants within the known exome [43], and STGD cases are no different in this regard. Which, if any, such variants in genes not currently associated with retinopathy in our sample contribute to disease onset and progression cannot be assessed at this time, nor can we rigorously address the potential of other genetic variants acting in a dominant fashion or in a digenic manner. The exome data provides an opportunity to make a concerted effort to re-analyze these data as new disease genes and genetic modifiers are implicated in retinal and macular dystrophies. In the meantime, several of our cases elude molecular diagnosis. While such cases represent a small proportion of the overall STGD participant population, they may eventually provide invaluable insight into the pathogenesis of STGD and the biology of the retina.
The disease phenotype of STGD can be quite variable, even amongst individuals carrying identical or putatively similar ABCA4 mutations. Age of onset, electroretinogram response, disease progression and disease severity are all clinically relevant parameters of phenotypic variability in these participants. While some genotype-phenotype correlations have been found for population-specific ABCA4 mutations [11], the majority of this variability remains unexplained. One major benefit of exome sequencing over targeted ABCA4 mutation screening is the potential for identifying disease-modifying alleles in other genes. If approached properly, clinical exome sequencing can enable such studies as a byproduct of providing highly sensitive molecular testing.
Knowledge of genome-wide functional variation may be particularly critical to enrollment of participants in future clinical trials. Restoration or replacement of ABCA4 protein function would likely be insufficient for a participant harboring disease-causing mutations in additional genes, such as seen with PRPH2 in this study. Thus the positive identification of two mutations in ABCA4 may not be a proper endpoint for molecular diagnosis in STGD. In contrast, exome sequencing can help place STGD participants into genomic context as it identifies both common and rare functional genetic variation.
Clinicians now have a multitude of options available to them for STGD mutation screening. Targeted approaches continue to grow in sensitivity and shrink in cost [20], but they will always be fundamentally limited to specific genes or alleles. From these small-scale results, we find very strong potential for STGD molecular diagnosis by exome sequencing. Presently, an approach following up array-based ABCA4 mutation screening with exome sequencing is a prudent and cost-effective alternative to direct re-sequencing. In the near future, exome sequencing and indeed whole genome sequencing will likely be sufficiently inexpensive (and potentially covered by health insurance), eliminating the need for STGD-specific mutation screening.
While important issues such as off-target findings and variants of unknown significance remain highly controversial in the field of genomic diagnostics, harnessing WES technology to provide molecular diagnosis for a specific subset of genes – essentially performing gene panels by exome analysis – largely circumvents these issues. If participants are properly consented, willing to participate, and educated properly about the risks, benefits, and scope of such testing, clinical molecular diagnosis and research can and should be pursued simultaneously. With sufficient sample sizes, a deeper understanding of locus heterogeneity and perhaps even modifier genes becomes possible. This interplay between clinical testing and research should provide better patient care and pave the way toward an improved understanding of Stargardt Disease and macular degeneration in general.
Conclusions
Genomic analysis is the most direct means to resolve the clinical overlap of inherited early onset macular degeneration disorders. Based upon the striking level of success in our most difficult to classify cases, clinical exome sequencing should be considered as a potential front line diagnostic tool for retinal and macular dystrophies with atypical presentations, as a follow-up after negative or unclear results from initial screening by targeted mutation analysis, Additionally, WES may prove crucial in ruling out rare mutations in genes that may confound the determination of causality before proceeding to gene-targeted therapies.
Abbreviations
STGD: Stargardt Disease; WES: whole exome sequencing; OD: right eye; OS: left eye; OCT: Optical coherence tomography; ERG: electroretinogram; Mb: megabases.
Competing interests
The authors claim no financial conflicts or competing interests relevant to this study.
Authors’ contributions
SPS analyzed the exome sequence data, and wrote and prepared the manuscript. YQG performed dideoxy DNA sequencing and analyzed the results. AM provided genetic counseling and pedigree analysis, and aided in the phenotypic assessment of participants. CO recruited study participants and aided in phenotypic assessment. ZC directed the preparation and high throughput sequencing of exomic libraries. SFN provided the means, expertise, and computational resources to perform and analyze exome sequencing data. SN performed and analyzed results of electroretinogram measurements. DBF was a co-investigator, involved in sample recruitment, conceptualization, and all dideoxy sequencing analysis. MBG, the principal investigator, conceived this project and contributed to all aspects of this work including clinical examination of participants, and interpretation of results. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
Figure S1. Autofluorescence (A,B) and infrared (C,D) imaging of participant STGD-01. Figure S2. Fundus photos of participant STGD-02. Figure S3. Autofluorescence (AF) imaging and optical coherence tomography (OCT) images for participant STGD-03. 50° AF image of OD (A) and OS (B) shows geographic atrophy, discrete autofluorescent flecks, and peripapillary sparing in both eyes. OCT of OD (C) shows severe retinal edema. Figure S4. Autofluorescent imaging of participant STGD-04. 30° AF image of OD (A) shows “stellate” pattern dystrophy. Pattern less clear in OS (B). Figure S5. Autofluorescent imaging of participant STGD-05. 50° AF imaging of both OD (A) and OS (B) shows possible “stellate” pattern dystrophy. Figure S6. Red-free fundus images of participant STGD-06. 30° fundus images of both OD (A) and OS (B) show wide-spread geographic atrophy of the macula with mild peripapillary sparing. Figure S7. Color fundus images of participant STGD-07. 50° color fundus images from both OD (A) and OS (B) show geographic atrophy with peripapillary sparing. Figure S8. Autofluorescence and optical coherence tomography images for participant STGD-08. 50° AF shows irregular geographic atrophy without peripapillary sparing or autofluorescent flecks in both OD (A) and OS (B). OCT shows an irregularly thickened contour of the retina in both OD (C) and OS (D). Figure S9. Fundus photos of OD (A) and OS (B) for participant STGD-08. Clear “horse-shoe” pattern of atrophy is observable in both eyes.
Table S1. Whole Exome Sequencing Details
Figure S1. Autofluorescence (A,B) and infrared (C,D) imaging of participant STGD-01.
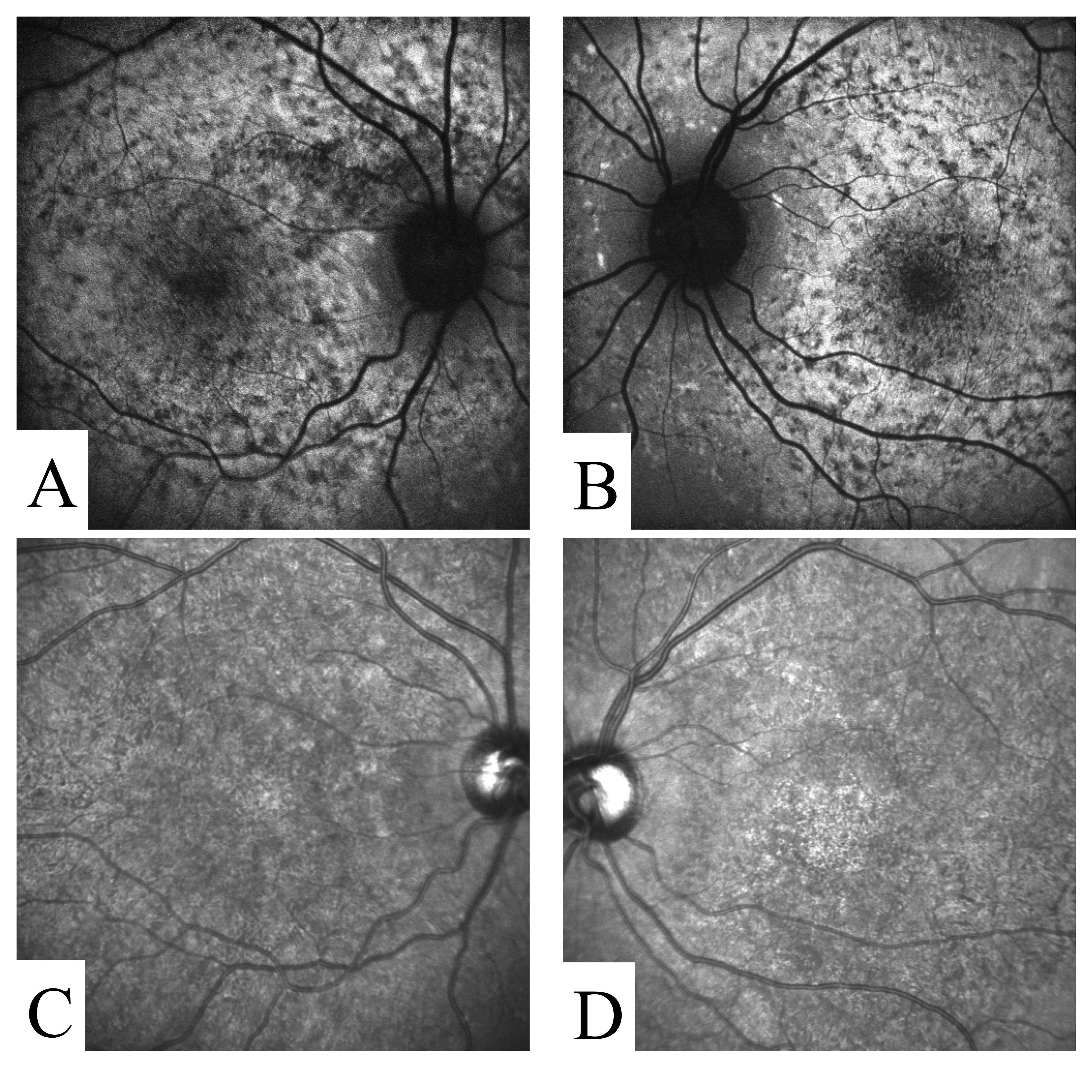{kind=link}
Figure S2. Fundus photos of participant STGD-02.
{kind=link}
Figure S3. Autofluorescence (AF) imaging and optical coherence tomography (OCT) images for participant STGD-03. 50° AF image of OD (A) and OS (B) shows geographic atrophy, discrete autofluorescent flecks, and peripapillary sparing in both eyes. OCT of OD (C) shows severe retinal edema.
{kind=link}
Figure S4. Autofluorescent imaging of participant STGD-04. 30° AF image of OD (A) shows “stellate” pattern dystrophy. Pattern less clear in OS (B).
{kind=link}
Figure S5. Autofluorescent imaging of participant STGD-05. 50° AF imaging of both OD (A) and OS (B) shows possible “stellate” pattern dystrophy.
{kind=link}
Figure S7. Color fundus images of participant STGD-07. 50° color fundus images from both OD (A) and OS (B) show geographic atrophy with peripapillary sparing.
{kind=link}
Figure S6. Red-free fundus images of participant STGD-06. 30° fundus images of both OD (A) and OS (B) show wide-spread geographic atrophy of the macula with mild peripapillary sparing.
{kind=link}
Figure S8. Autofluorescence and optical coherence tomography images for participant STGD-08. 50° AF shows irregular geographic atrophy without peripapillary sparing or autofluorescent flecks in both OD (A) and OS (B). OCT shows an irregularly thickened contour of the retina in both OD (C) and OS (D).
{kind=link}
Figure S9. Fundus photos of OD (A) and OS (B) for participant STGD-08. Clear “horse-shoe” pattern of atrophy is observable in both eyes.
{kind=link}
Contributor Information
Samuel P Strom, Email: strom@jsei.ucla.edu.
Yong-Qing Gao, Email: yonggao8963@gmail.com.
Ariadna Martinez, Email: amartinez@jsei.ucla.edu.
Carolina Ortube, Email: ortube@jsei.ucla.edu.
Zugen Chen, Email: zugenchen@mednet.ucla.edu.
Stanley F Nelson, Email: snelson@ucla.edu.
Steven Nusinowitz, Email: nusinowitz@jsei.ucla.edu.
Deborah B Farber, Email: farber@jsei.ucla.edu.
Michael B Gorin, Email: gorin@jsei.ucla.edu.
Acknowledgements
We would like to thank all of our study participants for enabling this research by contributing their valuable time, medical information, and genetic material. We thank Kevin Squire and Hane Lee for bioinformatics assistance. We thank Traci Toy for preparing genomic DNA libraries for exome sequencing. This work was made possible in part by generous donations from Harold and Pauline Price.
Funding
This research was supported by: The Foundation Fighting Blindness, Research to Prevent Blindness, and the Jules Stein Eye Institute Foundation. This publication was made possible in part by Grant Number 5 T32 GM008243 from the National Institutes of Health, National Institute of General Medical Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIGMS.
References
- Nasonkin I, Illing M, Koehler MR, Schmid M, Molday RS, Weber BH. Mapping of the rod photoreceptor ABC transporter (ABCR) to 1p21-p22.1 and identification of novel mutations in Stargardt's disease. Hum Genet. 1998;102(1):21–26. doi: 10.1007/s004390050649. [DOI] [PubMed] [Google Scholar]
- Rozet JM, Gerber S, Souied E, Perrault I, Châtelin S, Ghazi I, Leowski C, Dufier JL, Munnich A, Kaplan J. Spectrum of ABCR gene mutations in autosomal recessive macular dystrophies. Eur J Hum Genet. 1998;6(3):291–295. doi: 10.1038/sj.ejhg.5200221. [DOI] [PubMed] [Google Scholar]
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A. et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15(3):236–246. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- Coco RM, Tellería JJ, Sanabria MR, Rodríguez-Rúa E, García MT. PRPH2 (Peripherin/RDS) mutations associated with different macular dystrophies in a Spanish population: a new mutation. Eur J Ophthalmol. 2010;20(4):724–732. doi: 10.1177/112067211002000413. [DOI] [PubMed] [Google Scholar]
- Poloschek CM, Bach M, Lagrèze WA, Glaus E, Lemke JR, Berger W, Neidhardt J. ABCA4 and ROM1: implications for modification of the PRPH2-associated macular dystrophy phenotype. Invest Ophthalmol Vis Sci. 2010;51(8):4253–4265. doi: 10.1167/iovs.09-4655. [DOI] [PubMed] [Google Scholar]
- Petrukhin K, Koisti MJ, Bakall B, Li W, Xie G, Marknell T, Sandgren O, Forsman K, Holmgren G, Andreasson S. et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998;19(3):241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- Edwards AO, Donoso LA, Ritter R. A novel gene for autosomal dominant Stargardt-like macular dystrophy with homology to the SUR4 protein family. Invest Ophthalmol Vis Sci. 2001;42(11):2652–2663. [PubMed] [Google Scholar]
- Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z, Li Y, Metzker ML, Allikmets R, Zack DJ. et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001;27(1):89–93. doi: 10.1038/83817. [DOI] [PubMed] [Google Scholar]
- Vasireddy V, Wong P, Ayyagari R. Genetics and molecular pathology of Stargardt-like macular degeneration. Prog Retin Eye Res. 2010;29(3):191–207. doi: 10.1016/j.preteyeres.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Chen Y, Lillo C, Chien J, Yu Z, Michaelides M, Klein M, Howes KA, Li Y, Kaminoh Y. et al. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J Clin Invest. 2008;118(8):2908–2916. doi: 10.1172/JCI35891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonelli F, Testa F, Zernant J, Nesti A, Rossi S, Allikmets R, Rinaldi E. Genotype-phenotype correlation in Italian families with Stargardt disease. Ophthalmic Res. 2005;37(3):159–167. doi: 10.1159/000086073. [DOI] [PubMed] [Google Scholar]
- Passerini I, Sodi A, Giambene B, Mariottini A, Menchini U, Torricelli F. Novel mutations in of the ABCR gene in Italian patients with Stargardt disease. Eye (Lond) 2010;24(1):158–164. doi: 10.1038/eye.2009.35. [DOI] [PubMed] [Google Scholar]
- Paloma E, Martínez-Mir A, Vilageliu L, Gonzàlez-Duarte R, Balcells S. Spectrum of ABCA4 (ABCR) gene mutations in Spanish patients with autosomal recessive macular dystrophies. Hum Mutat. 2001;17(6):504–510. doi: 10.1002/humu.1133. [DOI] [PubMed] [Google Scholar]
- Aguirre-Lamban J, Riveiro-Alvarez R, Maia-Lopes S, Cantalapiedra D, Vallespin E, Avila-Fernandez A, Villaverde-Montero C, Trujillo-Tiebas MJ, Ramos C, Ayuso C. Molecular analysis of the ABCA4 gene for reliable detection of allelic variations in Spanish patients: identification of 21 novel variants. Br J Ophthalmol. 2009;93(5):614–621. doi: 10.1136/bjo.2008.145193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui T, Yamamoto S, Nakano K, Tsujikawa M, Morimura H, Nishida K, Ohguro N, Fujikado T, Irifune M, Kuniyoshi K. et al. ABCA4 gene mutations in Japanese patients with Stargardt disease and retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2002;43(9):2819–2824. [PubMed] [Google Scholar]
- Maia-Lopes S, Aguirre-Lamban J, Castelo-Branco M, Riveiro-Alvarez R, Ayuso C, Silva ED. ABCA4 mutations in Portuguese Stargardt patients: identification of new mutations and their phenotypic analysis. Mol Vis. 2009;15:584–591. [PMC free article] [PubMed] [Google Scholar]
- Hargitai J, Zernant J, Somfai GM, Vamos R, Farkas A, Salacz G, Allikmets R. Correlation of clinical and genetic findings in Hungarian patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2005;46(12):4402–4408. doi: 10.1167/iovs.05-0504. [DOI] [PubMed] [Google Scholar]
- Duno M, Schwartz M, Larsen PL, Rosenberg T. Phenotypic and genetic spectrum of Danish patients with ABCA4-related retinopathy. Ophthalmic Genet. 2012. [Epub ahead of print] [DOI] [PubMed]
- Jaakson K, Zernant J, Külm M, Hutchinson A, Tonisson N, Glavac D, Ravnik-Glavac M, Hawlina M, Meltzer MR, Caruso RC. et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003;22(5):395–403. doi: 10.1002/humu.10263. [DOI] [PubMed] [Google Scholar]
- Zernant J, Schubert C, Im KM, Burke T, Brown CM, Fishman GA, Tsang SH, Gouras P, Dean M, Allikmets R. Analysis of the ABCA4 gene by next-generation sequencing. Invest Ophthalmol Vis Sci. 2011;52(11):8479–8487. doi: 10.1167/iovs.11-8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audo I, Bujakowska KM, Leveillard T, Mohand-Said S, Lancelot ME, Germain A, Antonio A, Michiels C, Saraiva JP, Letexier M. et al. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J Rare Dis. 2012;7(1):8. doi: 10.1186/1750-1172-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Tosha C, Gorin MB, Nusinowitz S. Analysis of autofluorescent retinal images and measurement of atrophic lesion growth in Stargardt disease. Exp Eye Res. 2010;91(2):143–152. doi: 10.1016/j.exer.2010.03.021. [DOI] [PubMed] [Google Scholar]
- Clark MJ, Chen R, Lam HY, Karczewski KJ, Euskirchen G, Butte AJ, Snyder M. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol. 2011;29(10):908–914. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Heart Lung and Blood Association, National Human Genome Research Institute. SeattleSeq Annotation 131. University of Washington, Seattle, WA; 2011. [Google Scholar]
- González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88(4):440–449. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa F, Marini V, Rossi S, Interlandi E, Nesti A, Rinaldi M, Varano M, Garré C, Simonelli F. A novel mutation in the RDS gene in an Italian family with pattern dystrophy. Br J Ophthalmol. 2005;89(8):1066–1068. doi: 10.1136/bjo.2004.064188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleman TS, Cideciyan AV, Aguirre GK, Huang WC, Mullins CL, Roman AJ, Sumaroka A, Olivares MB, Tsai FF, Schwartz SB. et al. Human CRB1-associated retinal degeneration: comparison with the rd8 Crb1-mutant mouse model. Invest Ophthalmol Vis Sci. 2011;52(9):6898–6910. doi: 10.1167/iovs.11-7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier I, Sénéchal A, Dhaenens CM, Arndt C, Puech B, Defoort-Dhellemmes S, Manes G, Chazalette D, Mazoir E, Bocquet B. et al. Systematic Screening of BEST1 and PRPH2 in Juvenile and Adult Vitelliform Macular Dystrophies: A Rationale for Molecular Analysis. Ophthalmology. 2011;118(6):1130–1136. doi: 10.1016/j.ophtha.2010.10.010. Epub 2011 Jan 26. [DOI] [PubMed] [Google Scholar]
- Bonnefond A, Durand E, Sand O, De Graeve F, Gallina S, Busiah K, Lobbens S, Simon A, Bellanné-Chantelot C, Létourneau L. et al. Molecular diagnosis of neonatal diabetes mellitus using next-generation sequencing of the whole exome. PLoS One. 2010;5(10):e13630. doi: 10.1371/journal.pone.0013630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohocki MM, Daiger SP, Bowne SJ, Rodriquez JA, Northrup H, Heckenlively JR, Birch DG, Mintz-Hittner H, Ruiz RS, Lewis RA. et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat. 2001;17(1):42–51. doi: 10.1002/1098-1004(2001)17:1<42::AID-HUMU5>3.0.CO;2-K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KP, Yip SP, Cheung SC, Leung KW, Lam ST, To CH. Novel PRPF31 and PRPH2 mutations and co-occurrence of PRPF31 and RHO mutations in Chinese patients with retinitis pigmentosa. Arch Ophthalmol. 2009;127(6):784–790. doi: 10.1001/archophthalmol.2009.112. [DOI] [PubMed] [Google Scholar]
- Feist RM, White MF, Skalka H, Stone EM. Choroidal neovascularization in a patient with adult foveomacular dystrophy and a mutation in the retinal degeneration slow gene (Pro 210 Arg) Am J Ophthalmol. 1994;118(2):259–260. doi: 10.1016/s0002-9394(14)72913-7. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Kemp CM, Sheffield VC, Stone EM. Photoreceptor function in heterozygotes with insertion or deletion mutations in the RDS gene. Invest Ophthalmol Vis Sci. 1996;37(8):1662–1674. [PubMed] [Google Scholar]
- Trujillo MJ, Bueno J, Osorio A, Sanz R, Garcia-Sandoval B, Ramos C, Ayuso C. Three novel RDS-peripherin mutations (689delT, 857del17, G208D) in Spanish families affected with autosomal dominant retinal degenerations. Mutations in brief no. 147. Online. Hum Mutat. 1998;12(1):70. doi: 10.1002/(SICI)1098-1004(1998)12:1<70::AID-HUMU13>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Renner AB, Fiebig BS, Weber BH, Wissinger B, Andreasson S, Gal A, Cropp E, Kohl S, Kellner U. Phenotypic variability and long-term follow-up of patients with known and novel PRPH2/RDS gene mutations. Am J Ophthalmol. 2009;147(3):518–530. doi: 10.1016/j.ajo.2008.09.007. e511. [DOI] [PubMed] [Google Scholar]
- Boon CJ, van Schooneveld MJ, den Hollander AI, van Lith-Verhoeven JJ, Zonneveld-Vrieling MN, Theelen T, Cremers FP, Hoyng CB, Klevering BJ. Mutations in the peripherin/RDS gene are an important cause of multifocal pattern dystrophy simulating STGD1/fundus flavimaculatus. Br J Ophthalmol. 2007;91(11):1504–1511. doi: 10.1136/bjo.2007.115659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol. 2001;119(3):359–369. doi: 10.1001/archopht.119.3.359. [DOI] [PubMed] [Google Scholar]
- Downes SM, Fitzke FW, Holder GE, Payne AM, Bessant DA, Bhattacharya SS, Bird AC. Clinical features of codon 172 RDS macular dystrophy: similar phenotype in 12 families. Arch Ophthalmol. 1999;117(10):1373–1383. doi: 10.1001/archopht.117.10.1373. [DOI] [PubMed] [Google Scholar]
- den Hollander AI, Davis J, van der Velde-Visser SD, Zonneveld MN, Pierrottet CO, Koenekoop RK, Kellner U, van den Born LI, Heckenlively JR, Hoyng CB. et al. CRB1 mutation spectrum in inherited retinal dystrophies. Hum Mutat. 2004;24(5):355–369. doi: 10.1002/humu.20093. [DOI] [PubMed] [Google Scholar]
- Travis GH, Sutcliffe JG, Bok D. The retinal degeneration slow (rds) gene product is a photoreceptor disc membrane-associated glycoprotein. Neuron. 1991;6(1):61–70. doi: 10.1016/0896-6273(91)90122-G. [DOI] [PubMed] [Google Scholar]
- Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, Albrechtsen A, Andersen G, Cao H, Korneliussen T. et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet. 2010;42(11):969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Autofluorescence (A,B) and infrared (C,D) imaging of participant STGD-01. Figure S2. Fundus photos of participant STGD-02. Figure S3. Autofluorescence (AF) imaging and optical coherence tomography (OCT) images for participant STGD-03. 50° AF image of OD (A) and OS (B) shows geographic atrophy, discrete autofluorescent flecks, and peripapillary sparing in both eyes. OCT of OD (C) shows severe retinal edema. Figure S4. Autofluorescent imaging of participant STGD-04. 30° AF image of OD (A) shows “stellate” pattern dystrophy. Pattern less clear in OS (B). Figure S5. Autofluorescent imaging of participant STGD-05. 50° AF imaging of both OD (A) and OS (B) shows possible “stellate” pattern dystrophy. Figure S6. Red-free fundus images of participant STGD-06. 30° fundus images of both OD (A) and OS (B) show wide-spread geographic atrophy of the macula with mild peripapillary sparing. Figure S7. Color fundus images of participant STGD-07. 50° color fundus images from both OD (A) and OS (B) show geographic atrophy with peripapillary sparing. Figure S8. Autofluorescence and optical coherence tomography images for participant STGD-08. 50° AF shows irregular geographic atrophy without peripapillary sparing or autofluorescent flecks in both OD (A) and OS (B). OCT shows an irregularly thickened contour of the retina in both OD (C) and OS (D). Figure S9. Fundus photos of OD (A) and OS (B) for participant STGD-08. Clear “horse-shoe” pattern of atrophy is observable in both eyes.
Table S1. Whole Exome Sequencing Details
Figure S1. Autofluorescence (A,B) and infrared (C,D) imaging of participant STGD-01.
Figure S2. Fundus photos of participant STGD-02.
Figure S3. Autofluorescence (AF) imaging and optical coherence tomography (OCT) images for participant STGD-03. 50° AF image of OD (A) and OS (B) shows geographic atrophy, discrete autofluorescent flecks, and peripapillary sparing in both eyes. OCT of OD (C) shows severe retinal edema.
Figure S4. Autofluorescent imaging of participant STGD-04. 30° AF image of OD (A) shows “stellate” pattern dystrophy. Pattern less clear in OS (B).
Figure S5. Autofluorescent imaging of participant STGD-05. 50° AF imaging of both OD (A) and OS (B) shows possible “stellate” pattern dystrophy.
Figure S7. Color fundus images of participant STGD-07. 50° color fundus images from both OD (A) and OS (B) show geographic atrophy with peripapillary sparing.
Figure S6. Red-free fundus images of participant STGD-06. 30° fundus images of both OD (A) and OS (B) show wide-spread geographic atrophy of the macula with mild peripapillary sparing.
Figure S8. Autofluorescence and optical coherence tomography images for participant STGD-08. 50° AF shows irregular geographic atrophy without peripapillary sparing or autofluorescent flecks in both OD (A) and OS (B). OCT shows an irregularly thickened contour of the retina in both OD (C) and OS (D).
Figure S9. Fundus photos of OD (A) and OS (B) for participant STGD-08. Clear “horse-shoe” pattern of atrophy is observable in both eyes.