

We show that depletion of nuclear Like-Sm 8 (LSm8) dramatically increases processing body (P-body) number, provide the explanation that LSm8 acts via the alteration of the nuclear–cytoplasmic distribution of LSm2–7 proteins, and propose a model that P-bodies form via self-organization.
Abstract
Processing bodies (P-bodies) are dynamic cytoplasmic structures involved in mRNA degradation, but the mechanism that governs their formation is poorly understood. In this paper, we address a role of Like-Sm (LSm) proteins in formation of P-bodies and provide evidence that depletion of nuclear LSm8 increases the number of P-bodies, while LSm8 overexpression leads to P-body loss. We show that LSm8 knockdown causes relocalization of LSm4 and LSm6 proteins to the cytoplasm and suggest that LSm8 controls nuclear accumulation of all LSm2–7 proteins. We propose a model in which redistribution of LSm2–7 to the cytoplasm creates new binding sites for other P-body components and nucleates new, microscopically visible structures. The model is supported by prolonged residence of two P-body proteins, DDX6 and Ago2, in P-bodies after LSm8 depletion, which indicates stronger interactions between these proteins and P-bodies. Finally, an increased number of P-bodies has negligible effects on microRNA-mediated translation repression and nonsense mediated decay, further supporting the view that the function of proteins localized in P-bodies is independent of visible P-bodies.
INTRODUCTION
Cytoplasm of eukaryotic cells contains many different structures, and several of them are not encapsulated by a membrane, and their assembly and maintance are driven solely by protein–protein, RNA–protein, and, potentially, RNA–RNA interactions. Two of these nonmembrane cytoplasmic structures, stress granules and processing bodies (P-bodies; also called GW182 bodies or Dcp-bodies) are involved in mRNA metabolism (Eulalio et al., 2007a; Aizer and Shav-Tal, 2008; Franks and Lykke-Andersen, 2008). Stress granules form under stress conditions and accumulate mRNAs that are stalled in the process of translation initiation, but their exact role in the stress response is unclear (Balagopal and Parker, 2009). P-bodies contain numerous factors involved in mRNA degradation, namely decapping machinery, Like-Sm (LSm) 1–7 proteins, exonucleases and deadenylases, and several factors involved in RNA interference (RNAi) and nonsense-mediated decay (NMD) pathways (Eulalio et al., 2007a; Parker and Sheth, 2007; Balagopal and Parker, 2009).
P-bodies are highly mobile and dynamic structures constantly exchanging their content with surrounding cytoplasm (Andrei et al., 2005; Kedersha et al., 2005; Leung et al., 2006; Aizer et al., 2008). In yeast, P-body assembly is induced by ribonucleoprotein particles containing translationally repressed mRNAs, and further protein accumulation shows signs of hierarchy and interdependency; however, the exact molecular mechanism of P-body formation remains elusive (Andrei et al., 2005; Teixeira and Parker, 2007). LSm proteins are one of the factors that play a crucial role in formation and maintenance of P-body structure. In human cells, knockdown of several P-body proteins, including proteins from the LSm1–7 ring, LSm1 and LSm4, leads to reduction of P-bodies (Andrei et al., 2005; Kedersha et al., 2005). In addition, several studies showed that a prion-like self-aggregating domain of yeast LSm4p targets LSm4p to P-bodies and, together with Edc3p, is important for P-body formation (Decker et al., 2007; Mazzoni et al., 2007; Reijns et al., 2008). The role of LSm proteins in P-body formation was further strengthened by a recent finding that an LSm-binding partner, the deadenylation factor Pat1b, is able to induce P-body–like structures (Totaro et al., 2011).
In the cytoplasm, LSm1–7 proteins promote mRNA degradation, likely via interaction with the decapping machinery and Pat1b (Franks and Lykke-Andersen, 2008; Braun et al., 2010; Haas et al., 2010; Ozgur et al., 2010; Totaro et al., 2011). A similar LSm complex, in which LSm8 replaces LSm1, is localized exclusively in the nucleus, where it interacts with the U6 small nuclear RNA (snRNA) and promotes U4/U6 snRNA annealing (Achsel et al., 1999). In this work, we test the importance of nuclear LSm8 for the cellular distribution of LSm2–7 and formation of P-bodies.
RESULTS
To address the role of LSm8 in P-body formation, we transfected HeLa cells with a negative control small interfering RNA (siRNA), siRNAs against LSm1 as a positive control reducing the number of P-bodies (Andrei et al., 2005; Chu and Rana, 2006), and siRNA against LSm8. P-bodies were visualized by immunolocalization of two P-body markers, RNA helicase DDX6 (also called rck/p54), which is involved in suppression of RNA translation and mRNA degradation (Andrei et al., 2005; Weston and Sommerville, 2006; Flemr et al., 2010; Figure 1), and GW182, which is crucial for microRNA (miRNA)-mediated mRNA silencing (Eystathioy et al., 2003; Eulalio et al., 2009; Braun et al., 2011; Supplemental Figure S1A). LSm1 knockdown had only a minor effect on the number of P-bodies per cell, but the number of cells containing P-bodies was reduced twofold relative to cells treated with a negative control siRNA (Figure 1, B, D, and E). However, it should be noted that treatment with control siRNA reduced appearance of P-bodies with respect to nontreated cells (Figure S1B), which is consistent with previous observations (Serman et al., 2007). Surprisingly, knockdown of LSm8 strongly increased both P-bodies per cell and the number of cells containing P-bodies (Figure 1, C–E). In addition, we mapped influence of LSm8 knockdown on distribution of the stress granule marker eIF3b (Anderson and Kedersha, 2006). Depletion of LSm8 resulted in reduced appearance of stress granules, and eIF3b was evenly distributed throughout the cytoplasm (Figure 1G). Together these data showed that, in contrast to LSm proteins found in the LSm1–7 ring (Andrei et al., 2005; Kedersha et al., 2005), LSm8 reduction had a strong stimulatory effect on P-bodies.
FIGURE 1:

LSm8 knockdown induces formation of P-bodies. HeLa cells were treated with (A) control, (B) anti-LSm1, or (C) anti-LSm8 siRNAs, and a marker of P-bodies (DDX6) was visualized by indirect immunofluorescence (red) and the nucleus was visualized by DAPI staining (blue). Quantification of DDX6-labeled P-bodies revealed that LSm8 depletion increased the presence of cells containing microscopically detectable P-bodies (D) as well as the number of P-bodies per cell (E). The average of three experiments (each containing hundreds of cells) ± SEM is shown. * indicates significant changes with p ≤ 0.05. (F) The efficiency of siRNAs was tested by Western blotting. Snu114 protein served as a loading control. (G) For comparison of behavior of stress granules and P-bodies after LSm8 depletion, cells were treated with control or anti-LSm8 siRNA, and stress granules and P-bodies were visualized by immunolocalization of eIF3b and GW182, respectively. Scale bar: 10 μm.
How could nuclear LSm8 have such a strong influence on cytoplasmic structures? One of the potential mechanisms might involve relocalization of LSm2–7 proteins from the nucleus to the cytoplasm, where they could induce P-body assembly via the C-terminal domain of LSm4 (Decker et al., 2007; Mazzoni et al., 2007; Reijns et al., 2008). In yeast, LSm8p is important for nuclear localization of LSm7p, indicating that LSm8p might indeed influence localization of LSm2–7 proteins (Spiller et al., 2007). To test whether LSm8 is important for nuclear accumulation of LSm4 in human cells, we depleted LSm8 or LSm1 and localized endogenous LSm4 by immunodetection (Figure 2, A–D). Under normal conditions, LSm4 was found in the nucleus, where it accumulated in Cajal bodies, and in the cytoplasm, where it concentrated in P-bodies (Figure 2A; Ingelfinger et al., 2002; Stanek et al., 2003). Nuclear–cytoplasmic distribution was not affected upon LSm1 reduction, but in a manner similar to other P-body markers (DDX6 and GW182), cytoplasmic LSm4 did not concentrate in discrete foci but was dispersed throughout the cytoplasm (Figure 2B). LSm8 knockdown caused significant delocalization of LSm4 from the nucleus to the cytoplasm, where it accumulated in numerous foci that colocalized with the P-body marker GW182 (Figure 2, C and D). In addition, we screened distribution of LSm4 and LSm6 using high-throughput microscopy and assayed hundreds to thousands of cells in a single experiment. Distribution of LSm4 was analyzed in a stable cell line expressing green fluorescent protein (GFP)-LSm4 from a bacterial artificial chromosome (Figure 3A). The expression from the bacterial artificial chromosome has the advantage that endogenous promoters, exon/intron structure, and other regulatory elements are preserved, and we successfully utilized GFP-tagged proteins expressed from bacterial artificial chromosome for kinetic studies (Poser et al., 2008; Huranova et al., 2010; Novotny et al., 2011). The qualitative evaluation revealed that the nuclear:cytoplasmic ratio of LSm4 was reduced by almost 40% after LSm8 knockdown (Figure 3C). These data show that human LSm8 is required for nuclear localization of LSm4. To further support the concept that LSm8 is important for nuclear accumulation of LSm2–7 proteins, we established a stable cell line expressing yellow fluorescent protein (YFP)-LSm6 (Figure 3B) and analyzed cellular distribution of YFP-LSm6 after LSm8 depletion. LSm8 knockdown resulted in YFP-LSm6 relocalization to the cytoplasm to an extent similar to LSm4-GFP (Figure 3, B and D). Relocalization of LSm4 and LSm6 proteins was further confirmed by cell fractionation, which showed the reduction of nuclear concentration and increase of cytoplasmic signal after LSm8 depletion (Figure 3F). However, the LSm redistribution to the cytoplasm was less visible after cell fractionation compared with microscopical data. LSm proteins are small and can easily leak between the cytoplasm and nucleus during isolation of nuclear and cytoplasmic extracts, which could partially diminish relocalization after LSm8 knockdown. Thus we believe that microscopical analysis of LSm distribution was a more suitable approach in this particular case. Immunoprecipitation of LSm4-GFP and YFP-LSm6 with anti-GFP antibodies showed that both tagged proteins were incorporated into U4/U6 and U4/U6•U5 snRNPs (Figure 3E), indicating they are properly assembled into the LSm ring. Together these results suggest that LSm8 is important for nuclear localization of all LSm2–7 proteins.
FIGURE 2:

LSm4 delocalizes to the cytoplasm after LSm8 knockdown. HeLa cells were treated with control (A), anti-LSm1 (B), or anti-LSm8 (C and D) siRNAs. LSm4 was visualized by indirect immunofluorescence (red in A–C; green in D) and the nucleus was visualized by DAPI staining (blue). LSm8 knockdown resulted in LSm4 delocalization to numerous cytoplasmic dots that colocalized with a P-body marker GW182 (red in D). Scale bar: 10 μm.
FIGURE 3:

LSm4-GFP and YFP-LSm6 delocalize to the cytoplasm after LSm8 knockdown. HeLa cells stably expressing LSm4-GFP (A) or YFP-LSm6 (B) were treated with control or anti-LSm8 siRNA. Scale bar: 10 μm. (C and D) Relative distribution of LSm4-GFP (C) and YFP-LSm6 (D) after LSm8 knockdown was determined by high-throughput microscopy. The average of three experiments (each containing hundreds of cells) ± SEM is shown. * indicates significant changes with p ≤ 0.05. (E) Both LSm4-GFP and YFP-LSm6 were properly incorporated into U4/U6 and U4/U6•U5 snRNPs as indicated by immunoprecipitation using anti-GFP antibody followed by detection of copurified snRNAs by silver-staining. Note the enrichment of U4, U5, and U6 snRNAs in the immunoprecipitate fraction with respect to input. (F) Nuclear and cytoplasmic fractions were prepared from stable cell lines expressing either LSm4-GFP or YFP-LSm6 treated with control or anti-LSm8 siRNAs. LSm4-GFP and YFP-LSm6 were detected by anti-GFP antibodies, and the purity of fractions was confirmed by visualization of UBF (nuclear marker) and CypA (cytoplasmic marker).
Opposite effects of LSm1 and LSm8 knockdowns on P-bodies suggested that it was the balance between LSm8 and LSm1 that influenced P-body formation. To test this hypothesis, we analyzed effects of LSm1 or LSm8 overexpression on P-bodies. YFP-tagged LSm1 or LSm8 were transiently expressed in HeLa cells, and P-bodies were visualized by DDX6 immunostaining (Figure 4). YFP alone did not have any significant effect on P-bodies. Expression of YFP-LSm1 had a diverse effect on P-bodies in individual cells; cells with low-to-medium levels of YFP-LSm1 exhibited more P-bodies (Figure 4B) than did cells highly expressing YFP-LSm1 (Figure S2A). However, when we averaged out the whole cell population, no significant effect was observed (Figure 4E). On the contrary, overexpression of YFP-LSm8 resulted in a reduced number of P-bodies per cell and relocalization of DDX6 to stress granules (Figures 4, C and E, and S2B). Ectopic expression of YFP-LSm6 did not have any apparent effect on P-bodies (Figure 4, D and E), suggesting that the complete LSm1–7 ring is required for P-body formation.
FIGURE 4:

Overexpression of YFP-LSm8 disrupts P-bodies. HeLa cells were transfected with (A) YFP, (B) YFP-LSm1, (C) YFP-LSm8, and (D) YFP-LSm6. P-bodies were visualized by DDX6 immunodetection (red) and nuclei were visualized by DAPI staining (blue). (B) Low/middle expression of YFP-LSm1 induced formation of P-bodies. (C) In cells expressing YFP-LSm8, DDX6 often did not stain P-bodies and accumulated instead in structures resembling stress granules (arrows). (D) Expression of YFP-LSm6 did not significantly affect formation of P-bodies. Scale bar: 10 μm. (E) Quantification of P-bodies in cells transiently expressing YFP-tagged proteins. The average of three experiments (each containing hundreds of cells) ± SEM is shown.
Next we characterized newly formed P-bodies in the LSm8 knockdown cells. First, we determined residence time of three P-body components: DDX6, Ago2, and LSm4. Proteins were tagged with GFP and transiently expressed in HeLa cells. All three GPF fusion proteins properly localized to P-bodies in control cells and to the newly formed P-bodies in LSm8 knockdown cells (Figure 5). Dynamics of fusion proteins in P-bodies was monitored by fluorescence recovery after photobleaching (FRAP). Half-time for each experiment was determined after fitting the FRAP curve with a double exponential function (Figure 5 and Table 1). DDX6 and Ago2 exhibited a large immobile fraction, which indicated that a significant fraction of these proteins in P-bodies did not exchange with the surrounding cytoplasm. In addition, the immobile fraction partially increased after LSm8 depletion (Figure 5, A and B). The recovery of DDX6 and Ago2 in P-bodies was reduced after LSm8 knockdown, as indicated by longer half-times (Table 1), and this effect was more pronounced in the case of DDX6. Finally, we analyzed the dynamics of LSm4-GFP. LSm4-GFP delocalized from the nucleus to the cytoplasm in a manner similar to the endogenous LSm4 protein (Figure 5C). We did not detect any significant changes in the immobile fraction, but recovery was faster after depletion of LSm8. These data revealed that depletion of LSm8 stimulated association of DDX6 and Ago2 with P-bodies and increased exchange of LSm4 between P-bodies and the nucleoplasm.
FIGURE 5:

Mobility of P-body components is reduced after LSm8 depletion. Cells transiently expressing DDX6-GFP (A), Ago2-GFP (B), or LSm4-GFP (C) were treated with control (NC) or anti-LSm8 siRNAs. Scale bar: 10 μm. Fluorescence recovery was measured in P-bodies after GFP photobleaching in control and LSm8 siRNA-treated cells, and recovery curves were fitted to a double exponential function. Examples of recovery curves with fitted function are shown next to corresponding micrographs. Two of the tested P-body proteins, DDX6-GFP and Ago2-GFP, exhibited slower recovery after LSm8 knockdown (see Table 1 for half-times).
TABLE 1:
Residence time of DDX6-GFP and Ago2-GFP in P-bodies is prolonged after depletion of LSm8.
| Gene | siRNA | Half-time (s)a |
|---|---|---|
| DDX6-GFP | Negative control | 5.7 ± 3.1 |
| DDX6-GFP | Anti-LSm8 | 14.1 ± 4.4 |
| Ago2-GFP | Negative control | 2.2 ± 1.5 |
| Ago2-GFP | Anti-LSm8 | 5.1 ± 4.3 |
| LSm4-GFP | Negative control | 6.7 ± 3.5 |
| LSm4-GFP | Anti-LSm8 | 4.3 ± 2.6 |
aHalf-times determined by fitting FRAP curves and the average of 10 measurements with SD is shown.
To further characterize P-bodies newly formed after LSm8 depletion, we analyzed the functionality of the miRNA pathway. It has been suggested that microscopically visible P-bodies are not required for miRNA-mediated translation inhibition (Chu and Rana, 2006; Eulalio et al., 2007b) and that formation of P-bodies might be in fact seen as a consequence of miRNA activity. The increased number of P-bodies, presumably without having a direct effect on miRNA pathway, provided us with a unique opportunity to gain more insight into the relationship between LSm proteins, P-bodies, and miRNA activity. To test whether the increased number of P-bodies caused by LSm8 knockdown had any consequences for miRNA-mediated repression of translation, we used HeLa cell lines stably expressing a firefly luciferase reporter that carried four bulged binding sites for either let-7 (Pillai et al., 2005) or miR-30 (Ma et al., 2010) in its 3′ untranslated region (UTR) (Figure 6A). These reporters are naturally suppressed by corresponding endogenous miRNAs, and their expression increased sixfold (let-7 reporter) and 21-fold (miR-30 reporter) upon inhibition of the corresponding miRNA with inhibitory oligonucleotides (Figure 6D). The luciferase activity was measured 48 h after transfection with siRNAs against LSm1, LSm8, or the negative control siRNA. LSm1 knockdown did not have any significant effect on the firefly luciferase reporter activity containing either of the tested miRNA target sequences (Figure 6, B and C). LSm8 depletion partially reduced activity of the luciferase reporter carrying miR-30 with respect to the negative control (Figure 6B) but did not affect the reporter with let-7 binding sites (Figure 6C). Furthermore, there was no statistically significant difference in miR-30 reporter activity between LSm1 and LSm8 knockdowns that have opposite effects on P-bodies. At present, it is unclear why miR-30 and let-7 reporter constructs exhibit slightly different responses to LSm8 knockdown. Perhaps the effect on miR-30 reporter is reflecting more efficient posttranscriptional targeting of this particular reporter by endogenous miR-30. Importantly, the effects of LSm8 and LSm1 knockdowns on P-body numbers in the reporter cell lines were similar to HeLa cells used in previous assays (Figure S3). In any case, our results showed that reduction of P-bodies did not relieve miRNA-mediated repression, while the increased formation of P-bodies in LSm8 knockdown did not commonly correlate with a stronger miRNA-mediated repression.
FIGURE 6:
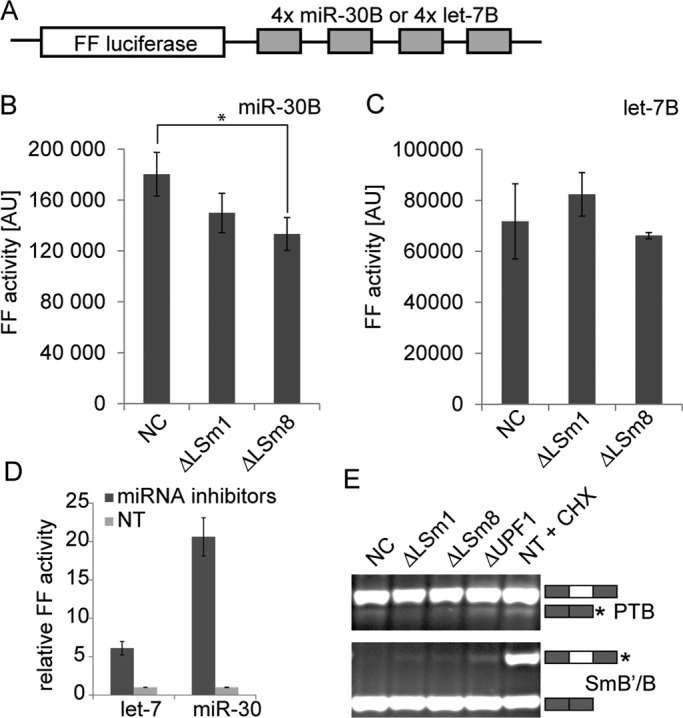
LSm8 knockdown does not affect RNAi or NMD pathway. (A) Schematic representation of mRNA used in translation assay. (B and C) Stable HeLa cell lines expressing either FF-4xmiR-30B or FF-4xlet-7B mRNAs were treated with anti-LSm1, anti-LSm8, or negative control (NC) siRNAs, and luciferase activity was measured 48 h after transfection. (B) Treatment with LSm8 siRNA reduced activity of the FF-4xmiR-30B reporter with respect to control cells (p ≤ 0.05). (C) No significant changes in luciferase activity were detected in the case of the FF-4xlet-7B reporter. (D) Stable cell lines expressing either FF-4xmiR-30B or FF-4xlet-7B mRNAs were treated with an miRNA-specific inhibitor, and luciferase activity was measured 48 h after treatment. Luciferase activity increased sixfold in the case of let-7 inhibition and 21-fold in the case of miR-30 inhibition relative to nontransfected cells (NT). (E) Depletion of LSm8 does not change activity of NMD pathway. Abundance of two NMD targets, PTB and SmB/B′, was tested by RT-PCR in control cells and cells transfected with anti-LSm1 or anti-LSm8 siRNAs. NMD-targeted splicing isoforms are marked by asterisks and visualized after cycloheximide treatment (CHX). No significant differences in NMD-targeted mRNAs were observed between cells treated with LSm8 and negative control siRNA.
Finally, we tested whether the increased number of P-bodies had any effect on the NMD pathway. Several NMD components have been shown to accumulate in P-bodies (Eulalio et al., 2007a; Parker and Sheth, 2007; Balagopal and Parker, 2009), yet the NMD pathway was not inhibited in cells devoid of visible P-bodies (Eulalio et al., 2007b; Stalder and Muhlemann, 2009). In this study, we analyzed the efficiency of NMD in cells with a high number of P-bodies formed after LSm8 knockdown. We detected two NMD targets that were created by alternative splicing of PTB and SmB/B′ in control cells and in cells depleted of LSm1 and LSm8 (Figure 6E). The PTB gene contains a 34-nucleotide exon (exon 11) that is normally included in the mRNA to encode full-length PTB protein. If exon 11 is skipped during splicing of the mRNA, the reading frame is shifted to introduce a premature termination codon that targets the transcript to the NMD pathway (Wollerton et al., 2004). Protein SmB/B′ self-regulates its expression by promoting the inclusion of a highly conserved alternative exon in its own pre-mRNA that targets the transcript for the nonsense-mediated mRNA decay (Saltzman et al., 2011). We did not detect any significant change in abundance of either of the NMD targets upon depletion of LSm8, which indicated that the increased number of P-bodies apparently did not influence the activity of the NMD pathway.
DISCUSSION
Our understanding of formation and maintenance of non–membrane-bound organelles is still far from complete. These structures are often very dynamic; they move rapidly within the cell and constantly exchange their components with the surrounding environment. In addition, factors that determine their size and number are largely unknown. In this study, we concentrated on cytoplasmic P-bodies and addressed the role of LSm proteins in P-body formation. Our data showed that depletion of LSm1 resulted in a reduced number of P-bodies. This is consistent with previous findings in human cells, which showed that removal of either LSm1 or LSm4 protein from the LSm1–7 ring reduced the P-body number (Andrei et al., 2005; Kedersha et al., 2005). Collectively these data illustrate the importance of the LSm1–7 ring for P-body formation and indicate that destabilization of the LSm1–7 ring has a negative effect on P-body assembly. At the same time, we showed that ectopic expression of only one component of the LSm1–7 ring at a time does not increase the number of P-bodies (Figure 4), suggesting that the whole ring is required for productive P-body assembly.
In contrast with Lsm1, LSm8 depletion has the opposite effect on P-bodies and significantly stimulates their formation. The underlying mechanism likely involves redistribution of LSm2–7 proteins to the cytoplasm, where they create, together with cytoplasmic LSm1, a nucleation core for other P-body components and induce aggregation of P-body factors (Figure 7). In this respect, the C-terminal domain of LSm4 was suggested to promote P-body assembly (Kedersha et al., 2005; Decker et al., 2007; Franks and Lykke-Andersen, 2008; Reijns et al., 2008). We speculate that P-body formation is driven by the self-organization principle that has been shown to be the main force in the assembly of nuclear nonmembrane structures (Kaiser et al., 2008). We propose that the increased number of the LSm1–7 rings that accumulate in the cytoplasm creates additional binding sites that capture other P-body components and nucleates formation of microscopically visible structures. The reduced mobility of Ago2 and DDX6 in P-bodies after LSm8 knockdown supports this hypothesis and suggests that these P-body proteins encounter an increased number of interaction sites within P-bodies that restrict their movement. The stronger effect on DDX6 mobility is consistent with the known interaction of DDX6 with LSm1–7 proteins via Pat1b (Ozgur et al.; Totaro et al., 2011), while no interaction between Ago2 and LSm1–7 proteins, either direct or indirect, has been identified to date. Conversely, the LSm4 dynamics was partially increased upon LSm8 depletion, which demonstrates that the reduce mobility of P-body components is not a general phenomenon of LSm8 knockdown and that binding sites for LSm proteins inside P-bodies become saturated upon LSm8 knockdown followed by redistribution of LSm2–7 proteins to the cytoplasm.
FIGURE 7:

Model representing the role of nuclear LSm8 in the localization of LSm2–7 and P body formation.
We show that LSm8 is essential for nuclear localization of LSm4 and LSm6, and thus most likely all of the LSm2–7 proteins in human cells. LSm proteins are smaller than the nuclear pore molecular weight cutoff and can apparently freely diffuse throughout the cell. This prediction is supported by the localization of overexpressed YFP-LSm1 and YFP-LSm8. Both proteins were distributed throughout the whole cell (Figures 4 and S2), which implies that their steady-state distribution depends on a number of interaction sites in the cytoplasm and the nucleus and not on a targeted transport to a particular cellular compartment. This further implies that LSm8 by itself does not contain a strong nuclear localization signal, which is consistent with the previous finding that the formation of the complete LSm2–8 ring and its interaction with the U6 snRNA is important for LSm8 nuclear localization (Spiller et al., 2007). Depletion of LSm1 does not cause redistribution of LSm4 to the nucleus, which indicates that LSm1 is not essential for cytoplasm localization of LSm proteins. Taken together, these data imply that the interaction of the complete LSm2–8 ring with the U6 snRNA is the critical determinant of LSm2–7 nuclear localization.
Consistent with results published by others, our data show that manipulation of P-body number has negligible effects on miRNA-mediated repression and the efficiency of the NMD pathway. The results are thus fully consistent with the generally accepted view that amplification or reduction of visible P-bodies does not significantly alter cellular functions. In this respect, P-bodies are markedly reminiscent of nuclear Cajal bodies, in which essential spliceosomal snRNPs are assembled, but Cajal bodies are not essential for cell proliferation in a cell culture (reviewed in Stanek and Neugebauer, 2006). However, we showed that the assembly of snRNP complexes in Cajal bodies is faster by an order of magnitude than in the nucleoplasm (Klingauf et al., 2006; Novotny et al., 2011), indicating that organelles like P-bodies or Cajal bodies might enhance the assembly kinetics of various multicomponent complexes (Matera et al., 2009). In addition, even though disruption of P-bodies does not result in any apparent defect in mRNA metabolism under normal laboratory conditions, P-bodies might become indispensable under stress conditions, as was recently suggested in the case of yeast cells (Lavut and Raveh, 2012). Similarly, Cajal bodies are essential in rapidly developing zebrafish embryos (Strzelecka et al., 2010), and their depletion reduces mouse fertility and viability (Walker et al., 2009). It would be interesting to test whether P-bodies are essential in mammals, and if so, under which conditions. The fact that the P-body component LSm1 was found to be elevated in several types of cancer and was shown to be required for cell transformation may indicate that they are (Watson et al., 2008).
MATERIALS AND METHODS
Cells and antibodies
HeLa cells and stable cell lines derived from HeLa cells were cultured in DMEM supplemented with 10% fetal calf serum, penicillin, and streptomycin (Invitrogen, Carlsbad, CA). HeLa cells stably expressing LSm4-GFP from bacterial artificial chromosome were prepared as described previously (Poser et al., 2008) and kindly provided by Ina Poser and Tony Hyman (MPI-CBG, Dresden, Germany). HeLa-FF-4xlet-7B and HeLa-FF-4xmiR-30B cells stably expressing luciferase reporters were produced by stable integration of pGL4 plasmid derivatives carrying 3′UTRs with four bulged let-7 (Pillai et al., 2005) or miR-30 (Ma et al., 2010) binding sites. Stable cell lines for experiments were selected based on high sensitivity to miRCURY LNA miRNA family inhibitors (Exiqon, Woburn, MA). The following antibodies were used for immunofluorescence microscopy: rabbit antibodies against LSm4 (Achsel et al., 1999), kindly provided by R. Lührmann (MPI, Göttingen, Germany); rabbit antibodies against DDX6 (Bethyl Laboratories, Montgomery, TX); human anti-GW182 (Eystathioy et al., 2003), kindly provided by M.J. Fritzler (University of Calgary, Calgary, Canada); mouse antibody against eIF3b (#sc-28857; Santa Cruz Biotechnology, Santa Cruz, CA); goat antibodies against TIA1 (#sc-1751; Santa Cruz Biotechnology); and secondary anti-mouse or anti-rabbit antibodies conjugated with DyLight549 and DyLight 488 (Jackson ImmunoResearch Laboratories, West Grove, PA) and Alexa Fluor 594 (Invitrogen). The following antibodies were used for Western blotting: rabbit antibodies against hSnu114 (Fabrizio et al., 1997), kindly provided by R. Lührmann; mouse anti-LSm8 antibody (#sc-81315; Santa Cruz Biotechnology); rabbit anti-UBF (#sc-9131; Santa Cruz Biotechnology); rabbit anti-cyclophiline A (CypA; #07-313; Upstate/Millipore, Billerica, MA); rabbit anti-LSm1 antibodies (Sigma-Aldrich, St. Louis, MO); and secondary anti-mouse, anti-goat, and anti-rabbit antibodies 800 (LI-COR Biosciences, Lincoln, NE). Goat anti-GFP antibodies were raised against bacterially expressed full-length EGFP and obtained from David Drechsel (MPI-CBG, Dresden, Germany).
siRNA
Preannealed siRNA duplexes were obtained from Ambion (Applied Biosystems, Bedford, MA). The sequence of siRNA against LSm8 siRNA was kindly provided by Konstantin Licht (Institute of Biochemistry, Justus-Liebig-University of Giessen, Germany): 5′-GGAUGAAAGCCAUGAACGAtt. Predesigned LSm1 siRNA was purchased from Ambion: 5′-GAAGGACACUUAUAGGCUUtt. The “negative control 1” siRNA from Ambion was used as a negative control. siRNAs were transfected using Oligofectamine (Invitrogen) according to the manufacture's protocol, and cells were analyzed 48 h after transfection. Hsa-let-7 F-I and hsa-miR-30 FI miRCURY LNA miRNA family inhibitors were obtained from Exiqon. HeLa-FF-4xlet-7B and HeLa-FF-4xmiR-30B cells plated in 24-well plates were transfected by 10 nM of LNA miRNA family inhibitor using Lipofectamine (Invitrogen) and assayed after 48 h.
Plasmids and DNA
LSm1 was cloned from HeLa cell total RNA by reverse transcriptase PCR (RT-PCR) using primers (LSm1-Forward: 5′-CCGGAATTCTGATGAACTATATGCCTGGC; LSm1-Reverse: 5′-CGCGGATCCCGGTACTCATCAAGAGTATC) and inserted into EYFP-C3 vector (Clontech Laboratories, Mountain View, CA) using EcoRI/BamHI restriction enzymes. YFP-LSm8 and YFP-LSm6 were described previously (Stanek et al., 2003; Stanek and Neugebauer, 2004). EGFP-FLAG-HA-DDX6 construct was kindly provided by Matyas Flemr (Institute of Molecular Genetics AS CR, Prague, Czech Republic). The following primers were used to clone mouse DDX6 coding sequence: DDX6-Forward: 5′-GCGTCGACAGCACGGCCAGAACAGAGAACCCTGT; and DDX6-Reverse: 5′AGCGGCCGCTTACGGTTTCTCGTCTTCTGCAGGCT. DDX6 cDNA was subsequently cloned into SalI and NotI sites of vector carrying the EGFP sequence, FLAG and hemagglutinin (HA) tags upstream of the cloning site (vector map available upon request). Firefly reporter construction: four bulged (B) binding sites for either hsa-let-7a or hsa-miR-30 miRNA were cloned into the 3′-UTR of firefly luciferase reporter driven by 3-phosphoglycerate kinase promoter (PGK) promoter (pPGK-FF) to obtain pPGK-FF-4xlet-7B and pPGK-FF-4xmiR-30B plasmids. Plasmid DNAs were introduced into cells using FuGene HD Transfection Reagent (Roche Diagnostics, Basel, Switzerland) according to the manufacturer's protocol 24 h before the experiment.
RT-PCR
RNA was isolated 48 h after siRNA transfection using Trizol Reagent (Invitrogen) accordingly to the manufacturer's protocol. In parallel, cycloheximide was added to the medium at the final concentration of 75 μg/ml, and RNA was isolated 16 h after the treatment. Reverse transcription was performed by SuperScriptIII (Invitrogen), and cDNA was amplified by Taq polymerase (MBI Fermentas, Biogen, Prague, Czech Republic) using primers: PTB-Forward: 5′-GCG GCA GGT CGG ATC GCC AT-3′; PTB-Reverse: 5′-TTG CCG TCC GCC ATC TGC ACT-3′; SmB-Forward: 5′-CAA GGC TTT TGA CAA GCA CA-3′; SmB-Reverse: 5′-GGG AGG AGG TCC CTC TAC TG-3′. DNA was visualized on 3% agarose gel using intercalate reagent GelStar (Cambrex Bio Science, Rockland, ME).
Immunofluorescence and image acquisition
Cells were fixed and labeled as described previously (Novotny et al., 2011). Images were acquired using the DeltaVision microscopic system (Applied Precision, Issaquah, WA) coupled to the Olympus IX70 microscope equipped with an oil-immersion objective (60×/1.4 numerical aperture [NA]) and restored using a measured point-spread function (SoftWorx; Applied Precision) as described previously (Novotny et al., 2011).
High-throughput microscopy
Samples were scanned using automated acquisition driven by Acquisition Scan^R program using Scan^R system (Olympus, Hamburg, Germany) equipped with oil-immersion objective 40×/1.3 NA. One hundred forty-four images were taken per each sample containing hundreds of cells. Each image was reconstructed from stacks of 10 optical sections with a 300-nm Z-step and automatically restored using a measured point-spread function implemented in Analysis Scan^R software (Olympus). Cellular compartments were automatically identified based on fluorescence intensity combined with compartment edge detection. Cell nuclei were visualized using 4′,6-diamidino-2-phenylindole (DAPI) staining; DDX6 was stained to visualize P-bodies. Assignment of individual P-bodies to a particular cell was based on a relative distance from nuclei. Total intensities, areas, and counts for each cellular object were obtained using the Analysis Scan^R software. Mean and SEM of three biological experiments were calculated. Statistical significance was analyzed by Student's t test.
Cell fractionation
The fractionation procedure was adopted from Wodrich et al. (2000). Cells were harvested 48 h after transfection, centrifuged at 200 × g for 5 min at 4°C, resuspended in hypotonic buffer A (10 mM HEPES-KOH, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol [DTT]), and incubated on ice for 10 min. Cells were then disrupted by vortexing for 20 s and centrifuged at 400 × g for 2 min at 4°C, and the supernatant was used as the cytosolic extract. The pellet was washed in cold PBS, spun at 400 × g for 2 min at 4°C, and resuspended in buffer B (20 mM HEPES, pH 7.9, 0.5 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT). After a 30-min incubation on ice, nuclei were centrifuged at 20,000 × g for 5 min at 4°C, and the supernatant was taken as the nuclear extract. UBF was used as a marker of nuclear fraction and CypA as a cytoplasmic marker, as previously described (Horejsi et al., 2012).
snRNP immunoprecipitation and Western blot analysis
Cells were grown on a 15-cm Petri dish for 28 h. Immunoprecipitation was performed as described previously (Huranova et al., 2009) using goat α-GFP antibodies. RNA was extracted using phenol/chloroform, resolved on a 7 M urea-denaturing polyacrylamide gel, and silver-stained. For protein analysis, cells were mixed with SDS-sample buffer to obtain a whole-cell lysate. Proteins were resolved on SDS–PAGE gel, blotted onto the PROTRAN membrane (Whatman, Maidstone, UK), and detected with primary and secondary antibodies conjugated with fluorophore dye and detected by Odyssey reader (LI-COR Biosciences).
Bioluminescence assay
Cells grown in 24-well plates were collected in 150 μl Passive Lysis Buffer (Promega, Madison, WI). The activity of FL was measured in 5-μl aliquots using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. The data were corrected to protein concentration (Bio-Rad Protein Assay, Bio-Rad Czech Republic, Prague, Czech Republic) and normalized to control transfection. Results from two independent experiments done in duplicate are shown (mean ± SEM).
FRAP
FRAP experiments were performed using the DeltaVision microscopic system, as described previously (Novotny et al., 2011). Because of the high mobility of P-bodies, tracking by means of thresholding was used to identify and follow the motion of P-bodies and determine the fluorescence intensity within them. Resulting recovery curves were computed as averages of 10 measurements for each studied case. Data were fitted by sum of two exponentials: I = I0 +a(1 − e−bt) +c(1 − e−dt), where I0 is the initial intensity after photobleaching and a, b, c, and d are fitted parameters. Intensity in half of the maximum of the recovery curve (I½) was calculated for each measurement. The half-time t½ corresponding to I½ was determined for individual curves by optimization in Matlab, using NLINFIT–nonlinear regression (Matlab, MathWorks, Natick, MA). Mean value and SD were calculated for each case.
Supplementary Material
Acknowledgments
We thank Matyas Flemr, Ina Poser, Tony Hyman, and Reinhard Lührmann for reagents; Konstantin Licht for siRNA sequences; Radek Malik for technical assistance with the luciferase assay; and Katerina Chalupnikova and members of Stanek's lab for helpful discussions. This work was supported by the Academy of Sciences of the Czech Republic (KAN200520801, AV0Z50520514, and AV0Z50390703), the Czech Science Foundation (204/07/0133 to M.B., P305/10/2215 to P.S., and P302/11/1910 to D.S.), and the grant agency of Charles University (153310 to I.N. and 18110 to K.P.). L.S.V., D.S., and P.S. are members of the Centrum of Excellence supported by the Czech Science Foundations (P305/12/G034).
Abbreviations used:
- B
bulged
- CypA
cyclophiline A
- DAPI
4′,6-diamidino-2-phenylindole
- DTT
dithiothreitol
- FRAP
fluorescence recovery after photobleaching
- GFP
green fluorescent protein
- HA
hemagglutinin
- LSm
Like-Sm
- miRNA
microRNA
- NA
numerical aperture
- NMD
nonsense-mediated decay
- P-body
processing body
- PGK
3-phosphoglycerate kinase promoter
- RNAi
RNA interference
- RT-PCR
reverse transcriptase PCR
- siRNA
small interfering RNA
- snRNA
small nuclear RNA
- UTR
untranslated region
- YFP
yellow fluorescent protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-02-0085) on August 8, 2012.
REFERENCES
- Achsel T, Brahms H, Kastner B, Bachi A, Wilm M, Luhrmann R. A doughnut-shaped heteromer of human Sm-like proteins binds to the 3′- end of U6 snRNA, thereby facilitating U4/U6 duplex formation in vitro. EMBO J. 1999;18:5789–5802. doi: 10.1093/emboj/18.20.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizer A, Brody Y, Ler LW, Sonenberg N, Singer RH, Shav-Tal Y. The dynamics of mammalian P body transport, assembly, and disassembly in vivo. Mol Biol Cell. 2008;19:4154–4166. doi: 10.1091/mbc.E08-05-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizer A, Shav-Tal Y. Intracellular trafficking and dynamics of P bodies. Prion. 2008;2:131–134. doi: 10.4161/pri.2.4.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. RNA granules. J Cell Biol. 2006;172:803–808. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei MA, Ingelfinger D, Heintzmann R, Achsel T, Rivera-Pomar R, Luhrmann R. A role for eIF4E and eIF4E-transporter in targeting mRNPs to mammalian processing bodies. RNA. 2005;11:717–727. doi: 10.1261/rna.2340405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balagopal V, Parker R. Polysomes, P bodies and stress granules: states and fates of eukaryotic mRNAs. Curr Opin Cell Biol. 2009;21:403–408. doi: 10.1016/j.ceb.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun JE, Huntzinger E, Fauser M, Izaurralde E. GW182 proteins directly recruit cytoplasmic deadenylase complexes to miRNA targets. Mol Cell. 2011;44:120–133. doi: 10.1016/j.molcel.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Braun JE, Tritschler F, Haas G, Igreja C, Truffault V, Weichenrieder O, Izaurralde E. The C-terminal α–α superhelix of Pat is required for mRNA decapping in metazoa. EMBO J. 2010;29:2368–2380. doi: 10.1038/emboj.2010.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CY, Rana TM. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol. 2006;4:e210. doi: 10.1371/journal.pbio.0040210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker CJ, Teixeira D, Parker R. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J Cell Biol. 2007;179:437–449. doi: 10.1083/jcb.200704147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A, Behm-Ansmant I, Izaurralde E. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol. 2007a;8:9–22. doi: 10.1038/nrm2080. [DOI] [PubMed] [Google Scholar]
- Eulalio A, Behm-Ansmant I, Schweizer D, Izaurralde E. P-body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol Cell Biol. 2007b;27:3970–3981. doi: 10.1128/MCB.00128-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A, Tritschler F, Izaurralde E. The GW182 protein family in animal cells: new insights into domains required for miRNA-mediated gene silencing. RNA. 2009;15:1433–1442. doi: 10.1261/rna.1703809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eystathioy T, Jakymiw A, Chan EK, Seraphin B, Cougot N, Fritzler MJ. The GW182 protein colocalizes with mRNA degradation associated proteins hDcp1 and hLSm4 in cytoplasmic GW bodies. RNA. 2003;9:1171–1173. doi: 10.1261/rna.5810203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Laggerbauer B, Lauber J, Lane WS, Luhrmann R. An evolutionarily conserved U5 snRNP-specific protein is a GTP-binding factor closely related to the ribosomal translocase EF-2. EMBO J. 1997;16:4092–4106. doi: 10.1093/emboj/16.13.4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemr M, Ma J, Schultz RM, Svoboda P. P-body loss is concomitant with formation of a messenger RNA storage domain in mouse oocytes. Biol Reprod. 2010;82:1008–1017. doi: 10.1095/biolreprod.109.082057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks TM, Lykke-Andersen J. The control of mRNA decapping and P-body formation. Mol Cell. 2008;32:605–615. doi: 10.1016/j.molcel.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas G, Braun JE, Igreja C, Tritschler F, Nishihara T, Izaurralde E. HPat provides a link between deadenylation and decapping in metazoa. J Cell Biol. 2010;189:289–302. doi: 10.1083/jcb.200910141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horejsi B, et al. Nuclear γ-tubulin associates with nucleoli and interacts with tumor suppressor protein C53. J Cell Physiol. 2012;227:367–382. doi: 10.1002/jcp.22772. [DOI] [PubMed] [Google Scholar]
- Huranova M, Hnilicova J, Fleischer B, Cvackova Z, Stanek D. A mutation linked to retinitis pigmentosa in HPRP31 causes protein instability and impairs its interactions with spliceosomal snRNPs. Hum Mol Genet. 2009;18:2014–2023. doi: 10.1093/hmg/ddp125. [DOI] [PubMed] [Google Scholar]
- Huranova M, Ivani I, Benda A, Poser I, Brody Y, Hof M, Shav-Tal Y, Neugebauer KM, Stanek D. The differential interaction of snRNPs with pre-mRNA reveals splicing kinetics in living cells. J Cell Biol. 2010;191:75–86. doi: 10.1083/jcb.201004030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelfinger D, Arndt-Jovin DJ, Luhrmann R, Achsel T. The human LSm1-7 proteins colocalize with the mRNA-degrading enzymes Dcp1/2 and Xrnl in distinct cytoplasmic foci. RNA. 2002;8:1489–1501. [PMC free article] [PubMed] [Google Scholar]
- Kaiser TE, Intine RV, Dundr M. De novo formation of a subnuclear body. Science. 2008;322:1713–1717. doi: 10.1126/science.1165216. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol. 2005;169:871–884. doi: 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingauf M, Stanek D, Neugebauer KM. Enhancement of U4/U6 small nuclear ribonucleoprotein particle association in Cajal bodies predicted by mathematical modeling. Mol Biol Cell. 2006;17:4972–4981. doi: 10.1091/mbc.E06-06-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavut A, Raveh D. Sequestration of highly expressed mRNAs in cytoplasmic granules, P-bodies, and stress granules enhances cell viability. PLoS Genet. 2012;8:e1002527. doi: 10.1371/journal.pgen.1002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Calabrese JM, Sharp PA. Quantitative analysis of Argonaute protein reveals microRNA-dependent localization to stress granules. Proc Natl Acad Sci USA. 2006;103:18125–18130. doi: 10.1073/pnas.0608845103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Flemr M, Stein P, Berninger P, Malik R, Zavolan M, Svoboda P, Schultz RM. MicroRNA activity is suppressed in mouse oocytes. Curr Biol. 2010;20:265–270. doi: 10.1016/j.cub.2009.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera AG, Izaguire-Sierra M, Praveen K, Rajendra TK. Nuclear bodies: random aggregates of sticky proteins or crucibles of macromolecular assembly? Dev Cell. 2009;17:639–647. doi: 10.1016/j.devcel.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni C, D'Addario I, Falcone C. The C-terminus of the yeast Lsm4p is required for the association to P-bodies. FEBS Lett. 2007;581:4836–4840. doi: 10.1016/j.febslet.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Novotny I, Blazikova M, Stanek D, Herman P, Malinsky J. In vivo kinetics of U4/U6·U5 tri-snRNP formation in Cajal bodies. Mol Biol Cell. 2011;22:513–523. doi: 10.1091/mbc.E10-07-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgur S, Chekulaeva M, Stoecklin G. Human Pat1b connects deadenylation with mRNA decapping and controls the assembly of processing bodies. Mol Cell Biol. 2010;30:4308–4323. doi: 10.1128/MCB.00429-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol Cell. 2007;25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W. Inhibition of translational initiation by Let-7 microRNA in human cells. Science. 2005;309:1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- Poser I, et al. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat Methods. 2008;5:409–415. doi: 10.1038/nmeth.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MA, Alexander RD, Spiller MP, Beggs JD. A role for Q/N-rich aggregation-prone regions in P-body localization. J Cell Sci. 2008;121:2463–2472. doi: 10.1242/jcs.024976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzman AL, Pan Q, Blencowe BJ. Regulation of alternative splicing by the core spliceosomal machinery. Gene Dev. 2011;25:373–384. doi: 10.1101/gad.2004811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serman A, Le Roy F, Aigueperse C, Kress M, Dautry F, Weil D. GW body disassembly triggered by siRNAs independently of their silencing activity. Nucleic Acids Res. 2007;35:4715–4727. doi: 10.1093/nar/gkm491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiller MP, Reijns MA, Beggs JD. Requirements for nuclear localization of the Lsm2-8p complex and competition between nuclear and cytoplasmic Lsm complexes. J Cell Sci. 2007;120:4310–4320. doi: 10.1242/jcs.019943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder L, Muhlemann O. Processing bodies are not required for mammalian nonsense-mediated mRNA decay. RNA. 2009;15:1265–1273. doi: 10.1261/rna.1672509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanek D, Neugebauer KM. Detection of snRNP assembly intermediates in Cajal bodies by fluorescence resonance energy transfer. J Cell Biol. 2004;166:1015–1025. doi: 10.1083/jcb.200405160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanek D, Neugebauer KM. The Cajal body: a meeting place for spliceosomal snRNPs in the nuclear maze. Chromosoma. 2006;115:343–354. doi: 10.1007/s00412-006-0056-6. [DOI] [PubMed] [Google Scholar]
- Stanek D, Rader SD, Klingauf M, Neugebauer KM. Targeting of U4/U6 small nuclear RNP assembly factor SART3/p110 to Cajal bodies. J Cell Biol. 2003;160:505–516. doi: 10.1083/jcb.200210087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strzelecka M, Trowitzsch S, Weber G, Luhrmann R, Oates AC, Neugebauer KM. Coilin-dependent snRNP assembly is essential for zebrafish embryogenesis. Nat Struct Mol Biol. 2010;17:403–409. doi: 10.1038/nsmb.1783. [DOI] [PubMed] [Google Scholar]
- Teixeira D, Parker R. Analysis of P-body assembly in Saccharomyces cerevisiae. Mol Biol Cell. 2007;18:2274–2287. doi: 10.1091/mbc.E07-03-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totaro A, Renzi F, La Fata G, Mattioli C, Raabe M, Urlaub H, Achsel T. The human Pat1b protein: a novel mRNA deadenylation factor identified by a new immunoprecipitation technique. Nucleic Acids Res. 2011;39:635–647. doi: 10.1093/nar/gkq797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MP, Tian L, Matera AG. Reduced viability, fertility and fecundity in mice lacking the Cajal body marker protein, coilin. PLoS One. 2009;4:e6171. doi: 10.1371/journal.pone.0006171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PM, Miller SW, Fraig M, Cole DJ, Watson DK, Boylan AM. CaSm (LSm-1) overexpression in lung cancer and mesothelioma is required for transformed phenotypes. Am J Respir Cell Mol Biol. 2008;38:671–678. doi: 10.1165/rcmb.2007-0205OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston A, Sommerville J. Xp54 and related (DDX6-like) RNA helicases: roles in messenger RNP assembly, translation regulation and RNA degradation. Nucleic Acids Res. 2006;34:3082–3094. doi: 10.1093/nar/gkl409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodrich H, Schambach A, Krausslich HG. Multiple copies of the Mason-Pfizer monkey virus constitutive RNA transport element lead to enhanced HIV-1 Gag expression in a context-dependent manner. Nucleic Acids Res. 2000;28:901–910. doi: 10.1093/nar/28.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollerton MC, Gooding C, Wagner EJ, Garcia-Blanco MA, Smith CW. Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol Cell. 2004;13:91–100. doi: 10.1016/s1097-2765(03)00502-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.