

Cells respond to stress by inhibition of protein synthesis and subsequent assembly of stress granules (SGs). Cold shock is identified as a novel trigger of SG assembly in yeast and mammals. Cells actively suppress protein synthesis by parallel pathways to induce SG formation and ensure cellular survival at low temperatures.
Abstract
Cells respond to different types of stress by inhibition of protein synthesis and subsequent assembly of stress granules (SGs), cytoplasmic aggregates that contain stalled translation preinitiation complexes. Global translation is regulated through the translation initiation factor eukaryotic initiation factor 2α (eIF2α) and the mTOR pathway. Here we identify cold shock as a novel trigger of SG assembly in yeast and mammals. Whereas cold shock–induced SGs take hours to form, they dissolve within minutes when cells are returned to optimal growth temperatures. Cold shock causes eIF2α phosphorylation through the kinase PERK in mammalian cells, yet this pathway is not alone responsible for translation arrest and SG formation. In addition, cold shock leads to reduced mitochondrial function, energy depletion, concomitant activation of AMP-activated protein kinase (AMPK), and inhibition of mTOR signaling. Compound C, a pharmacological inhibitor of AMPK, prevents the formation of SGs and strongly reduces cellular survival in a translation-dependent manner. Our results demonstrate that cells actively suppress protein synthesis by parallel pathways, which induce SG formation and ensure cellular survival during hypothermia.
INTRODUCTION
The rate of protein synthesis in cells is tightly regulated. In response to various forms of stress, cells reduce global translation, by which they prevent further protein damage, reallocate their resources to repair processes, and ensure cellular survival. Most types of stress cause translation inhibition through phosphorylation of the α subunit of the translation initiation factor eukaryotic initiation factor 2 (eIF2), which delivers initiator tRNAiMet to the small 40S ribosomal subunit (Holcik and Sonenberg, 2005). In mammals, eIF2α phosphorylation is mediated via the four kinases HRI, PERK, GCN2, and PKR, whereas yeast contains only GCN2. Phosphorylated eIF2 no longer dissociates from its GDP exchange factor eIF2B, which prevents recharging of the eIF2-GTP-tRNAiMet ternary complex and inhibits translation initiation. As a consequence of polysome disassembly, stalled translation preinitiation complexes accumulate and aggregate into cytosolic stress granules (SGs; Kedersha et al., 2002).
In addition to eIF2α phosphorylation, translation is controlled by the mTOR pathway (Wullschleger et al., 2006). When insulin signaling, nutrients, or energy availability is low, inhibition of mTOR attenuates translation through the eIF4E-binding proteins (4EBPs) and S6-kinase 1 (S6K1; Holz et al., 2005; Richter and Sonenberg, 2005).
Inhibition of protein synthesis is intimately linked to the assembly of SGs, which contain poly(A)-mRNA, small, but not large, ribosomal subunits, and translation initiation factors eIF3, eIF4E, and eIF4G and the cytoplasmic poly(A)-binding protein (PABP; Kedersha et al., 2002). RNA-binding SG proteins such as TIA1, TIAR, and G3BP participate in the aggregation process driving SG assembly (Kedersha et al., 1999, 2002; Tourriere et al., 2003; Gilks et al., 2004). It is important to note that SGs form as a consequence of translation arrest, but they are not required for the arrest of translation (Anderson and Kedersha, 2008). Current thinking is that SGs help keep preinitiation complexes assembled and by sequestering specific proteins play a role in cellular survival and signal transduction (Kim et al., 2005; Arimoto et al., 2008; Takahara and Maeda, 2012). In Saccharomyces cerevisiae, SG-like aggregates, also termed EGP bodies, were described under different conditions including glucose deprivation and heat shock (Hoyle et al., 2007; Buchan et al., 2008; Grousl et al., 2009). Their composition is similar to that of mammalian SGs, yet yeast SGs lack eIF3 and small ribosomal subunits under some conditions.
Although mechanisms controlling translation have been studied extensively for endoplasmic reticulum (ER) stress, oxidative stress, heat shock, amino acid starvation, and exposure to double-stranded RNA (Holcik and Sonenberg, 2005), very little is known about mechanisms controlling translation during temperature downshift. Mammalian cells exposed to hypothermia show a progressive reduction of global translation rates (Roobol et al., 2009). In yeast, factors supporting protein synthesis, including ribosomal proteins, are down-regulated at temperatures <10°C (Homma et al., 2003; Murata et al., 2006), yet the mechanism responsible for cold shock–induced translation suppression remains largely unexplored. The adaptive response to low temperatures is of preeminent importance in organisms that cannot control body temperature. Even in thermoregulating warm-blooded animals, temperatures in the skin of extremities can drop to <10ºC (Heus et al., 1995), and the body temperature in small hibernating mammals reaches 2ºC during torpor (Carey et al., 2003).
In this study, we identify cold shock as a condition under which both mammalian cells and S. cerevisiae form SGs. Through our analysis of eIF2α phosphorylation, mTOR signaling, and AMP-activated protein kinase (AMPK) activation, we provide evidence that translation suppression and SG formation promote the survival of mammalian cells at low temperatures.
RESULTS
Cold shock induces SGs, translation arrest, and polysome disassembly in mammalian cells
To test whether mammalian cells react to hypothermia with a translation arrest response, we first analyzed African green monkey COS7 kidney cells because of their excellent imaging properties. On shift to a range of temperatures <37ºC, immunofluorescence (IF) staining of the translation initiation factor eIF3B revealed extensive formation of cytoplasmic granules after 10 h at 10ºC, whereas granules did not form at 30 or 20°C (Figure 1A). COS7 cells did not form eIF3B granules at 4ºC either (Figure 1A and Supplemental Figure S1A) and in fact detached from the plate after 10 h at 4ºC, indicative of cell death. In contrast, COS7 cells remained adherent for up to 24 h at 10ºC. This suggested that cells mount a stress response specific for adaptation to temperatures around 10ºC. The eIF3B granules observed at 10ºC were distinctly smaller and more numerous than SGs in cells subjected to arsenite-induced oxidative stress (Figure 1B). Poly(A)-mRNA accumulated in the same granules, as determined by colocalization with eIF3B (Figure 1C). Cold shock–induced granules also contain eIF4G (Figure 1D), eIF2α (Figure 1E), and the RNA-binding proteins G3BP (Figure 1F), PABP (Figure 1G), HuR, and TIA1 (unpublished data), confirming that these are bona fide SGs. Cold shock–induced SGs were also detected in human Du145, HeLa, and Huh7 cells, as well as in mouse embryonic fibroblasts (MEFs; Supplemental Figure S1B), indicating that our observation was not restricted to a particular cell line.
FIGURE 1:

Cold shock induces SGs and represses translation in mammalian cells. (A) COS7 cells were grown at 37°C or incubated at 30, 20, or 10°C for 10 h or 4°C for 8 h. Subcellular localization of eIF3B was determined by IF staining, followed by wide-field fluorescence microscopy. (B) As positive control for SG formation, cells were treated with 500 μM Na arsenite for 1 h. (C) COS7 cells exposed to cold shock were stained by fluorescence in situ hybridization for poly(A) mRNA and counterstained for eIF3B by IF. (D–F) COS7 cells exposed to cold shock at 10°C for 10 h were stained for the SG markers eIF4G (D), eIF2α (E), and G3BP (F) in combination with eIF3B. (G) COS7 cells were transiently transfected with a vector encoding GFP-PABP, exposed to cold shock, and fixed. Localization of GFP-PABP was analyzed in relation to endogenous eIF3B. (H) To quantify SG formation over time, the relative number of cells containing SGs was determined. Average values ± SE from four experiments are shown. (I) COS7 cells were exposed to cold shock at 10°C or kept at 37°C for different time periods, as indicated. Newly synthesized proteins were labeled with [35S]methionine/cysteine. Cell lysates were separated on 5–20% polyacrylamide gels and stained with colloidal Coomassie (bottom) or visualized by autoradiography (top). (J) COS7 cells were labeled as in I, and translation rates were quantified by measuring incorporation of [35S]methionine/cysteine into precipitated protein using a scintillation counter. Values of counts per minute (cpm) normalized to the total amount of proteins are depicted in the graph. (K) Polysome profiles were recorded from COS7 cells either grown under control conditions at 37°C or subjected to cold shock at 10°C for up to 10 h. To determine the percentage of polysomal ribosomes, the area below the polysomal part of the curve was divided by the area below the subpolysomal and polysomal parts of the curve and represented as average ± SD in the bar graph.
In following the kinetics of SG formation after cold shock, we detected the first granules after 4 h at 10ºC in 26% of COS7 cells (Supplemental Figure S1C; quantification in Figure 1H). This number increased progressively and reached a maximum of 93% SG-positive cells after 10 h of cold shock. SGs then persisted for 24 h, the last time point analyzed. Video microscopy showed that SGs remain assembled during 10ºC hypothermia (Supplemental Movie S1), which is different from oscillatory SGs induced by virus infection (Ruggieri et al., 2012).
To directly assess translation rates, we labeled COS7 cells with [35S]methionine/cysteine. Incorporation into nascent proteins was reduced to <10% already after 1 h at 10ºC compared with cells grown at 37ºC, and remained at this low level during the entire cold shock period (Figure 1, I and J). Sucrose gradient density centrifugation showed a progressive loss of polysomes during cold shock (Figure 1K), confirming that protein synthesis is suppressed. The percentage of polysome-associated ribosomes, which reflects the proportion of ribosomes engaged in translation, was found to drop from 67% at 37ºC to 8% after 10 h at 10ºC (bar graph in Figure 1K). We concluded that translation inhibition is accompanied by polysome disassembly during cold shock, which leads to the progressive assembly of SGs.
Cold shock induces SGs in S. cerevisiae
Next we examined whether the translational arrest in response to cold shock is conserved in S. cerevisiae. Using the SG marker Pub1 genomically tagged with green fluorescent protein (GFP), we observed prominent granules when cells were shifted from 30 to 10ºC for 4 h but not when shifted to 15ºC (Figure 2A; full time course in Supplemental Figure S2, A–C). SGs were strongly induced after 4 h at 0ºC and persisted for at least 24 h (Figure 2B). The SG markers Pab1-mCherry (Figure 2B) and poly(A)-mRNA (Figure 2C) also localized to these granules.
FIGURE 2:

Cold shock induces SGs in S. cerevisiae. (A) A genomically tagged yeast strain expressing Pub1-GFP was grown under control conditions at 30°C or incubated at 15 or 10°C for 4 h. Subcellular localization of Pub1-GFP was analyzed by confocal microscopy. (B) Yeast was either grown under control conditions at 30°C or exposed to cold shock at 0°C for 4 and 24 h. Localization of genomically tagged Pub1-GFP (green) and Pab1-mCherry (red) was analyzed by confocal microscopy. (C) Spheroplasts were prepared from a genomically tagged yeast strain expressing Pab1-myc after it was either grown under control conditions or exposed to cold shock at 0°C for 4 h. Poly(A) RNA (green) was localized by fluorescence in situ hybridization, and Pab1-myc (red) was visualized by IF staining. (D) The formation of SGs over time was quantified using automated image analysis. Two parameters were measured: the number of SGs per cell and optical section (yellow bars) and the percentage of Pub1-GFP signal in SGs (brown bars). Average values ± SE from three experiments are shown.
Time-course experiments (Supplemental Figure S2. A and B) and automated image analysis (Figure 2D) showed that the earliest SGs were formed after 2 h of incubation at 10 or 0ºC and that their number increased steadily during the first 8 h. Between 8 and 24 h of cold shock, the granules became fewer, yet larger and more elongated as Pub1-GFP continued to accumulate in the aggregates. An example of automated cell and foci detection is provided in Supplemental Figure S3. As in mammalian cells, the slow appearance of cold shock–induced SGs in yeast is in stark contrast to other types of stress such as heat shock or glucose deprivation, where SGs are formed within 10–20 min (Hoyle et al., 2007; Buchan et al., 2008; Grousl et al., 2009).
Given that yeast SGs show a tight connection to processing (P)-bodies (Buchan et al., 2008)—cytoplasmic foci at which mRNA decay enzymes are concentrated—we also visualized the P-body markers Dcp2 and Pat1 (Supplemental Figure S4, A and B). Both Dcp2-GFP and Pat1-GFP formed numerous foci after 4 h at 0ºC, yet only some of them colocalized with the SG marker Pab1-mCherry, indicative of physically separated SGs and P-bodies. On the other hand, colocalization became pronounced after 24 h at 0ºC, suggesting that the large aggregates during prolonged hypothermia arise from fusions between SGs and P-bodies.
Rapid SG disassembly during recovery from cold shock
Given the unusually slow kinetics of SG assembly during cold shock, we wanted to investigate the fate of these SGs upon return to optimal growth temperatures. First we analyzed a Pub1-GFP–expressing S. cerevisiae strain in which SGs had been induced by keeping cells for 4 h at 0ºC. Very much to our surprise, SGs disappeared within 2.5 min after returning cells to 30ºC (Figure 3, A and B), and polysomes were fully assembled within 5 min (Figure 3, C and D). Of interest, recovery from cold shock occurs faster than recovery from glucose starvation, for which it takes ∼10 min for polysomes to fully reassemble (Supplemental Figure S5).
FIGURE 3:

Rapid disassembly of SGs upon recovery from cold shock in S. cerevisiae. (A) Cells expressing Pub1 genomically tagged with GFP were exposed to cold shock at 0°C for 4 h and then returned to 30°C. Cells were fixed at 30-s intervals, and Pub1-GFP was visualized by confocal microscopy. Four optical sections were used for deconvolution, and the resulting maximum projection is depicted in the images. (B) The disassembly of yeast SGs over time was quantified using automated image analysis. Two parameters were measured: the number of SGs per cell and optical section (yellow bars) and the percentage of Pub1-GFP signal in SGs (brown bars). Average values ± SE from three experiments are shown. (C) Polysome profiles were recorded from yeast during recovery from cold shock. (D) The percentage of polysomal ribosomes was quantified as in Figure 1K, average ± SD.
In mammalian COS7 cells, SGs disappeared 5–10 min after returning from a 10ºC cold shock to 37ºC (Figure 4, A and B), whereas the reassembly of polysomes was rather slow (Figure 4C). Quantification showed that 1 h after return to 37ºC, polysomal ribosomes had reached only two-thirds of the level in control cells, and full recovery was observed after 6 h. Thus, whereas SGs disassemble within minutes in both yeast and mammalian cells during recovery from cold shock, the reassembly of polysomes occurs within minutes in yeast, yet takes several hours in COS7 cells. This suggested that in COS7 cells, SG disassembly is not the rate-limiting step for the resumption of translation after return to optimal growth temperatures.
FIGURE 4:

Rapid disassembly of mammalian SGs upon recovery from cold shock. (A) SG disassembly was monitored in COS7 cells that were subjected to cold shock at 10ºC for 10 h and then returned to 37ºC for 5, 10, 15, and 20 min. SGs were visualized by fluorescence wide-field microscopy after IF staining for eIF3B. (B) To quantify SG disassembly over time, the relative number of COS7 cells containing SGs was determined. Average values ± SE from four experiments are shown. (C) Polysome profiles were recorded from COS7 cells that were grown under control conditions at 37°C or subjected to cold shock at 10°C for 10 h and then allowed to recover at 37°C for the indicated time points. In the bar graph, the percentage of polysomal ribosomes was quantified as in Figure 1K, average ± SD.
eIF2α phosphorylation alone is not responsible for cold shock–induced translation arrest
Our next goal was to characterize the signaling pathways that control translation during cold shock. We first tested whether phosphorylation of eIF2α at serine 51 is involved. In COS7 cells and MEFs, we observed an increase in phospho-eIF2α levels 2–4 h after reducing the temperature to 10ºC, and eIF2α phosphorylation persisted until 24 h (Figure 5, A and B). The slow increase in eIF2α phosphorylation paralleled the progressive suppression of translation (Figure 1K) and the slow induction of SGs (Figure 1H). During recovery from cold shock, we found that eIF2α remained highly phosphorylated for 30 min and was fully dephosphorylated only after 2 h upon return to 37ºC (Figure 5C). Although SGs disassembled much more rapidly within 5–10 min (Figure 4B), the kinetics of translation reinitiation (Figure 4C) appeared to follow eIF2α dephosphorylation.
FIGURE 5:

Cold shock induces PERK-dependent eIF2α phosphorylation. (A) COS7 cells were subjected to cold shock at 10°C for up to 12 h (lanes 1–7), and total protein lysates were analyzed by Western blotting for phospho(S51)-eIF2α and total eIF2α. α-Tubulin serves as loading control. As a positive control, COS7 cells were treated for 1 h with 500 μM Na arsenite (lane 8). (B) Wild-type (eIF2α-SS) MEFs were subjected to cold shock at 10°C (lanes 1–8) and analyzed by Western blotting as in A. For control, eIF2α-SS and eIF2α-AA MEFs were treated for 1 h with 500 μM Na arsenite (lanes 9 and 10). (C) COS7 cells were subjected to cold shock at 10°C for 10 h and returned to 37°C. During the recovery phase, phospho(S51)-eIF2α levels were monitored as in A. (D) Wild-type and PERK ko MEFs were subjected to cold shock (CS) at 10°C for 10 h (lanes 5 and 6) and analyzed by Western blotting as in A. As controls, cells were treated for 1 h with 500 μM Na arsenite (lanes 3 and 4) or 2 mM DTT (lanes 7 and 8). (E) Polysome profiles were recorded from wt and PERK ko MEFs. Cells were grown under control conditions, subjected to cold shock at 10°C for 10 h, or treated with 2 mM DTT for 1 h. Polysome profiles were recorded as in Figure 1K; the bar graph shows quantification of two independent experiments.
Four eIF2α kinases—HRI, PERK, GCN2, and PKR—mediate translational arrest in response to different types of stress. By analyzing knockout (ko) MEFs of each of these kinases, we identified PERK to be responsible for eIF2α phosphorylation during cold shock (Figure 5D, lanes 5 and 6). PERK is known to be activated by ER stress (Harding et al., 1999), and this was confirmed by the failure of PERK ko MEFs to phosphorylate eIF2α in response to treatment with dithiothreitol (DTT; lanes 7 and 8). In contrast to DTT-induced ER stress, cold shock caused SG formation (unpublished data) and translation suppression in both wild-type (wt) and PERK ko MEFs (Figure 5E). This suggested that PERK activation is not the only pathway by which translation is inhibited during cold shock.
We further tested knock-in MEFs in which both eIF2α alleles were replaced either with wt eIF2α-SS encoding for serine at position 51 or with mutant eIF2α-AA encoding for nonphosphorylatable alanine at this position (Scheuner et al., 2001). Similar to the PERK MEFs, both eIF2α-SS and eIF2α-AA MEFs showed massive polysome disassembly (Figure 6A) and SG formation (Figure 6, B and C) upon cold shock. As a control, we subjected the cells to oxidative stress induced by arsenite and observed that the eIF2α-AA MEFs were resistant to translation inhibition and did not form SGs (Figure 6, A–C), as reported previously (McEwen et al., 2005).
FIGURE 6:
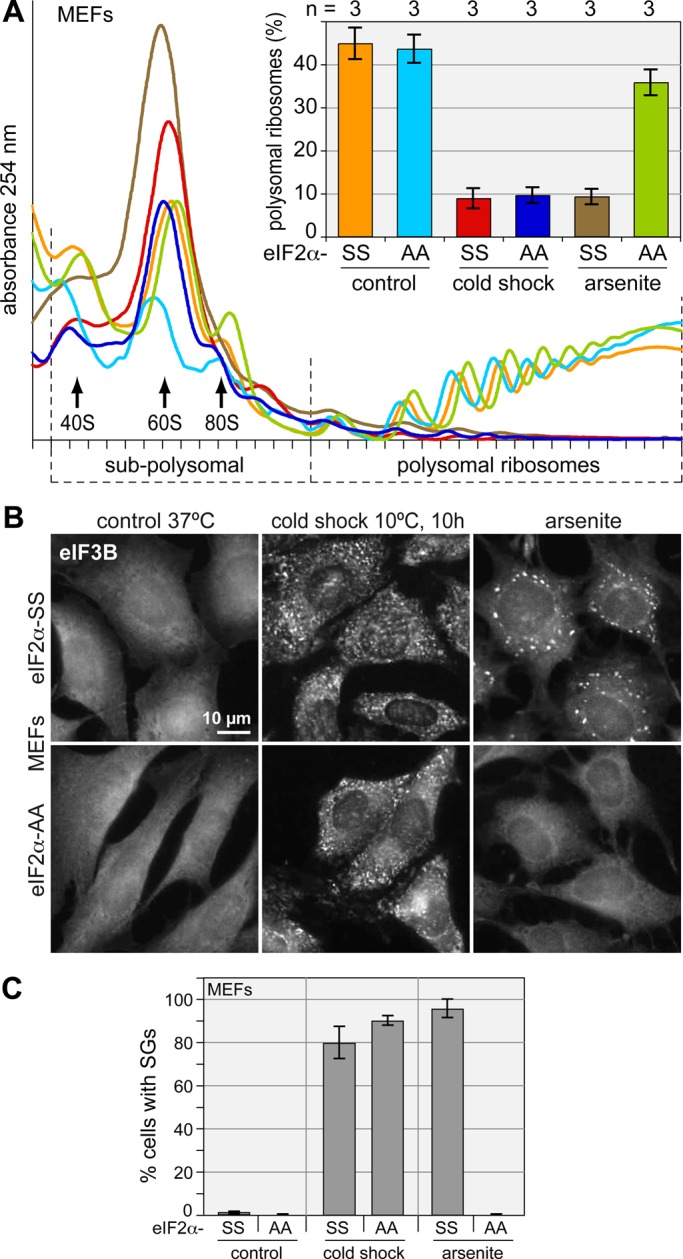
Cold shock–induced translation arrest and SG formation in the absence of phospho-eIF2α in mammalian cells. (A) Polysome profiles were recorded from eIF2α-SS and eIF2α-AA MEFs. Cells were grown under control conditions, treated with 500 μM Na arsenite for 1 h, or subjected to cold shock at 10°C for 10 h. Polysomal ribosomes were quantified as in Figure 1K; the bar graph shows average values ± SE from three independent experiments. (B) SG formation was monitored in eIF2α-SS and eIF2α-AA MEFs grown at 37ºC (control), subjected to cold shock for 10 h at 10ºC, or treated for 1 h with 500 μM Na arsenite. SGs were visualized by IF microscopy after staining for eIF3B. (C) The percentage of cells containing SGs was quantified; average values ± SE are based on three experiments.
Given that S. cerevisiae contains a single eIF2α kinase, Gcn2, we then addressed the role of eIF2α phosphorylation in yeast using a gcn2Δ strain. In both wt and gcn2Δ cells, polysomes were disassembled to a similar degree upon cold shock (Figure 7, A and B). In addition, gcn2Δ cells responded to cold shock by robust induction of SGs, just like wt cells (Figure 7, C and D). Hence, eIF2α phosphorylation appears not to be required for cold shock–induced translation arrest, both in yeast and mammalian cells. These data point toward an additional, possibly redundant, pathway that suppresses translation during cold shock.
FIGURE 7:

Gcn2-independent translation arrest and SG formation upon cold shock in yeast. (A) Polysome profiles were recorded from wt S. cerevisiae and a gcn2Δ strain. Cells were grown under control conditions or subjected to cold shock at 0°C for 1 and 4 h. (B) Translation was quantified as in Figure 3D. (C) Subcellular localization of Pab1-GFP was analyzed by confocal microscopy in the gcn2Δ strain. (D) SGs were quantified in wt and gcn2Δ S. cerevisiae using automated image analysis, as in Figure 2D. Average values ± SE from three experiments are shown.
mTOR inhibition during cold shock
Another pathway by which cells attenuate translation occurs through inhibition of mTOR activity (Wullschleger et al., 2006). In the absence of mTOR signaling, translation is inhibited by dephosphorylation of the mTOR targets 4EBP and S6K1. Dephosphorylated 4EBP prevents eIF4E from recruiting 43S preinitiation complex factors (Richter and Sonenberg, 2005). On cold shock, we observed dephosphorylation of 4EBP1 (Figure 8A, lanes 1 and 2), as well as increased binding of 4EBP1 to eIF4E, using a cap-Sepharose pull-down assay (lanes 4 and 5). At the same time, binding of eIF4AI, eIF3B, and PABP was reduced, and equal changes were observed when cells were treated with the mTOR inhibitor rapamycin (lane 6). By recording polysome profiles, we observed that at 37°C, 10 h of rapamycin treatment reduced polysomal ribosomes moderately from 69 to 41% (Supplemental Figure S6). After 10 h of 10°C cold shock, translation was suppressed to a much greater extent, with only 7% polysomal ribosomes remaining. This fraction was not reduced further by rapamycin, suggesting that mTOR is fully inhibited during cold shock.
FIGURE 8:

Cold shock causes mTOR inhibition. (A) Huh7 cells were grown under control conditions at 37°C, subjected to 10°C cold shock for 10 h, or treated with 0.2 μM rapamycin for 1 h. Cells were lysed, and the cytoplasmic fraction (input, lanes 1–3) was incubated with 7-methyl-GTP (cap) Sepharose. Proteins retained by cap-Sepharose (lanes 4–6) were visualized by Western blotting using antibodies against eIF4E, 4EBP1, eIF4AI, eIF3B, and PABP. (B) 4EBP1+2 double ko MEFs, as well as wt counterparts, were grown under control conditions or subjected to 10 h of cold shock at 10°C. Polysome profiles were recorded as in Figure 1K; the bar graph shows quantification of two independent experiments. (C) Huh 7 cells were exposed to cold shock at 10°C for 10 h or kept under control conditions at 37°C. Cell lysates were loaded onto 17.5–50% sucrose gradients and separated by ultracentrifugation. After fractionation, protein extracts were resolved on 5–20% polyacrylamide gels. eIF4G, eIF4E, eIF4A, eIF3B, PABP, and rpL7 were detected by Western blotting.
mTOR activates translation at least in part through phosphorylation of 4EBP proteins (Richter and Sonenberg, 2005). 4EBP1 and 4EBP2 double-ko MEFs lack 4EBP activity since the third member of the family, 4EBP3, is not expressed in these cells (Dowling et al., 2010). The suppressive effect of mTOR inhibition on translation was previously shown to be much weaker in 4EBP1+2 ko MEFs than in wt MEFs (Dowling et al., 2010). During 10°C cold shock, however, translation suppression was as efficient in 4EBP1+2 ko MEFs as in wt counterparts (Figure 8B). Similarly, SG formation was not affected by deletion of EAP1 and CAF20, the two 4EBP paralogues in yeast (Supplemental Figure S7). These results suggested that 4EBP proteins alone do not account for cold shock–induced repression of translation.
In yeast, eIF4A dissociates from preinitiation complexes upon glucose starvation and shifts to the free fraction in polysome gradients (Castelli et al., 2011). We therefore asked whether such an alternative mechanism might also cause translation suppression during cold shock. However, none of eIF4G, eIF4E, eIF4AI, and eIF3B showed a prominent change in its sucrose gradient distribution upon cold shock in mammalian Huh7 cells (Figure 8C). Only PABP and ribosomal protein L7 shifted from polysomal toward lighter fractions, consistent with the disassembly of polysomes. We inferred that preinitiation complexes, at large, remain assembled during cold shock, whereas mTOR inhibition reduces the specific association of eIF4E with eIF4AI, eIF3B, and PABP.
Cold shock causes energy depletion and AMPK activation
Because mTOR is controlled by the nutrient and energy status of the cell (Wullschleger et al., 2006), mTOR inhibition indicated that energy availability might be a critical factor during cold shock. Indeed, we found that ATP levels dropped progressively during cold shock in COS7 cells (Figure 9A). After 8 h at 10°C, ATP levels were <60% of those in control cells and close to the level observed after treating cells for 1 h with the mitochondrial uncoupler carbonyl cyanide trifluoromethoxyphenylhydrazone (FCCP).
FIGURE 9:

Cold shock causes energy depletion and AMPK activation. (A) COS7 cells were grown under control conditions at 37°C, subjected to cold shock at 10°C for up to 8 h, or treated with FCCP (5 μM) for 1 h. ATP levels in the cellular lysates were measured using recombinant firefly luciferase and represented as percentage of control. Shown are average values ± SE from n = 7 (cold shock) or n = 4 (FCCP) independent experiments. (B) COS7 cells were grown under control conditions at 37°C or subjected to cold shock at 10°C for 10 h. Cells were labeled with Mitotracker Orange CM-H2TMRos 1 h before fixation and analyzed by fluorescence microscopy. (C) COS7 cells were grown under control conditions at 37°C or subjected to cold shock (CS) at 10°C for 9 h and then labeled for 1 h with Mitotracker Orange CM-H2TMRos at 10°C. The intensity of mitotracker staining was measured by flow cytometry. (D) COS7 cells were grown under control conditions at 37°C or subjected to cold shock at 10°C for up to 24 h. Total protein lysates were analyzed by Western blotting for phospho(T172)-AMPK, total AMPK, and eIF3A as loading control. (E) COS7 cells were subjected to cold shock at 10°C for 10 h and returned to 37°C. During the recovery phase, phospho(T172)-AMPK levels were monitored as in D.
As the major source of ATP, mitochondria were stained with mitotracker and found to undergo prominent morphological changes in cold-shocked COS7 cells. Whereas mitochondria formed an elongated tubular network under control conditions, they were arranged in circular-shaped clumps after 10 h at 10°C (Figure 9B). These clumps did not colocalize with SGs (unpublished data) and may reflect mitochondrial fragmentation. In addition, we measured a drop in mitotracker staining by flow cytometry in cold-shocked cells (Figure 9C). Given that mitotracker retention depends on the mitochondrial membrane potential, this suggested that hypothermia causes reduced mitochondrial function.
When ATP levels inside the cell are low, the AMP:ATP ratio rises and stimulates AMPK, by which its α subunit becomes phosphorylated at threonine 172 (Hardie, 2007). AMPK in turn phosphorylates numerous proteins and thereby orchestrates cellular responses to reduced energy supply. Consistent with reduced ATP levels, we observed strong activation of AMPK in COS7 cells in response to cold shock (Figure 9D). During recovery from cold shock, AMPK was dephosphorylated 15–30 min after return of cells to 37ºC (Figure 9E), which is after SG disassembly (Figure 4B) but before eIF2α dephosphorylation (Figure 5C) and full resumption of translation (Figure 4C).
AMPK inhibition prevents SG formation and attenuates translation repression
To further address the role of AMPK, we used compound C, a cell-permeable inhibitor of AMPK (Zhou et al., 2001). Treatment of COS7 cells with compound C reduced phosphorylation of raptor, an AMPK target and integral component of the TOR complex 1 (TORC1), at both 37 and 10°C (Supplemental Figure S8A). We found that compound C prevented the formation of SGs at both 4 and 8 h of cold shock (Figure 10A). Similarly, compound C attenuated cold shock–induced translation repression (Figure 10, B and C). Quantification of the polysome profiles revealed that compound C causes greater than twofold increase in polysomal ribosomes after both 4 and 8 h of 10°C cold shock (Figure 10D). These data suggested that activation of AMPK during cold shock contributes to translation suppression. To validate these results, we analyzed MEFs lacking both isoforms of the catalytic α subunit of AMPK (Laderoute et al., 2006) and found a subtle derepression of translation in these cells (Supplemental Figure S8B). The difference between wt and AMPKα1+2 ko MEFs was weaker than the effect of compound C, suggesting that compound C might exert its effect not only through AMPK inhibition.
FIGURE 10:

AMPK and Scr kinase inhibitors attenuate SG formation, translation inhibition, and cell survival during cold shock. (A) COS7 cells were subjected to cold shock at 10°C for 4 or 8 h and simultaneously treated with 20 μM compound C. Cells were fixed and stained for eIF3B by IF. (B, C) Polysome profiles were recorded from COS7 cells subjected to cold shock at 10°C for (B) 4 h and (C) 8 h, in either the absence or presence of 20 μM compound C. (D) Translation was quantified in cold-shocked cells, in either the absence or presence of 20 μM compound C, by measuring the percentage of polysomal ribosomes as in Figure 1K. Average values ± SE from three experiments are shown. (E) COS7 cells were subjected to cold shock at 10°C for 8 h and simultaneously treated with 20 μM Src inhibitor-1. Cells were fixed and stained for eIF3B by IF. (F) Cell death was measured by the uptake of propidium iodide in unpermeabilized cells using flow cytometry. Before propidium iodide staining, COS7 cells were grown under control conditions at 37°C, treated for 10 h with 20 μM compound C or 20 μM Src inhibitor-1, or subjected to cold shock at 10°C for up to 10 h, in the absence or presence of 20 μM compound C or 20 μM Src inhibitor-1. The percentage of damaged or dead, that is, propidium iodide–positive cells is presented as average value ± SE, n = 5. (G) In addition to compound C (20 μM), cells were exposed to the translation inhibitor cycloheximide (CHX, 10 μg/ml) or puromycin (puro, 1 μg/ml) during the entire 10-h cold shock. Cell death was measured as described above; shown are average values ± SE, n = 6.
Because the kinase Src is also supposed to be inhibited by compound C (Bain et al., 2007), we tested whether a different antagonist, Src inhibitor-1, would affect SG formation as well. Indeed, we found that cold shock–induced SGs were strongly reduced in cells treated with Src inhibitor-1 (Figure 10E). Taken together, these results suggested that different targets of compound C, including AMPK and Src kinase, contribute to translation repression and SG formation in response to cold shock.
SG formation and translation repression are linked to cellular survival during cold shock
Finally, we tested whether formation of SGs and translation arrest are important for cellular survival under cold shock conditions. COS7 cells tolerate hypothermia surprisingly well, with <10% of damaged or dead cells even after 10 h of 10°C cold shock (Figure 10F). In stark contrast, cells showed massive cell death when exposed to the same cold shock in the presence of compound C or Src inhibitor-1, resulting in 86 or 82% dead cells, respectively. Under 37°C control conditions, 10 h of treatment with compound C or Src inhibitor-1 augmented the percentage of damaged or dead cells only minimally, from 4 to 7%. Of importance, the lethal effect of compound C during cold shock was antagonized by simultaneous inhibition of translation with cycloheximide or puromycin (Figure 10G). As expected for a translation elongation inhibitor that prevents ribosome release, cycloheximide blocked the assembly of SGs under cold shock conditions (Supplemental Figure S9). Taken together, these results demonstrate that translation arrest is essential for cellular survival under conditions of cold shock.
DISCUSSION
In this article, we describe an essential stress response pathway by which cells adapt to cold conditions through the repression of global protein synthesis and subsequent formation of SGs. Adaptation to low temperatures is physiologically important to all poikilotherm organisms, which need to adjust their metabolism to ambient temperature. Moreover, the body temperature in some small hibernating mammals drops to 2–10ºC (Carey et al., 2003), the range in which we observed translation repression and SG formation. Indeed, translation is strongly attenuated in hibernating animals, and loss of polysomes was observed in corresponding tissue extracts (Carey et al., 2003). AMPK activation also occurs in hibernating animals (Horman et al., 2005), suggesting that the regulatory response we describe at the cellular level might apply to the biology of hibernation. Even in nonhibernating mammals, the skin temperature in extremities can drop to <10ºC upon exposure to cold (Heus et al., 1995). In the medical context, cold storage at 4ºC is the routine method to preserve organs before transplantation and reduce reperfusion damage. Besides inhibiting tissue-degrading enzymes, there is evidence that AMPK activation contributes to the beneficial effects of cold organ storage (Bouma et al., 2010).
Our study identified cold shock as a novel, evolutionary conserved inducer of SGs in mammalian cells and yeast. These SGs contain all characteristic components, including poly(A)-mRNA, PABP/Pab1, and TIA1/Pub1 and, in mammalian cells, eIF2α, eIF3B, and eIF4G (Figures 1 and 2). Cold shock–induced SGs appear only after keeping cells for several hours at low temperatures yet dissolve very rapidly within minutes of return to optimal growth temperatures (Figures 3 and 4). This is in stark contrast to heat shock– or arsenite-induced oxidative stress, in which SGs appear after 15–30 min but take 30–90 min to disassemble when the stressor is removed (Kedersha et al., 1999, 2000). Given that ATP levels drop slowly (Figure 9) and polysomes disassemble progressively during cold shock (Figure 1), it is plausible that the threshold for SG formation is met only after several hours at low temperature. Rapid SG resolution may reflect the fact that nucleic acids and proteins are well preserved in the cold and that enzymatic activities will increase instantly upon temperature upshift. In contrast, biomolecules suffer considerable damage during oxidative stress or heat shock, in which recovery requires repair processes or de novo synthesis, and therefore takes longer.
Our analysis points toward several mechanisms that suppress translation under cold shock conditions (Figure 11). First, every enzymatic process is temperature dependent, which implies that cold shock will force cellular enzymes, including those involved in translation, to work below their temperature optimum. Second, ATP levels drop during cold shock, presumably because of compromised mitochondrial function (Figure 9). Because protein synthesis is considered to be the most energy-consuming process in the cell, using 22–30% of the cellular energy (Buttgereit and Brand, 1995; Choo et al., 2010), it is particularly sensitive to ATP availability.
FIGURE 11:
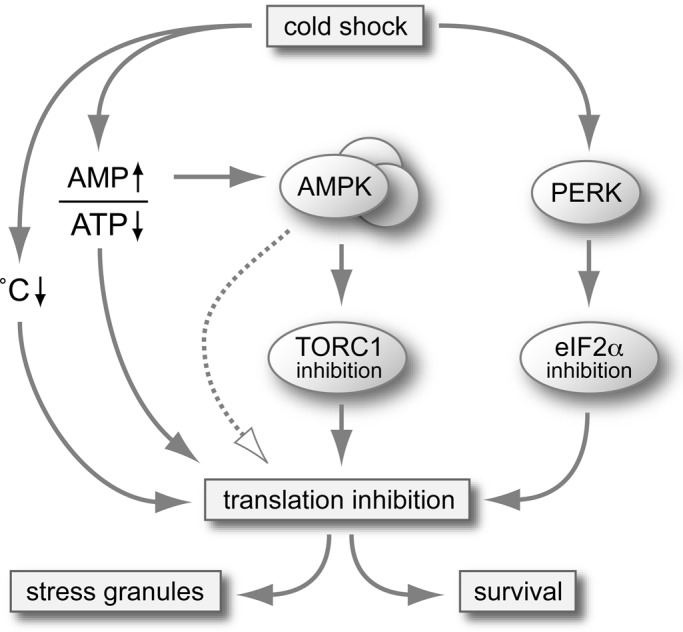
Mechanisms suppressing translation in mammalian cells exposed to cold shock. The model depicts parallel pathways that contribute to cold shock–induced translation suppression. Passive mechanisms include generally reduced enzymatic activities at lower temperatures and reduced mitochondrial function with consecutive drop in ATP levels. Active mechanisms include activation of AMPK, inhibition of TORC1, and PERK-dependent phosphorylation of eIF2α and may involve additional pathways. Translation suppression promotes cell survival under conditions of cold shock.
Of importance, passive mechanisms are not solely responsible for cold shock–induced translation repression. Our findings 1) that cold shock causes eIF2α phosphorylation in a PERK-dependent manner (Figure 5), 2) that the mTOR pathway is inhibited (Figure 8), and 3) that an AMPK inhibitor interferes with translation suppression (Figure 10) demonstrate participation of active mechanisms. Although PERK is well established as an ER stress–responsive kinase (Harding et al., 1999), it also activated by hypoxia (Koumenis et al., 2002). Given that the secretory pathway is particularly sensitive to low temperatures (Saraste et al., 1986), we speculate that proteins accumulate in the ER and cause activation of PERK under cold shock conditions. The involvement of both eIF2α and TORC1 would further explain why interfering with either of the two pathways in the PERK ko MEFs (Figure 5), the eIF2α-AA knock-in MEFs (Figure 6), or the 4EBP1+2 ko MEFs (Figure 8) did not affect translation suppression during cold shock. AMPK was proposed to activate eEF2 kinase and thereby inhibit translation elongation (Horman et al., 2002), which may add to the redundancy of mechanisms suppressing translation at low temperatures.
Our results indicate that the cold shock response serves to adjust cellular energy consumption. By actively reducing the rate of translation, cells are able to save ATP and thereby improve their capacity to survive. In line with this idea, pharmacological inhibition of AMPK and Src kinase caused massive cell death during cold shock (Figure 10). Of importance, blocking translation with cycloheximide or puromycin strongly reduced the lethal effect of compound C. Under glucose starvation, translation inhibitors were also found to enhance survival by reducing ATP consumption (Choo et al., 2010).
We further noticed that cell death induced by compound C and Src inhibitor-1 at 10ºC correlated with lack of SG formation (Figure 10), which may suggest that SGs play a role in cell survival. Indeed, SGs were previously found to serve an anti-apoptotic function by sequestering RACK1 after DNA damage, which prevents RACK1 from activating the proapoptotic MTK1 kinase (Arimoto et al., 2008). Under cold shock conditions, however, cycloheximide treatment not only saved cells from compound C–induced cell death, but it also prevented the assembly of SGs (Supplemental Figure S9). We conclude that cold shock–induced SGs are not required for survival, whereas translation suppression is critical for cells to adapt their energy metabolism and ensure survival under conditions of low temperatures.
MATERIALS AND METHODS
Yeast strains and culture
S. cerevisiae strains used in this study are listed in Supplemental Table S1. Standard yeast genetic techniques were applied throughout this study. Genomic tagging with mCherry, EGFP, and myc was carried out as described (Janke et al., 2004), and transformations were performed using the lithium acetate method (Schiestl and Gietz, 1989). For all experiments, cells were grown to OD600 0.4–0.6 in synthetic complete dextrose (SCD) medium containing 2% glucose at 30°C. To induce cold shock, cells were harvested by centrifugation, resuspended in fresh, ice-cold medium, and incubated on ice with continuous shaking for different time periods. For recovery experiments, cells were directly transferred from ice to a shaking water bath prewarmed to 30°C. For glucose depletion experiments, cells were grown to exponential phase, filtered using a vacuum pump, washed, and resuspended in SC medium lacking glucose, and incubated at 30°C for 10 min. For recovery experiments, glucose was added to starved cells to a final concentration of 2%.
Mammalian cell culture
HeLa, Huh7, and COS7 cells, as well as MEFs, were cultured in DMEM containing 10% fetal bovine serum (FBS; PAA Laboratories, Pasching, Austria), 2 mM l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin (all PAN Biotech, Aidenbach, Germany) at 37°C in 5% CO2. DU145 cells were maintained in RPMI 1640 medium supplemented with 10% FBS, l-glutamine, penicillin, and streptomycin. eIF2α-SS and eIF2α-AA MEFs (Scheuner et al., 2001) were a kind gift from R. Kaufman (University of Michigan Medical Center, Ann Arbor, MI). 4EBP1+2 ko MEFs (Dowling et al., 2010) were kindly provided by Nahum Sonenberg (McGill University, Montreal, Canada), AMPKα1+2 ko MEFs (Laderoute et al., 2006) were a kind gift from Benoit Viollet (Institut Cochin, Université Paris Descartes, Paris, France), and PERK ko MEFs (Harding et al., 2003) were kindly provided by David Ron (New York University School of Medicine, New York, NY). Plasmid pSRα-HA-GFP-PABP (p2024) was described previously (Kedersha et al., 2000) and was transfected into COS7 cells using polyethyleneimine (1 mg/ml, pH 7.0; Polysciences Europe, Eppelheim, Germany). Cells were replated onto glass coverslips 12 h before cold shock.
IF microscopy
Mammalian cells were grown on glass coverslips and fixed at 4ºC in 4% paraformaldehyde for 10 min before cell membranes were permeabilized in –20°C cold methanol for 10 min. Phosphate-buffered saline (PBS) containing 0.1% sodium azide and 5% horse serum was used for blocking and antibody dilution. Cy3- or Cy2-conjugated secondary donkey antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) were used for detection of primary antibodies. Cells were mounted onto glass slides using a solution of 14% polyvinyl alcohol (P8136; Sigma-Aldrich, St. Louis, MO) and 30% glycerol in PBS. SGs were counted on an upright epifluorescence microscope (BX60; Olympus, Tokyo, Japan). Images were acquired at the Nikon Imaging Center, Heidelberg, on an upright epifluorescence microscope (90i; Nikon, Melville, NY) using an electron-multiplying charge-coupled device (EM-CCD) camera (Hamamatsu, Hamamatsu, Japan) and at the Imaging Facility of the Zentrum für Molekulare Biologie der Universität Heidelberg using the Olympus Xcellence Pointfrap microscope and a Hamamatsu Orca-R2 camera.
Yeast cells were grown at 30°C, centrifuged at room temperature, resuspended in ice-cold SCD, and incubated in ice water for the indicated time periods. Cells were fixed with 4% paraformaldehyde and washed three times before microscopy. To visualize the entire cell volume, optical sections at a distance of 0.2 μm were acquired on a spinning disk confocal microscope (UltraView ERS [PerkinElmer, Waltham, MA] on a TE2000 inverted microscope [Nikon]) equipped with an EM-CCD camera (Hamamatsu) at the Nikon Imaging Center, Heidelberg. The entire stack was used for deconvolution using Huygens software (Scientific Volume Imaging, Hilversum, Netherlands). A maximum-intensity projection of three optical sections was generated for image representation, and contrast was enhanced using ImageJ software (National Institutes of Health, Bethesda, MD). For quantitative analysis, single-plane images were acquired and normalized to the exposure time.
Automated image analysis
An algorithm to calculate cell number, cell area, signal intensity within the cell, number of aggregates, area of aggregates, and signal intensity within aggregates from micrographs of S. cerevisiae was written in Matlab software (MathWorks, Natick, MA). Images were first normalized according to exposure times before cell and nuclear boundaries were determined. For the detection of aggregates in the cytoplasm, the local baseline fluorescence for each pixel within cells was calculated using opening by reconstruction, and aggregates were defined as pixels at which the fluorescence increase is >0.75 times the local baseline. The algorithm is available upon request.
Fluorescence in situ hybridization
Yeast spheroplasts were prepared as described previously (Finger et al., 1993), spotted onto poly-l-lysine–coated glass slides, and stored in 70% ethanol at −20°C. Acetylation was performed according to Schwab et al. (1998). Briefly, the cells were washed once with 0.1 M triethanolamine, pH 8.0, incubated in 0.1 M triethanolamine/0.25% acetic anhydride for 10 min, and washed with PBS and 2× SSC. Hybridization with Alexa 488–conjugated oligo(dT)50 (Invitrogen, Carlsbad, CA) was performed overnight at 37°C in hybridization buffer containing 20% formamide, 2× SSC, 0.1% Triton X-100, 2% blocking reagent (Roche, Indianapolis, IN), and 10% dextran sulfate. After sequential washes with 2× SSC, 0.2× SSC, and PBS, samples were incubated for 1 h at room temperature in blocking buffer containing 1% blocking solution and 0.1% Triton X-100 in PBS. Cells were incubated with α-myc antibody (9E10, 1:500; Roche) overnight at 4°C in blocking buffer and further stained with 2 μg/ml Hoechst 33422 dye and Cy3-labeled secondary antibody diluted 1:500 in blocking buffer for 1 h at room temperature. Finally, cells were mounted in PBS containing 12.5% polyvinyl alcohol (Sigma-Aldrich), 25% glycerol, and 0.25% sodium azide.
Mammalian cells were fixed as described for IF, washed in 2× SSC, and incubated in a 1:5000 dilution of Alexa 555–coupled oligo(dT)50 probe (100 pmol/μl; Invitrogen) in hybridization buffer (1 mg/ml yeast RNA, 20% formamide, 2 mg/ml BSA, 0.1 g/ml dextran sulfate, 1× SSC) for 1 h at room temperature. Cells were then washed three times in 2× SSC at room temperature before mounting.
Mitotracker staining
Cells were subjected to cold shock at 10°C for 10 h or kept under control conditions. At 1 h before the end of the treatment, 0.1 μM Mitotracker Orange CM-H2TMRos (Molecular Probes, Eugene, OR) was added to the cell culture. Cells were then collected by trypsinization and centrifugation, resuspended in PBS containing 2% fetal bovine serum, and analyzed on a FACSCanto II (BD Biosciences, Heidelberg, Germany) flow cytometer using Flowjo software (Tree Star, Ashland, OR). For microscopy, cells were grown on glass coverslips, stained with mitotracker as described, and fixed with 4% paraformaldehyde and −20°C methanol. Images were taken with a Leica DM5000B epifluorescence microscope (Leica, Wetzlar, Germany) and a CCD camera (Andor Technology, South Windsor, CT).
Western blot analysis
Cells were lysed in SDS sample buffer with 100 mM DTT. Proteins were resolved on 5–20% polyacrylamide gradient Tris-glycine gels and transferred onto nitrocellulose membranes of 0.2 μm pore size (Peqlab, Erlangen, Germany) for Western blotting. Membranes were then blocked in 5% horse serum in PBS containing 0.1% sodium azide, incubated with antibodies diluted in the same solution, and washed in 150 mM NaCl, 50 mM Tris, pH 7.5, and 1% Tween-20. Horseradish peroxidase–coupled secondary antibodies (Jackson ImmunoResearch Laboratories) in combination with Western Lightning enhanced chemiluminescence substrate (PerkinElmer) were used for detection.
Antibodies
The following antibodies were used for Western blot and IF: mouse monoclonal antibodies against G3BP1 (TT-Y, sc-81940; Santa Cruz Biotechnology, Santa Cruz, CA) and eIF2α (ab5369; Abcam, Cambridge, MA); polyclonal rabbit antibodies against phospho(S51)-eIF2α (KAP-CP131; Stressgen, San Diego, CA), eIF4GI (2498; Cell Signaling Technology, Frankfurt am Main, Germany), monoclonal rabbit phospho(T172)-AMPK (40H9, #2535; Cell Signaling Technology), phospho(S792)-raptor (#2083; Cell Signaling Technology); monoclonal rabbit antibodies against AMPKα (23A3, #2603; Cell Signaling Technology), 4EBP1 (53H11, #9644; Cell Signaling Technology), phospho(T37/46)-4EBP1 (#2855; Cell Signaling Technology), and raptor (#2280; Cell Signaling Technology); polyclonal goat antibody against eIF3B (sc-16377; Santa Cruz Biotechnology); and rat monoclonal antibody against α-tubulin (ab6160; Abcam).
Polysome analysis
Mammalian cells were treated with Na arsenite (500 μM, 1 h), rapamycin (0.2 μM, 1 h), or DTT (2 mM, 1 h) or were exposed to cold shock in either the absence or presence of compound C (20 μM) for the times indicated. Before lysis, 100 μg/ml cycloheximide (Sigma-Aldrich) was added to cells for 5 min, and cells were washed with cold PBS containing 100 μg/ml cycloheximide. Cells were harvested by scraping and lysed in 0.2 ml lysis buffer containing 15 mM Tris, pH 7.4, 15 mM MgCl2, 300 mM NaCl, 1% Triton X-100, 100 μg/ml cycloheximide, 500 μg/ml heparin, 0.2 U/ml RNasin (Promega, Madison, WI), 0.1% 2-mercaptoethanol, and EDTA-free protease inhibitor (Roche). Lysates were cleared by centrifugation at 10,000 rpm for 10 min at 4°C. Supernatants were loaded onto linear gradients of 17.5–50% sucrose in 15 mM Tris, pH 7.4, 15 mM MgCl2, and 300 mM NaCl and centrifuged in a SW60 rotor at 35,000 rpm for 2.5 h at 4°C. Fractions were eluted from the top of the gradient using a Teledyne Isco (Lincoln, NE) gradient elution system; polysome profiles were obtained by measuring absorbance at 254 nm.
Yeast cultures were treated with 100 μg/ml cycloheximide on ice for 5 min, collected by centrifugation, washed once with ice-cold dH2O and resuspended in ice-cold yeast polysome lysis buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.5, 50 mM KCl, 10 mM MgCl2, 300 mM NaCl, 1% TritonX-100, protease inhibitor cocktail [Roche], 1 mM phenylmethylsulfonyl fluoride, 1 mM DTT, 100 μg/ml cycloheximide, and 50 U/ml Superasein [Ambion, Austin, TX]). Cell suspensions were then frozen in drops in liquid nitrogen and pulverized by mixer milling (MM 400; Retsch, Newtown, PA). Lysates were clarified by centrifugation at 14,000 × g for 10 min, and the amount corresponding to 1 mg of total RNA was loaded on 7–47% sucrose gradients. Gradients were centrifuged in a SW40 rotor at 35,000 rpm for 2.5 h and processed as described for mammalian cells. For quantification, “empty” gradients loaded with buffer alone were recorded using the same settings. Normalization was carried out by subtracting “empty gradient” values from “sample gradient” values, and the areas under the curve corresponding to the amount of polysomal and total ribosomes were determined by integration. Polysomal ribosomes were divided by total ribosomes as a measure for the translation rate.
Polysome fractionation
Huh7 cells were kept under control conditions at 37°C or exposed to cold shock at 10°C for 10 h. Before lysis, cells were treated with 100 μg/ml cycloheximide for 5 min, washed with cold PBS containing 100 μg/ml cycloheximide, harvested by scraping, and lysed in 0.2 ml of lysis buffer containing 10 mM HEPES, 2.5 mM MgCl2, 62.5 mM KCl, 1% NP-40, 100 μg/ml cycloheximide, 500 μg/ml heparin, 0.2 U/ml RNasin, 1 mM DTT, and EDTA-free protease inhibitor (Roche). Lysates were cleared by centrifugation at 10,000 rpm for 10 min at 4°C and subjected to sucrose density gradient centrifugation as described. After fractionation, proteins were precipitated using acetone and resuspended in SDS sample buffer.
[35S]methionine/cysteine labeling
COS7 cells were seeded 8–12 h before incubation in methionine- and cysteine-free DME medium supplemented with 5% fetal bovine serum (PAA Laboratories), 2 mM l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin (all PAN Biotech) for 1 h. Then 200 μCi of 35S-labeled methionine and cysteine (EasyTag; PerkinElmer) was added to each dish, and cells were simultaneously exposed to cold shock at 10°C or kept at 37°C for different periods of time. As a control, one dish was treated with 500 μM Na arsenite for 1 h. Cells were then washed with PBS, collected, and solubilized in 150 μl of lysis buffer containing 15 mM Tris, pH 7.4, 15 mM MgCl2, 300 mM NaCl, and 1% Triton X-100. After centrifugation at 9000 rpm for 3 min, proteins were precipitated out of the supernatants by spotting 20 μl of each lysate onto Whatman paper and soaking in 5% trichloroacetic acid followed by acetone. The 35S incorporation was measured in 4 ml of Econofluor-2 (PerkinElmer) using a scintillation counter (LS 6000IC; Beckman Coulter, Brea, CA). For normalization, the total protein concentration of each sample was determined using the bicinchoninic acid protein assay reagent kit (Sigma-Aldrich). For each sample, 100 ng of total protein was resolved on a 5–20% polyacrylamide gradient Tris-glycine gel and stained using colloidal Coomassie. The gel was dried at 80°C for 2 h under vacuum, and 35S incorporation into newly synthesized proteins was detected by autoradiography using a phosphoimager (FLA-7000; FujiFilm, Tokyo, Japan).
Cap pull-down assay
Huh7 cells were seeded in 15-cm dishes and allowed to adhere for 8–12 h. To account for the reduced cell mass after cold shock, two dishes were subjected to 10°C cold shock for 10 h, one was treated with 0.2 μM rapamycin for 1 h, and one dish was kept under control conditions. Cells were lysed in 0.6 ml of cap binding buffer containing 50 mM Tris-HCl (pH 7.0), 100 mM NaCl, 1 mM EDTA, 0.5% NP-40, and complete protease inhibitors (Roche). Lysates were cleared via centrifugation at 25,000 × g for 15 min at 4°C. A 50-μl amount of 7-methyl-GTP–conjugated Sepharose beads (GE Healthcare, Piscataway, NJ) was added to each sample. After rotation at 4°C for 2 h, beads were washed four times in a buffer containing 15 mM Tris-HCl (pH 7.2), 100 mM NaCl, 1 mM EDTA, and 0.1% NP-40. Proteins were eluted in SDS sample buffer and separated on a 5–20% polyacrylamide gradient gel.
Measurement of ATP levels
After cold shock, cells were collected by trypsinization, and cell pellets were weighed on a fine balance before lysis in 20 mM Tris (pH 7.4), 0.1 mM EDTA, 2.67 mM MgSO4, and 33.3 mM DTT. Lysates were incubated on ice for 5–10 min and centrifuged at 13,000 rpm for 1 min. For ATP measurements, 20 μl of serially diluted lysates was added to 80 μl of lysis buffer containing 1 nM of firefly luciferase recombinantly expressed in E. coli and 500 μM luciferin. Light emission was measured with a luminometer (Lumat LB 9507; Berthold Technologies, Bad Wildbad, Germany). Results were normalized to the weight of the cell pellets and calculated as percentage of control.
Cell viability assay
Cells were subjected to cold shock for different time periods and treated with 20 μM compound C (Calbiochem, La Jolla, CA) or 20 μM Src inhibitor-1 (S2075; Sigma-Aldrich) where indicated. Cells were collected from the media and combined with those recovered from the dish by trypsinization, resuspended in 10 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM MgCl2, and 1.8 mM CaCl2 and stained for 10 min on ice with 50 μg/ml propidium iodide (AppliChem, Darmstadt, Germany). Propidium iodide uptake was measured by flow cytometry using a FACSCanto II (BD Biosciences).
Supplementary Material
Acknowledgments
We thank Silke Druffel-Augustin (University of Heidelberg) for technical assistance, Aurelio Teleman (German Cancer Research Center, Heidelberg) and Lys Guibride for critical comments on the manuscript, Randal Kaufman for generously providing the eIF2α-AA and eIF2α-SS MEFs, David Ron for the PERK ko MEFs, Nahum Sonenberg for the 4EBP1+2 ko MEFs, Benoit Viollet for the AMPKα1+2 ko MEFs, and Nancy Kedersha (Harvard Medical School, Boston, MA) for the GFP-PABP plasmid, as well as Ulrike Engel (Nikon Imaging Center, University of Heidelberg, Heidelberg, Germany) and Holger Lorenz (Imaging Facility Zentrum für Molekulare Biologie, University of Heidelberg) for help with confocal, wide-field, and live-imaging microscopy. This work was supported by Young Investigator Grant HZ-NG-210 from the Helmholtz Gemeinschaft to G.S., Research Grants STO 859/2-1 and SFB 1036 from the Deutsche Forschungsgemeinschaft to G.S. and B.B., a bridging project grant from the German Cancer Research Center–Zentrum für Molekulare Biologie der Universität Heidelberg Alliance to G.S. and B.B., and a fellowship from the Heidelberg Molecular Life Sciences initiative to P.B.
Abbreviations used:
- AMPK
AMP-activated protein kinase
- cpm
counts per minute
- CS
cold shock
- DTT
dithiothreitol
- 4EBP
eIF4E-binding protein
- eIF
eukaryotic translation initiation factor
- EM-CCD
electron-multiplying charge-coupled device
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IF
immunofluorescence
- ko
knockout
- MEF
mouse embryonic fibroblast
- mTOR
mammalian target of rapamycin
- PABP
poly(A)-binding protein
- P-body
processing body
- PBS
phosphate-buffered saline
- SCD
synthetic complete dextrose
- SG
stress granule
- wt
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-04-0296) on August 8, 2012.
*These authors contributed equally to this work.
REFERENCES
- Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci. 2008;33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, Takekawa M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat Cell Biol. 2008;10:1324–1332. doi: 10.1038/ncb1791. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouma HR, Ketelaar ME, Yard BA, Ploeg RJ, Henning RH. AMP-activated protein kinase as a target for preconditioning in transplantation medicine. Transplantation. 2010;90:353–358. doi: 10.1097/TP.0b013e3181e7a3aa. [DOI] [PubMed] [Google Scholar]
- Buchan JR, Muhlrad D, Parker R. P bodies promote stress granule assembly in Saccharomyces cerevisiae. J Cell Biol. 2008;183:441–455. doi: 10.1083/jcb.200807043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttgereit F, Brand MD. A hierarchy of ATP-consuming processes in mammalian cells. Biochem J. 1995;312:163–167. doi: 10.1042/bj3120163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey HV, Andrews MT, Martin SL. Mammalian hibernation: cellular and molecular responses to depressed metabolism and low temperature. Physiol Rev. 2003;83:1153–1181. doi: 10.1152/physrev.00008.2003. [DOI] [PubMed] [Google Scholar]
- Castelli LM, et al. Glucose depletion inhibits translation initiation via eIF4A loss and subsequent 48S preinitiation complex accumulation, while the pentose phosphate pathway is coordinately up-regulated. Mol Biol Cell. 2011;22:3379–3393. doi: 10.1091/mbc.E11-02-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo AY, Kim SG, Vander Heiden MG, Mahoney SJ, Vu H, Yoon SO, Cantley LC, Blenis J. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol Cell. 2010;38:487–499. doi: 10.1016/j.molcel.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling RJ, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. 2010;328:1172–1176. doi: 10.1126/science.1187532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger A, Knop M, Wolf DH. Analysis of two mutated vacuolar proteins reveals a degradation pathway in the endoplasmic reticulum or a related compartment of yeast. Eur J Biochem. 1993;218:565–574. doi: 10.1111/j.1432-1033.1993.tb18410.x. [DOI] [PubMed] [Google Scholar]
- Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell. 2004;15:5383–5398. doi: 10.1091/mbc.E04-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grousl T, et al. Robust heat shock induces eIF2alpha-phosphorylation-independent assembly of stress granules containing eIF3 and 40S ribosomal subunits in budding yeast, Saccharomyces cerevisiae. J Cell Sci. 2009;122:2078–2088. doi: 10.1242/jcs.045104. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harding HP, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Heus R, Daanen HA, Havenith G. Physiological criteria for functioning of hands in the cold: a review. Appl Ergon. 1995;26:5–13. doi: 10.1016/0003-6870(94)00004-i. [DOI] [PubMed] [Google Scholar]
- Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol. 2005;6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–580. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Homma T, Iwahashi H, Komatsu Y. Yeast gene expression during growth at low temperature. Cryobiology. 2003;46:230–237. doi: 10.1016/s0011-2240(03)00028-2. [DOI] [PubMed] [Google Scholar]
- Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol. 2002;12:1419–1423. doi: 10.1016/s0960-9822(02)01077-1. [DOI] [PubMed] [Google Scholar]
- Horman S, Hussain N, Dilworth SM, Storey KB, Rider MH. Evaluation of the role of AMP-activated protein kinase and its downstream targets in mammalian hibernation. Comp Biochem Physiol B Biochem Mol Biol. 2005;142:374–382. doi: 10.1016/j.cbpb.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Hoyle NP, Castelli LM, Campbell SG, Holmes LE, Ashe MP. Stress-dependent relocalization of translationally primed mRNPs to cytoplasmic granules that are kinetically and spatially distinct from P-bodies. J Cell Biol. 2007;179:65–74. doi: 10.1083/jcb.200707010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, et al. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J, Anderson P. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell. 2002;13:195–210. doi: 10.1091/mbc.01-05-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol. 2000;151:1257–1268. doi: 10.1083/jcb.151.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol. 1999;147:1431–1442. doi: 10.1083/jcb.147.7.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WJ, Back SH, Kim V, Ryu I, Jang SK. Sequestration of TRAF2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol Cell Biol. 2005;25:2450–2462. doi: 10.1128/MCB.25.6.2450-2462.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–5347. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen E, Kedersha N, Song B, Scheuner D, Gilks N, Han A, Chen JJ, Anderson P, Kaufman RJ. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J Biol Chem. 2005;280:16925–16933. doi: 10.1074/jbc.M412882200. [DOI] [PubMed] [Google Scholar]
- Murata Y, et al. Genome-wide expression analysis of yeast response during exposure to 4 degrees C. Extremophiles. 2006;10:117–128. doi: 10.1007/s00792-005-0480-1. [DOI] [PubMed] [Google Scholar]
- Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005;433:477–480. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- Roobol A, Carden MJ, Newsam RJ, Smales CM. Biochemical insights into the mechanisms central to the response of mammalian cells to cold stress and subsequent rewarming. FEBS J. 2009;276:286–302. doi: 10.1111/j.1742-4658.2008.06781.x. [DOI] [PubMed] [Google Scholar]
- Ruggieri A, et al. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe. 2012;12:71–85. doi: 10.1016/j.chom.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste J, Palade GE, Farquhar MG. Temperature-sensitive steps in the transport of secretory proteins through the Golgi complex in exocrine pancreatic cells. Proc Natl Acad Sci USA. 1986;83:6425–6429. doi: 10.1073/pnas.83.17.6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Schiestl RH, Gietz RD. High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr Genet. 1989;16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- Schwab MH, Druffel-Augustin S, Gass P, Jung M, Klugmann M, Bartholomae A, Rossner MJ, Nave KA. Neuronal basic helix-loop-helix proteins (NEX, neuroD, NDRF): spatiotemporal expression and targeted disruption of the NEX gene in transgenic mice. J Neurosci. 1998;18:1408–1418. doi: 10.1523/JNEUROSCI.18-04-01408.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahara T, Maeda T. Transient sequestration of TORC1 into stress granules during heat stress. Mol Cell. 2012;47:242–252. doi: 10.1016/j.molcel.2012.05.019. [DOI] [PubMed] [Google Scholar]
- Tourriere H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J Cell Biol. 2003;160:823–831. doi: 10.1083/jcb.200212128. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Zhou G, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.