

EMT requires cooperation of the EGF/Ras with the TGF-β signaling pathways in a multistep process. ERF, a bona fide Ras-Erk effector, inhibits TGF-β–induced EMT via Semaphorin-7a repression, and Semaphorin-7a induction is required for EMT progress. These data provide new insights into the Ras–TGF-β interconnection.
Abstract
Epithelial-to-mesenchymal transition (EMT) is a key process in cancer progression and metastasis, requiring cooperation of the epidermal growth factor/Ras with the transforming growth factor-β (TGF-β) signaling pathway in a multistep process. The molecular mechanisms by which Ras signaling contributes to EMT, however, remain elusive to a large extent. We therefore examined the transcriptional repressor Ets2-repressor factor (ERF)—a bona fide Ras–extracellular signal-regulated kinase/mitogen-activated protein kinase effector—for its ability to interfere with TGF-β–induced EMT in mammary epithelial cells (EpH4) expressing oncogenic Ras (EpRas). ERF-overexpressing EpRas cells failed to undergo TGF-β–induced EMT, formed three-dimensional tubular structures in collagen gels, and retained expression of epithelial markers. Transcriptome analysis indicated that TGF-β signaling through Smads was mostly unaffected, and ERF suppressed the TGF-β–induced EMT via Semaphorin-7a repression. Forced expression of Semaphorin-7a in ERF-overexpressing EpRas cells reestablished their ability to undergo EMT. In contrast, inhibition of Semaphorin-7a in the parental EpRas cells inhibited their ability to undergo TGF-β–induced EMT. Our data suggest that oncogenic Ras may play an additional role in EMT via the ERF, regulating Semaphorin-7a and providing a new interconnection between the Ras- and the TGF-β–signaling pathways.
INTRODUCTION
Epithelial-to-mesenchymal transition (EMT) is a highly conserved, fundamental process in embryogenesis and cancer during which epithelial cells disassemble, acquire a fibroblastic–mesenchymal phenotype, digest basement membranes, and transmigrate to surrounding tissues (Thiery, 2003). EMT is involved in trophoblast differentiation, gastrulation movements, and emigration of neural crest cells from the neural tube. Formation of the heart, the musculoskeletal system, and the peripheral nervous system also involve this process. EMT also has a role in tissue reorganization and wound healing in the adult (Sun et al., 2000; Shook and Keller, 2003). Processes similar to EMT occur in pathological situations such as the acquisition of an invasive, metastatic phenotype in tumors of epithelial origin. During late steps of carcinogenesis, EMT allows malignant cells to lose their epithelial polarity, digest the basement membrane and invade surrounding tissues, intravasate into the bloodstream, and colonize distant tissues (Vernon and LaBonne, 2004; Hugo et al., 2007).
EMT manifests through multiple cellular and molecular changes that lead to loss of epithelial polarity and cell–cell adhesion, transdifferentiation to a mesenchymal (fibroblastoid) cell phenotype, and induction of cell motility/invasiveness. Loss of E-cadherin—the transmembrane adhesion molecule responsible for the formation of adherence junctions (Christofori and Semb, 1999)—largely contributes to loss of epithelial polarity. Concomitantly, EMT involves repression of other epithelial-specific proteins and de novo expression of mesenchymal proteins. Massive cytoskeletal reorganization and induction of the expression of metalloproteases during EMT lead to acquisition of cellular motility and the ability to digest and transmigrate through basement membranes (Boyer et al., 2000).
Because of its crucial importance in physiological and pathological events, EMT has been intensely studied using several different epithelial cell systems from various tissues in which EMT can be induced. This work has yielded some conflicting results because of the different properties of the cells used and the different culture conditions (Grunert et al., 2003; Jechlinger et al., 2003). Contradictions were minimized through use of cell models that allow the formation of organotypic structures consisting of fully polarized cells under near-physiological, three-dimensional cultures, for example, collagen gels (Grunert et al., 2003; Jechlinger et al., 2003).
In most cellular models, EMT is induced by external stimuli. Transforming growth factor-β (TGF-β) regulates multiple morphogenetic events, as well as migration of normal and cancerous cells, and is a key inducer of EMT. TGF-β, however, requires cooperation with the RTK/Ras or other signaling pathways since it causes cell cycle arrest and apoptosis in cells lacking oncogenic Ras (Moustakas and Heldin, 2007).
A large number of diverse transcription factors have been reported to induce molecular changes essential for EMT. Slug (Bolos et al., 2003), Snail (Cano et al., 2000), SIP-1 (Comijn et al., 2001), Twist (Yang et al., 2004), E12/E47 (Moreno-Bueno et al., 2006), and dEF1 (Eger et al., 2005) contribute to EMT by repressing E-cadherin, leading to the disruption of intercellular junctions. c-Jun (Fialka et al., 1996), c-Fos (Eger et al., 2004), the nuclear complex β-catenin/LEF-1 (Kim et al., 2002), and Ets-1 (Delannoy-Courdent et al., 1998) have been shown to elicit EMT, and NF-κB appears to be necessary for the induction and maintenance of EMT in Ras-transformed epithelial cells (Huber et al., 2004). Although transcriptions factors inducing EMT have been extensively studied, with the exception of Id2 (Kowanetz et al., 2004), transcription factors inhibiting this process have not been described.
ERF (Ets2-repressor factor) is an ets-domain gene with transcriptional repressor activity that functions as a downstream effector of the Ras/extracellular signal-regulated kinase (Erk) pathway. In its nuclear, nonphosphorylated form, ERF can inhibit cell-cycle progression and suppresses ets- and ras-induced tumorigenicity in fibroblasts (Sgouras et al., 1995; Le Gallic et al., 1999, 2004; Polychronopoulos et al., 2006), whereas Fli-1/ERF hybrid proteins can suppress transformation of Ewing's sarcoma cells (Athanasiou et al., 2000). Phosphorylation of ERF through Erk/mitogen-activated protein kinase (MAPK) signaling causes its nuclear-to-cytoplasmic translocation, where it has distinct but largely elusive functions. Homozygous deletion of Erf in mice leads to embryonic lethality at day 10 due to trophoblast stem cell differentiation and placental defects (Papadaki et al., 2007).
We recently showed that ERF mediates ERF-induced epithelial cell migration via early growth response-1 regulation (Tarcic et al., 2012), linking ERF to a key aspect of EMT. In this study, we endeavored to address the possible role of ERF, as a downstream effector of the Ras/ERK pathway, in the induction/maintenance of EMT beyond the motility effect. We used expression of wild-type and mutated forms of ERF in the fully polarized mammary epithelial cell line EpH4 expressing oncogenic Ras (EpRas), which undergo EMT upon exposure to TGF-β. These cells were analyzed both on plastic and in three-dimensional cultures (Janda et al., 2002b) for their ability to undergo EMT in response to TGF-β. Analysis of cell morphology and proliferation and expression of cellular/molecular epithelial and mesenchymal markers indicated that forced ERF expression can inhibit TGF-β–induced EMT. Of interest, ERF inhibits EMT independent of its c-Myc–associated ability to inhibit cell proliferation (Verykokakis et al., 2007), suggesting that Ras/MAPK signaling regulates EMT and proliferation via different mechanisms. Transcriptome and genetic analysis of the ERF-expressing lines indicated that Semaphorin-7a/CDw108 (Lange et al., 1998) may be a key, Ras/ERF-dependent regulator, modifying the cellular response to TGF-β signaling during EMT. This is the first example that events downstream of ERK/MAPK signaling are causally related to EMT, providing additional insights into the need for hyperactivated Ras/MAPK signaling in EMT (Janda et al., 2002a).
RESULTS
ERF inhibits EMT
To investigate the possible role of ERF as a Ras/Erk mediator during EMT, we introduced wild-type (wt) and mutant ERF into EpRas cells. The ERFm1-7 mutant carries Ser/Thr-to-Ala mutations in seven potential Erk phosphorylation sites and exhibits constitutive nuclear localization, whereas the ERF-FSF/FKF mutant carries mutations that inhibit the ERF–ERK interaction and thus decrease Erk signaling to ERF. After drug selection, cell clones named Ep-ERF, Ep-M1-7, and Ep-FSF/FKF, respectively, were isolated and expanded, and the expression of ERF and altered phosphorylation was verified by immunoblotting with the S17S anti–Erf-specific polyclonal antibody (Figure 1A). Near-confluent monolayers of the clones growing on plastic dishes were treated with TGF-β for 5 d to determine affects on morphology. Whereas all cell clones showed the expected cobblestone, epithelial morphology in the absence of TGF-β, the control vector–expressing EpRas cells (and less clearly the nonphosphorylatable ERFm1-7 clone) adopted a spindle-like fibroblastoid morphology, suggesting that these cells undergo EMT. The Ep-ERF and Ep-FSF/FKF clones, however, predominantly maintained an epithelial morphology (Figure 1B). To examine whether corresponding EMT marker proteins were expressed in these TGF-β–resistant phenotypes, we determined the levels of the epithelial marker E-cadherin and the mesenchymal marker fibronectin. Consistent with their epithelial morphology, Ep-ERF and Ep-FSF/FKF cells retained high E-cadherin levels upon TGF-β treatment. Fibronectin levels were elevated in the absence of TGF-β and exhibited a small increase upon treatment. Control and Ep-M1-7 cells, however, underwent a seemingly TGF-β–induced EMT, that is, they lost E-cadherin and gained fibronectin expression (Figure 1C). These data suggest that expression of wt-ERF and Erk-interaction–deficient ERF attenuated TGF-β–induced EMT in EpRas cells with respect to both morphology and EMT marker expression.
FIGURE 1:
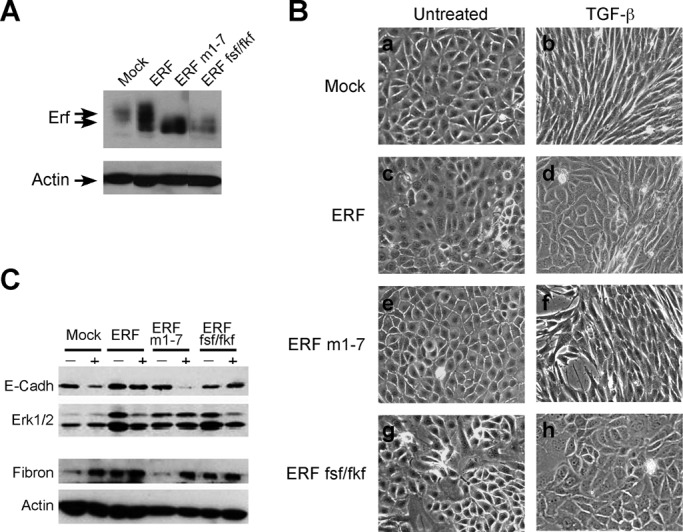
Characterization of the EpRas cell clones overexpressing wt or mutated ERF. (A) Selected clones of EpRas cells transfected with empty vector, wt ERF, the M1-7 ERF mutation, or the FSF/FKF ERF mutation were examined for ERF expression by immunoblotting using S17S Erf-specific antibody (top) and an actin-specific antibody (bottom) as a loading control. The lanes shown are not consecutive on the gel. All clones expressed 3- to 20-fold higher amounts of ERF compared with the vector control. (B) Phase contrast images of the mentioned clones were grown in the absence (a, c, e, g) or presence (b, d, f, h) of TGF-β for 5 d. Vector-transfected cells acquired a spindle-like morphology after exposure to TGF-β (b), whereas ERF-expressing cells maintained to a large extent their epithelial morphology (d, f, h). (C) The levels of E-cadherin (top) and fibronectin (second from bottom) in cells cultured for 5 din the presence or absence of TGF-β were examined by immunoblotting. Erk2/1 (second from top) and actin (bottom) levels were used respectively as loading controls. Vector-transfected and ERFm1-7–expressing cells show decreased E-cadherin and increased fibronectin levels after treatment with TGF-β. In contrast, E-cadherin and fibronectin levels in wt ERF– and ERF-FSF/FKF–expressing cells remained constant.
Epithelial cells like EpH4/EpRas, however, fail to fully polarize on plastic dishes (Grunert et al., 2003). We therefore analyzed the aforementioned cells on porous supports better suited to attain full epithelial polarity (Ojakian et al., 1997; Grunert et al., 2003; Jechlinger et al., 2003). Again, TGF-β–treated Ep-ERF and Ep-FSF/FKF cells failed to adopt a fibroblastoid morphology, whereas control cells underwent TGF-β–induced EMT, and Ep-M1-7 cells showed an intermediate phenotype (Supplemental Figure S1A). When analyzed for epithelial and mesenchymal markers by immunofluorescence, all clones showed membrane-localized E-cadherin and cytoplasmic fibronectin in the absence of TGF-β (Figure 2, A and B). After exposure to TGF-β for 5 d, the vector control cells completely lost E-cadherin and deposited fibronectin extracellularly (Figure 2, A and B, b). In contrast, TGF-β–treated Ep-FSF/FKF and Ep-ERF cells maintained plasma membrane expression of E-cadherin and cytoplasmic localization of fibronectin (Figure 2, A and B, d and h). Again, the Ep-M1-7 cells showed an intermediate phenotype with cytoplasmic E-cadherin (Figure 2A, f) and enhanced but not extracellularly deposited fibronectin (Figure 2B, f).
FIGURE 2:

ERF-expressing EpRas cells resist TGF-β–induced EMT. (A, B) Vector-transfected EpRas cells (a, b) and clones expressing wt ERF (c, d), ERFm1-7 (e, f), or ERF-FSF/FKF (g, h) were grown for 6 d on porous support in the absence (a, c, e, g) or presence (b, d, f, h) of 5 ng/ml TGF-β. The cells were stained for E-cadherin (A) or fibronectin (B) with the respective specific antibody (red) and TOPRO-3 for DNA (blue) and analyzed by confocal microscopy. Vector-transfected cells lost E-cadherin expression (A, b) and secreted fibronectin (B, b) as a result of TGF-β treatment. In contrast, wt ERF– (d) and ERF-FSF/FKF–expressing cells (h) maintained basolateral E-cadherin expression and mostly cytoplasmic fibronectin. ERFm1-7–expressing cells have cytoplasmic E-cadherin (A, f) and high levels of cytoplasmic fibronectin (B, f). (C) The same cells were cultured in three-dimensional collagen gels in serum-free media for 5 d in the absence (a, c, e, g) or presence (b, d, f, h) of 5 ng/ml TGF-β and stained for E-cadherin (red) and DNA (blue). Vector-transfected cells failed to express E-cadherin after treatment with TGF-β (b). In contrast, all ERF-expressing cells maintained basolateral expression of E-cadherin (d, f, h).
To analyze the function of ERF in cells growing under more physiological conditions, we seeded cells into serum-free collagen gels where EpRas cells can form hollow, tubular, or alveolar organotypic structures consisting of fully polarized cells (Oft et al., 1996; Montesano et al., 1998). In the absence of TGF-β all cell lines formed compact, tubular structures (Supplemental Figure S1B). Treatment of the cultures with TGF-β induced an EMT-like response in vector-transfected cells, forming unordered strands of spindle-shaped cells. (Supplemental Figure S1B, b). In contrast, Ep-ERF, Ep-M1-7, and Ep-FSF/FKF cells maintained their compact structure morphology in the presence of TGF-β (Supplemental Figure S1B, d, f, and h), indicating their inability to undergo TGF-β–induced EMT. These findings were confirmed by immunostaining of the collagen gel structures for E-cadherin. In the absence of TGF-β the compact structures formed by all cell lines exhibited plasma membrane expression of E-cadherin (Figure 2C, left). After 5 d of TGF-β treatment, the empty vector control cells had completely lost E-cadherin expression, whereas the compact structures formed by Ep-ERF, Ep-M1-7, and Ep-FSF/FKF cells retained plasma membrane E-cadherin expression (Figure 2C, d, f, and h). Similarly, the cortical localization of actin was changed to cytoplasmic stress fibers only in TGF-β–treated control cells, whereas this treatment did not alter cortical actin expression in the ERF-expressing clones (Supplemental Figure S2). Of interest, in EpRas cells growing on collagen gels, ERF exhibited an increased nuclear localization, as evidenced by the accumulation of the nonphosphorylated form of ERF and by immunofluorescence (Supplemental Figure S3), supporting the apparently enhanced EMT block under these conditions. These data suggested to us that overexpression of either wt or mutated ERF in EpRas cells may inhibit their ability to undergo EMT in response to TGF-β signaling.
Increased motility is one of the hallmarks of cells undergoing EMT (Moustakas and Heldin, 2007). We recently showed that ERF may be required for increased motility. Thus we analyzed the migratory capacity of ERF-expressing cell lines in “wound-healing” assays in vitro. EpRas and EpRas-derived cell lines were cultured to confluency in the presence of TGF-β for 3 d, the cell monolayers were scratched in a defined manner, and closure of the “wound” was observed 15 h later. With the exception of Ep-M1-7 cells, all cell lines exhibited comparable, very slow “wound” closure (Figure 3A). The apparent decreased “healing” of Ep-ERFm1-7 cells could be due to the previously suggested function of cytoplasmic ERF in motility (Tarcic et al., 2102) or the antiproliferative effects of nuclear ERF (Le Gallic et al., 1999). Indeed, Ep-M1-7 cells exhibited a significantly lower proliferation rate (Figure 3B), which could account for the observed delay in wound closure. To distinguish between the two possibilities, we determined cell motility by Transwell cell migration assays. An apparent increased motility observed for Ep-wt ERF and Ep-ERFm1-7 cells was not statistically significant. However, migration of Ep-ERF-FSF/FKF cells was significantly slower than that of both the parental cells and the other ERF clones (Figure 3C). The effect of ERF-FSF/FKF may reflect changes at the level of available Erk protein due to loss of Erf–Erk interaction. These data suggest that ERF overexpression may have an indirect effect on cell motility, independent of its ability to inhibit mesenchymal transition.
FIGURE 3:
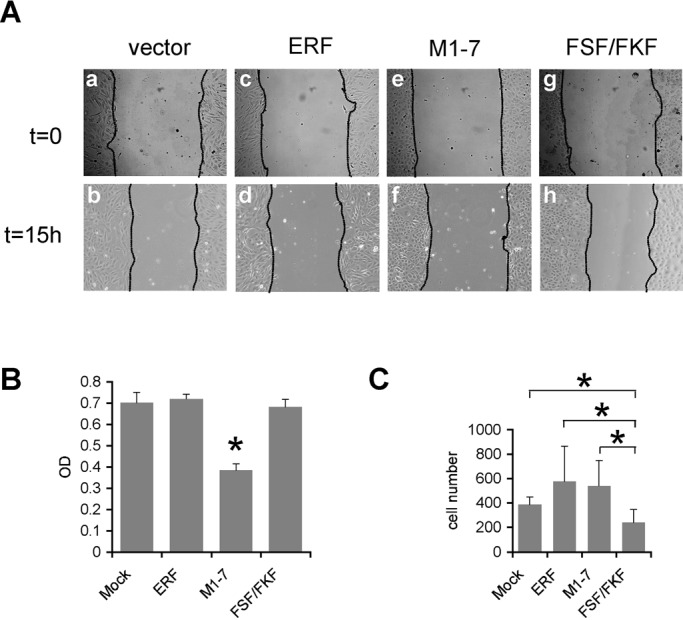
ERF mutations decreased EpRas cell motility. (A) Phase contrast images of vector-transfected (a, b) and ERF-expressing cells (c–h) subjected to “wound-healing” assay in the presence of TGF-β. With the exception of ERFm1-7–expressing cells (f), gap closure for all cell lines was comparable. (B) Cell proliferation rate as assessed colorimetrically with MTT. All cell lines had comparable proliferation rates, except the ERFm1-7–expressing cells, which exhibited statistically significant reduced proliferation compared with all other lines (asterisk). (C) Transwell motility assay of EpRas cells treated with TGF-β for 24 h and induced to migrate using serum as a chemoattractant. A minimum of three random fields from each Transwell were scored for cell presence. The average of four independent experiments is shown. The two-tailed homoscedastic p-values of standard Student's test for wtERF and ERFm1-7 compared with vector-transfected cells were 0.113 and 0.103, respectively. For ERF-FSF/FKF–expressing cells the p-values against Mock-, wtERF-, and ERFm1-7–expressing cells were 0.021, 0.018, and 0.011, respectively. Statistically significant differences are indicated by an asterisk.
We tested whether inhibition of the TGF-β–induced EMT could be attributed to impaired TGF-β signaling, examining the expression of EMT marker genes, targets of TGF-βR signaling (Cano et al., 2000; Dong et al., 2002; Kowanetz et al., 2004). Vector-transfected control cells undergoing EMT showed significant up-regulation of Snail and c-Myc but loss of Id2. All ERF-wt/mutant clones showed a similar up-regulation or down-regulation (Supplemental Figure S4), with the exception of Snail, whose up-regulation was somewhat suppressed by wtERF and ERF-FSF/FKF. We were also unable to detect any changes in Smad2/3 (unpublished data), suggesting that ERF may not affect the TGF-β signaling pathway directly.
ERF-induced transcriptional changes
To identify changes in gene expression that could account for the inhibition of EMT by ERF, we used transcriptome expression profiling. Parental EpRas cells, as well as wt, M1-7, and FSF/FKF ERF–overexpressing cells, were compared under normal growth conditions for 2-h and 4-d exposure to TGF-β from two independent experiments. Unsupervised clustering analysis showed that the 2-h TGF-β samples are clustered together and flanked by the untreated and 4-d-treated samples (Supplemental Figure S5). However, clonal and experimental variation was also evident. Two-way analysis of variance was used to identify genes with at least twofold expression difference and p ≤ 0.05, among cell lines and TGF-β exposure conditions, yielding a significant number of genes altered across cell lines and conditions.
We reasoned that a common subset of genes may be responsible for the resistance to EMT exhibited by all ERF clones. This subset could be distinct from the role of Erf in motility or proliferation. Thus we inquired for genes that were different between the parental EpRas cells and each of the three ERF lines in pairwise comparisons under all conditions used. We identified 7 genes that were different between the parental and all the ERF cell lines in the absence of TGF-β, 11 genes in cells exposed to TGF-β for 2 h, and 30 genes in cells exposed to TGF-β for 4 d (Supplemental Figure S6A). Based on the phenotypic similarities of all ERF clones, this limited list was furthered filtered for genes that were common in any two or all three populations and were also affected by TGF-β treatment in the parental EpRas cells but not the ERF cell lines (Supplemental Figure S6B). Only one gene, Semaphorin-7a, was lower in all ERF clones, was induced in the parental cells after 4 d of TGF-β exposure, and failed to be increased in all three ERF lines. Semaphorin-7a (Sema7a) was the only family member of the semaphorin family that was induced by TGF-β in EpRas cells (Supplemental Figure S7A). Among the known semaphorin effectors—integrins and plexins—only Integrin-α5 was induced by TGF-β, but this was also true in the ERF clones (Supplemental Figure S7B). Of interest, Plexin-C1, an established Semaphorin-7a receptor, is not expressed or induced in the parental EpRas cells and ERF clones, suggesting that Sema7a may involve a distinct set of effectors in EMT. Sema7a has been already suggested to affect TGF-β signaling independent of Smad3 (Kang et al., 2007; Gan et al., 2011) and thus could be a reason for the observed inhibition of EMT by ERF.
Semaphorin-7a is required for EMT
To determine the possible role of Sema7a in the ERF-induced inhibition of EMT, we analyzed the expression pattern of Semaphorin-7a in all EpRas clones during TGF-β treatment, using semiquantitative PCR. Consistent with the microarray data, Sema7a was induced in parental EpRas cells, whereas in all ERF-expressing clones semaphorin levels were considerably decreased and failed to respond to TGF-β treatment (Figure 4A). We also examined the ability of ERF to repress transcription of a reporter gene driven by the Sema7a promoter when cotransfected into a heterologous system. Indeed, a twofold-to-threefold inhibition was observed in the presence of ERF (Figure 4B), suggesting that Erf may affect the expression level of Semaphorin-7a, consistent with the plethora of ets-binding sites in the Sema7a promoter region. Treatment of EpRas cells with a Mek1/2 inhibitor results in the dramatic decrease of Sema7a mRNA levels but not that of other TGF-β–induced genes (Figure 4C), supporting the hypothesis that Erf may regulate Sema7a expression.
FIGURE 4:

Semaphorin-7a inhibition mediates the Erf EMT phenotype. (A) Semiquantitative PCR analysis of the Sema7a mRNA levels among the different EpRas lines in the absence or presence of 5 ng/ml TGF-β for 2 h or 4 d. Sema7a levels for each sample were normalized to CPH and compared with the RNA of the parental EpRas cells growing in normal media. Sema7a mRNA levels are lower in all ERF clones and fail to elevate after 4 d of TGF-β treatment. (B) Luciferase assays from Ref1 cells cotransfected with the pGL3-Sema7a reporter, empty vector, or an ERF-expressing plasmid and pRSV-LacZ as transfection efficiency control. Luciferase activity was normalized to β-galactosidase activity, and they were all compared with vector-transfected cells. Three independent experiments in duplicate were evaluated, indicating a transcriptional repression by ERF. The cartoon shows the structure of the plasmid used with the putative ets-binding sites marked by circles. (C) Semiquantitative PCR analysis of the Sema7a and Snai1 mRNA levels in the parental EpRas cells in the presence or absence of TGF-β and the Mek1/2 inhibitor U0126. Cells were exposed to TGF-β for 5 d and to U0126 twice 16 and 2 h before harvest. The values were normalized to the values of the untreated parental cells. (D) Confocal images of Ep-ERFm1-7 cells expressing either the hygromycin resistance gene or hygromycin and Semaphorin-7a and growing on untreated glass coverslips. Cells were exposed to 5 ng/ml TGF-β for 4 d, fixed, and stained with an anti–E-cadherin antibody (red) and TOPRO-3, a DNA-intercalating dye (blue). Cells overexpressing Semaphorin-7a (bottom) undergo EMT as determined by the loss of E-cadherin. Similar results were obtained from EpRas-ERF sema7a–expressing cells.
We then examined the contribution of Sema7a decrease in the ERF-induced resistance to EMT. We reintroduced Sema7a into the wt ERF– and ERFm1-7–expressing EpRas cells, the two most divergent cell lines, as well as into the parental EpRas cells. Stable cell lines overexpressing Sema7a were selected by hygromycin B, and Sema7a expression was verified by quantitative PCR (Supplemental Figure S8). The response of Sema7a-expressing cells to TGF-β–induced EMT was determined by morphological changes and E-cadherin expression. EpERF and EpM1-7 clones expressing only the hygromycin resistance gene were resistant to EMT, like the parental clones. In contrast, Sema7a expression in these clones reestablished the EMT phenotype in response to TGF-β treatment (Figure 4D). Sema7a overexpression had no apparent effect on the TGF-β response of the EpRas parental cells (Supplemental Figure S9). These data suggest that the Sema7a inhibition by ERF could be contributing to the EMT resistance phenotype.
To determine whether Semaphorin-7a expression is required for TGF-β–induced EMT in EpRas cells independent of ERF, we quenched its expression via small interfering RNA (siRNA) and determined the response to TGF-β treatment. Cell lines expressing 2- to 10-fold lower Sema7a mRNA maintained epithelial morphology and E-cadherin expression after 5 d treatment with TGF-β (Figure 5A), recapitulating the effect of ERF overexpression. This was true for six of seven cell lines tested (Figure 5B), strongly suggesting that in EpRas epithelial cells, Semaphorin-7a expression is required for the manifestation of TGF-β–inducted EMT. Furthermore, cells with decreased Sema7a levels also failed to show increased motility in the presence of TGF-β, another indicator of EMT (Figure 5C). Collectively these data suggest the ERF may effect epithelial-to-mesenchymal transition, modulating the levels of Semaphorin-7a.
FIGURE 5:

Semaphorin-7a is required for EMT. (A) Phase contrast (left) and confocal images (right) of EpRas cells expressing either a scrambled siRNA or a Semaphorin-7a–specific siRNA and growing on untreated glass coverslips. Cells were exposed to 5 ng/ml TGF-β for 5 d, fixed, and stained with an anti–E-cadherin antibody (green) and TOPRO-3, a DNA-intercalating dye (blue). Loss of Semaphorin-7a (bottom) inhibits EMT as determined by the E-cadherin presence. (B) Graphic representation of Sema7a mRNA levels quantified by real-time quantitative PCR (white bars) and the fraction of E-cadherin–positive cells (gray bars) after 5 d of exposure to TGF-β. The E-cadherin–positive cells were quantified by averaging the independent fields of view for each cell line, stained as in A. (C) Wound-healing assay of parental EpRas cells and clones expressing either a scramble or Sema7a siRNA in the presence or absence of TGF-β. Wounds were photographed at 0, 6, and 16 h, and closure was measured at identical points. The 16-h time point is shown. Statistically significant differences (t test, p < 0.05) are indicated by an asterisk.
DISCUSSION
EMT is a key developmental process with a clear role in carcinoma progression and metastasis and has been extensively studied in multiple systems, albeit sometimes with conflicting results. In most but not all systems, TGF-β is essential for EMT (Waerner et al., 2006). In almost all cases, however, oncogenic or elevated Ras signaling is essential too (Waerner et al., 2006; Lahsnig et al., 2008). In addition to these, multiple other signaling pathways and transcriptional regulators contribute to EMT, often dependent on cell type and culture conditions (Huber et al., 2005; Waerner et al., 2006), thus hindering comprehensive analysis of key mechanism in EMT. The use of the Ets-related transcriptional repressor Erf, an established effector of the Ras-induced Erk/MAPK pathway essential for EMT (Janda et al., 2002a), creates the possibility to evaluate direct and indirect roles of transcriptional control during EMT induction. Employment of diverse culture methods allowed us to test EMT induction under conditions in which extracellular and attachment factors would vary. Finally transcriptome analysis allowed us to identify factors downstream of Erf, which may be involved in regulation of EMT by Erf. Our data suggest that ERF expression can inhibit TGF-β–induced EMT, primarily by blocking Semaphorin-7a expression and its induction by TGF-β, and that both Erf and Semaphorin-7a may have a role in regulating EMT.
We recently showed (Tarcic et al., 2102) that cytoplasmic Erf may have a role in epithelial cell motility, whereas the antiproliferative effect was one of the first identified functions of nuclear Erf. These activities may interfere with EMT and enhance or quench the apparent phenotype. A 5- to 10-fold overexpression of wt or mutated ERF in EpRas cells—an established system with which to analyze EMT—was sufficient to affect their ability to undergo TGF-β–induced EMT, although the phenotype was affected by different facets of Erf function. The nuclear and Erk interaction-competent ERFm1-7 exhibited decreased cellular proliferation and limited resistance to EMT when cells were grown on plastic, whereas ERF-FSF/FKF, which is also nuclear but unable to interact with Erks, exhibited somewhat decreased motility and the strongest EMT resistance. Wild-type ERF exhibited intermediate EMT resistance and no motility effects on plastic. When the cells were grown in serum-free three-dimensional collagen cultures, wt and ERF mutants showed a comparable level of EMT inhibition, although ERFm1-7 structures on collagen were considerably smaller, a possibly due to its antiproliferative effect. The increased nuclear localization of Erf in cells growing in collagen suggests that transcriptional inhibition may be the primary mode of action by which Erf inhibits TGF-β–induced EMT. In contrast, the motility differences appear to be primarily associated with the ability of Erf to interact with Erks, although a transcriptional component cannot be excluded.
The similarities in the transcription profile changes shared by all ERF clones support the hypothesis that Erf may affect the EMT program at the transcriptional level both directly and indirectly (Supplemental Figure S6A). It is of interest that overexpression of wt Erf in a cell with activated Ras/Erk pathway may have transcriptional effects, since Erf is predominantly cytoplasmic. However, a proportional increase of nuclear Erf, due to its overexpression may be sufficient to elicit transcriptional responses. In addition, global Erf-binding-site analysis indicated that Erf could be found bound at a number of sites on the chromatin in the presence of activated Erk (unpublished observations), suggesting possible transcriptional effects under these conditions. Finally, it is also conceivable that high levels of cytoplasmic Erf affect gene transcription indirectly.
The expression profiling data and the analysis of TGF-βR-signaling target genes indicated that the TFG-β/Smad pathway remains intact after ERF overexpression (Supplemental Figures S4 and S10 and unpublished data). C-Myc, a known Erf target that was recently implicated in EMT in vivo (Trimboli et al., 2008), also appears unaffected. However, a considerable number of genes were differentially expressed in the parental cells and the ERF clones. We reasoned that genes relevant to EMT would show differential expression in all pairwise comparisons between parental cells and ERF clones—preferably in more than one condition—and should be up-regulated or down-regulated by TGF-β in the parental cells and at the same time less so if at all in the ERF clones. A small number of genes fulfilled these criteria. Some of the identified genes were previously found to be involved in EMT or TGF-β signaling, like Cadherin 10 and Forkhead F2 (Aitola et al., 2000; Walker et al., 2008). Both genes were up-regulated in response to TGF-β in the parental EpRas cells, but they were also up-regulated in two (FoxF2) or all (Cdh10) ERF lines. It is conceivable that modulation of these genes contributed to differences among the different ERF mutations used, but they could not account for the EMT resistance observed by all the ERF lines. Sema7a emerged as only gene that fulfilled all our criteria. It was induced by TGF-β in the parental cells but not in any of the ERF-expressing cell lines, decreased in the absence of TGF-β in all the ERF lines compared with the parental cells, and was lower in all the ERF lines in the presence of TGF-β compared with the parental cells
Semaphorins are extracellular and/or membrane-associated proteins that regulate lymphocyte and neuronal development (Kruger et al., 2005; Suzuki et al., 2008), as well as cancer (Neufeld et al., 2005; Bielenberg and Klagsbrun, 2007). They bind to and signal through plexins and integrins and carry out diverse cell-type- and protein-specific functions (Zhou et al., 2008). Semaphorin-7a, the sole member of a family resembling viral semaphorin-like proteins, has also been implicated in lymphocyte (Czopik et al., 2006) and neuronal development (Pasterkamp et al., 2007). Of interest, Sema7a was found to be regulated by TGF-β and required for Smad3-independent TGF-β signaling in pulmonary fibrosis (Kang et al., 2007). Semaphorin-7a expression appears to be strictly dependent on Erk activity. ERF inhibits Sema7a transcription in transient transfection assays, and reexpression of Sema-7a in ERF-expressing EpRas cells reinstates EMT (Figure 4). Erf-independent inhibition of Semaphorin-7a in EpRas cells abrogates their ability to undergo EMT (Figure 5). Thus Semaphorin-7a appears to play a key role in the process.
In TGF-β–induced pulmonary fibrosis, which likely does not involve hyperactive Ras signaling, Semaphorin-7a protects the cells from undergoing apoptosis through activation of the phosphatidylinositol 3-kinase (PI3K) pathway (Kang et al., 2007). It was not surprising that Sema7a had no detectable effect on survival of EpRas cells (unpublished observations), since EpRas cells are strongly protected from apoptosis through cooperative Erk and PI3K hyperactivation (Janda et al., 2002a). Recent observations suggest that Sema7a plays a key role in cell motility through its interaction with integrin-β1 (Messina et al., 2011) and in metastasis via Plexin-C1 signaling (Lazova et al., 2009; Scott et al., 2009). Our data suggest that Sema7a may have an analogous function in the manifestation of EMT, although they likely implicate different receptors since Plexin-C1 is not expressed in EpRas cells.
It is unclear whether Erf regulates Sema7a transcription directly or indirectly. Promoter assays suggest a possible direct regulation, and the Sema7a dependence on Erk activity favors a direct regulation by Erf. However, the observed inhibition, when compared with the transcriptional repression of Erf on other promoters in transient assays (Sgouras et al., 1995), is rather limited. In addition, both nuclear and nuclear-shuttling forms of Erf exhibit limited differences. Finally, we were not able to detect statistically significant differences of Erf binding on the Sema7a genomic region via chromatin immunoprecipitation assays (unpublished observations). Thus an indirect regulation cannot be excluded, and further experiments are needed to decipher the exact mechanism of Sema7a regulation by Erf.
In conclusion, our data suggest that the strict requirement of hyperactive Ras signaling for TGF-β–induced EMT (Janda et al., 2002b) may be only partially due to the protection from TGF-β–induced apoptosis through PI3K signaling and that hyperactive Erk/MAPK signaling may also be essential for EMT because it abolishes repression of genes required for EMT, such as Semaphorin-7a.
MATERIALS AND METHODS
Cell culture and transfection
EpRas cells were grown in DMEM (Invitrogen, Carlsbad, CA) supplemented with 4% fetal bovine serum (FBS; Hyclone Laboratories, Logan, UT), 2 mM l-glutamine, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7, and 2 mM penicillin/streptomycin (Invitrogen). Ref1 cells were cultured in DMEM supplemented with 10% FBS and 2 mM penicillin/streptomycin. EpRas stable cell lines were established by cotransfecting the pBabe plasmid carrying a puromycin resistance cassette and a pSG5 plasmid expressing ERF or one of its mutants, in a 1:5 ratio. pSG5 plasmids encoding wt ERF (Sgouras et al., 1995), M1-7 ERF (Le Gallic et al., 1999), and FSF/FKF ERF (Polychronopoulos et al., 2006) were previously described. Cells (105) were seeded in 35-mm plates and transfected with 0.8 μg of plasmid DNAs and 4 μl of Lipofectamine (Invitrogen). The cells were selected with 1.5 μg/ml puromycin. Resistant clones were isolated, and ERF expression was verified by immunoblotting.
EpRas, EpERF, and EpM1-7 cells were cotransfected with pGK-Hygro and pCMV-SPOT6-Sema7a (imaGenes, Berlin, Germany) as described and selected with 250 mg/ml hygromycin B. Resulting clones where further selected for 1 wk in the presence of G418, puromycin, and hygromycin to ensure expression of all the transgenes (Ras, Erf, and Sema7a).
EpRas cells were transfected with the pLKO. 1-Semaphorin-7a-siRNA vector (Thermo Scientific, Waltham, MA) and selected with 4 mg/ml puromycin. Resulting clones where tested for the presence of the transgene and the Sema7a mRNA levels.
The pGL3-Sema7a reporter plasmid was generated by PCR of mouse genomic DNA, using the 5′-TTCCcGGGTTGTTCAGGTGACTGC-3′ and 5′-AGCCCtcGAGCGACA GCGGCAAGC-3′ primers and subsequent cloning of the 902–base pair DNA fragment between the SmaI and XhoI sites of the pGL3-basic vector (Promega, Madison, WI ). This DNA segment was sequence verified and corresponds to the Sema7a genomic region from −895 base pair to +6 with respect to the mRNA start. Ref1 cells were transiently transfected as previously described (Sgouras et al., 1995).
Serum-free three-dimensional cultures and growth on porous support have been described (Oft et al., 1996, 1998). See Supplemental Materials and Methods for details.
Immunofluorescence and immunoblotting
Cells on coverslips, porous filters, or collagen gels were fixed, stained with the appropriate antibody, and visualized by confocal microscopy. Subconfluent cultures were used for extracting and analyzing proteins by immunoblotting as previously described (Le Gallic et al., 1999). See Supplemental Materials and Methods for details. The following antibodies were used: rabbit polyclonal antibody S17S against ERF (Sgouras et al., 1995) and rabbit polyclonal antibodies against p42/p44 MAPK (Cell Signaling Technology, Beverly, MA), actin (Sigma-Aldrich, St. Louis, MO), and fibronectin (Santa Cruz Biotechnology, Santa Cruz, CA); mouse monoclonal antibody against E-cadherin (BD Transduction Laboratories, Lexington, KY); horseradish peroxidase anti-rabbit and anti-mouse (Jackson ImmunoResearch Laboratories, West Grove, PA); and S47 conjugated anti-rabbit and anti-mouse (Pierce, Rockford, IL) goat antibodies.
Proliferation and motility assays
Cellular proliferation was assessed colorimetrically with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) cell proliferation kit according to the manufacturer's indications (Roche, Indianapolis, IN). In wound-healing assays 105 cells were seeded in 35-mm plates and allowed to grow for 3 d in the presence of 5 ng/ml TGF-β, scratched, and monitored microscopically. Transwell cell migration assay was performed in chemotaxis chambers (Corning Costar, Cambridge, MA), and 5% FBS was used as the chemoattractant. Cells were fixed, stained with 4′,6-diamidino-2-phenylindole (DAPI), and analyzed by fluorescence microscopy. See Supplemental Materials and Methods for details.
mRNA analysis
RNA was extracted from TGF-β–treated or untreated cells and was reverse transcribed and subjected to semiquantitative PCR with primers specific for c-Myc, Snai1, Id2, Pai1, Sema7a, and Cph as normalization control. For transcriptome analysis Affymetrix GeneChip Mouse Gene 1.0ST DNA arrays (Affymetrix, Santa Clara, CA) were used, and the data were analyzed using the Partek Genomic Suite 6.3 software (Partek, St. Louis, MO). See Supplemental Materials and Methods for details.
Supplementary Material
Acknowledgments
We are grateful to D. Kafetzopoulos (Institute of Molecular Biology and Biotechnology) for help with the DNA microarrays, G. Papagiannakis (Institute of Molecular Biology and Biotechnology) for help with the Affymetrix scans, and the members of the Beug lab for helpful hints to and discussions with M.A. This work was supported by European Union Grant HPRN-CT-2000-00083 to G.M. and H.B., Greek Ministry of Education grants PYTHAGORAS II KA2092 to G.M. and HERAKLEITOS II KA3396 to A.Z., an EMBO short-term fellowship to M.A., and grants from the Austrian Science Foundation (FWF SFB028; FWF P17699-B12) and the Austrian Research Promotion Agency (Project Number 814.184) to H.B. In Memoriam Hartmut Beug (1945–2011).
Abbreviations used:
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
Dulbecco's modified Eagle's medium
- EMT
epithelial-to-mesenchymal transition
- ERF
Ets2-repressor factor
- FBS
fetal bovine serum
- MAPK
mitogen-activated protein kinase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide
- RTK
receptor tyrosine kinase
- TGF-β
transforming growth factor-β
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-04-0276) on August 8, 2012.
*Present address: Institut National de la Santé et de la Recherche Médicale U1065, 06204 Nice Cedex 3, France.
REFERENCES
- Aitola M, Carlsson P, Mahlapuu M, Enerback S, Pelto-Huikko M. Forkhead transcription factor FoxF2 is expressed in mesodermal tissues involved in epithelio-mesenchymal interactions. Dev Dyn. 2000;218:136–149. doi: 10.1002/(SICI)1097-0177(200005)218:1<136::AID-DVDY12>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Athanasiou M, LeGallic L, Watson DK, Blair DG, Mavrothalassitis G. Suppression of the Ewing's sarcoma phenotype by FLI1/ERF repressor hybrids. Cancer Gene Ther. 2000;7:1188–1195. doi: 10.1038/sj.cgt.7700220. [DOI] [PubMed] [Google Scholar]
- Bielenberg DR, Klagsbrun M. Targeting endothelial and tumor cells with semaphorins. Cancer Metastasis Rev. 2007;26:421–431. doi: 10.1007/s10555-007-9097-4. [DOI] [PubMed] [Google Scholar]
- Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. 2003;116:499–511. doi: 10.1242/jcs.00224. [DOI] [PubMed] [Google Scholar]
- Boyer B, Vallâes AM, Edme N. Induction and regulation of epithelial-mesenchymal transitions. Biochem Pharmacol. 2000;60:1091–1099. doi: 10.1016/s0006-2952(00)00427-5. [DOI] [PubMed] [Google Scholar]
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- Christofori G, Semb H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem Sci. 1999;24:73–76. doi: 10.1016/s0968-0004(98)01343-7. [DOI] [PubMed] [Google Scholar]
- Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267–1278. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- Czopik AK, Bynoe MS, Palm N, Raine CS, Medzhitov R. Semaphorin 7A is a negative regulator of T cell responses. Immunity. 2006;24:591–600. doi: 10.1016/j.immuni.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Delannoy-Courdent A, Mattot V, Fafeur V, Fauquette W, Pollet I, Calmels T, Vercamer C, Boilly B, Vandenbunder B, Desbiens X. The expression of an Ets1 transcription factor lacking its activation domain decreases uPA proteolytic activity and cell motility, and impairs normal tubulogenesis and cancerous scattering in mammary epithelial cells. J Cell Sci. 1998;111:1521–1534. doi: 10.1242/jcs.111.11.1521. [DOI] [PubMed] [Google Scholar]
- Dong C, Zhu S, Wang T, Yoon W, Goldschmidt-Clermont PJ. Upregulation of PAI-1 is mediated through TGF-β/Smad pathway in transplant arteriopathy. J Heart Lung Transplant. 2002;21:999–1008. doi: 10.1016/s1053-2498(02)00403-5. [DOI] [PubMed] [Google Scholar]
- Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 2005;24:2375–2385. doi: 10.1038/sj.onc.1208429. [DOI] [PubMed] [Google Scholar]
- Eger A, Stockinger A, Park J, Langkopf E, Mikula M, Gotzmann J, Mikulits W, Beug H, Foisner R. beta-Catenin and TGFbeta signalling cooperate to maintain a mesenchymal phenotype after FosER-induced epithelial to mesenchymal transition. Oncogene. 2004;23:2672–2680. doi: 10.1038/sj.onc.1207416. [DOI] [PubMed] [Google Scholar]
- Fialka I, Schwarz H, Reichmann E, Oft M, Busslinger M, Beug H. The estrogen-dependent c-JunER protein causes a reversible loss of mammary epithelial cell polarity involving a destabilization of adherens junctions. J Cell Biol. 1996;132:1115–1132. doi: 10.1083/jcb.132.6.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Y, et al. Role of semaphorin 7a signaling in transforming growth factor beta1-induced lung fibrosis and scleroderma-related interstitial lung disease. Arthritis Rheum. 2011;63:2484–2494. doi: 10.1002/art.30386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003;4:657–665. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569–581. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW. Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374–383. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, Beug H, Grèunert S. Ras and TGFβ cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol. 2002a;156:299–313. doi: 10.1083/jcb.200109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda E, Litos G, Grunert S, Downward J, Beug H. Oncogenic Ras/Her-2 mediate hyperproliferation of polarized epithelial cells in 3D cultures and rapid tumor growth via the PI3K pathway. Oncogene. 2002b;21:5148–5159. doi: 10.1038/sj.onc.1205661. [DOI] [PubMed] [Google Scholar]
- Jechlinger M, Grunert S, Tamir IH, Janda E, Ludemann S, Waerner T, Seither P, Weith A, Beug H, Kraut N. Expression profiling of epithelial plasticity in tumor progression. Oncogene. 2003;22:7155–7169. doi: 10.1038/sj.onc.1206887. [DOI] [PubMed] [Google Scholar]
- Kang HR, Lee CG, Homer RJ, Elias JA. Semaphorin 7A plays a critical role in TGF-β1-induced pulmonary fibrosis. J Exp Med. 2007;204:1083–1093. doi: 10.1084/jem.20061273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol Int. 2002;26:463–476. doi: 10.1006/cbir.2002.0901. [DOI] [PubMed] [Google Scholar]
- Kowanetz M, Valcourt U, Bergstrom R, Heldin CH, Moustakas A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor beta and bone morphogenetic protein. Mol Cell Biol. 2004;24:4241–4254. doi: 10.1128/MCB.24.10.4241-4254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger RP, Aurandt J, Guan KL. Semaphorins command cells to move. Nat Rev Mol Cell Biol. 2005;6:789–800. doi: 10.1038/nrm1740. [DOI] [PubMed] [Google Scholar]
- Lahsnig C, Mikula C, Petz M, Zulehner M, Schneller G, van Zijl DF, Huber H, Csiszar A, Beug H, Mikulits W. ILEI requires oncogenic for the epithelial to mesenchymal transition of hepatocytes and liver carcinoma progression. Oncogene. 2008;28:638–650. doi: 10.1038/onc.2008.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C, Liehr T, Goen M, Gebhart E, Fleckenstein B, Ensser A. New eukaryotic semaphorins with close homology to semaphorins of DNA viruses. Genomics. 1998;51:340–350. doi: 10.1006/geno.1998.5256. [DOI] [PubMed] [Google Scholar]
- Lazova R, Gould Rothberg BE, Rimm D, Scott G. The Semaphorin 7A receptor Plexin C1 is lost during melanoma metastasis. Am J Dermatopathol. 2009;31:177–181. doi: 10.1097/DAD.0b013e318196672d. [DOI] [PubMed] [Google Scholar]
- Le Gallic L, Sgouras D, Beal G Jr, Mavrothalassitis G. Transcriptional repressor ERF is a Ras/mitogen-activated protein kinase target that regulates cellular proliferation. Mol Cell Biol. 1999;19:4121–4133. doi: 10.1128/mcb.19.6.4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gallic L, Virgilio L, Cohen P, Biteau B, Mavrothalassitis G. ERF nuclear shuttling, a continuous monitor of Erk activity that links it to cell cycle progression. Mol Cell Biol. 2004;24:1206–1218. doi: 10.1128/MCB.24.3.1206-1218.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina A, et al. Dysregulation of Semaphorin7A/β1-integrin signaling leads to defective GnRH-1 cell migration, abnormal gonadal development and altered fertility. Hum Mol Genet. 2011;20:4759–4774. doi: 10.1093/hmg/ddr403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesano R, Soriano JV, Fialka I, Orci L. Isolation of EpH4 mammary epithelial cell subpopulations which differ in their morphogenetic properties. In Vitro Cell Dev Biol Anim. 1998;34:468–477. doi: 10.1007/s11626-998-0080-3. [DOI] [PubMed] [Google Scholar]
- Moreno-Bueno G, et al. Genetic profiling of epithelial cells expressing e-cadherin repressors reveals a distinct role for snail, slug, and e47 factors in epithelial-mesenchymal transition. Cancer Res. 2006;66:9543–9556. doi: 10.1158/0008-5472.CAN-06-0479. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007;98:1512–1520. doi: 10.1111/j.1349-7006.2007.00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld G, Shraga-Heled N, Lange T, Guttmann-Raviv N, Herzog Y, Kessler O. Semaphorins in cancer. Front Biosci. 2005;10:751–760. doi: 10.2741/1569. [DOI] [PubMed] [Google Scholar]
- Oft M, Heider KH, Beug H. TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. 1998;8:1243–1252. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E. TGF-βeta1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996;10:2462–2477. doi: 10.1101/gad.10.19.2462. [DOI] [PubMed] [Google Scholar]
- Ojakian GK, Nelson WJ, Beck KA. Mechanisms for de novo biogenesis of an apical membrane compartment in groups of simple epithelial cells surrounded by extracellular matrix. J Cell Sci. 1997;110:2781–2794. doi: 10.1242/jcs.110.22.2781. [DOI] [PubMed] [Google Scholar]
- Papadaki C, Alexiou M, Cecena G, Verykokakis M, Bilitou A, Cross JC, Oshima RG, Mavrothalassitis G. Transcriptional repressor erf determines extraembryonic ectoderm differentiation. Mol Cell Biol. 2007;27:5201–5213. doi: 10.1128/MCB.02237-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasterkamp RJ, Kolk SM, Hellemons AJ, Kolodkin AL. Expression patterns of semaphorin7A and plexinC1 during rat neural development suggest roles in axon guidance and neuronal migration. BMC Dev Biol. 2007;7:98. doi: 10.1186/1471-213X-7-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polychronopoulos S, Verykokakis M, Yazicioglu MN, Sakarellos-Daitsiotis M, Cobb MH, Mavrothalassitis G. The transcriptional ETS2 repressor factor associates with active and inactive Erks through distinct FXF motifs. J Biol Chem. 2006;281:25601–25611. doi: 10.1074/jbc.M605185200. [DOI] [PubMed] [Google Scholar]
- Scott GA, McClelland LA, Fricke AF, Fender A. Plexin C1, a receptor for semaphorin 7a, inactivates cofilin and is a potential tumor suppressor for melanoma progression. J Invest Dermatol. 2009;129:954–963. doi: 10.1038/jid.2008.329. [DOI] [PubMed] [Google Scholar]
- Sgouras DN, Athanasiou MA, Beal GJ Jr, Fisher RJ, Blair DG, Mavrothalassitis GJ. ERF: an ETS domain protein with strong transcriptional repressor activity, can suppress ets-associated tumorigenesis and is regulated by phosphorylation during cell cycle and mitogenic stimulation. EMBO J. 1995;14:4781–4793. doi: 10.1002/j.1460-2075.1995.tb00160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shook D, Keller R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech Dev. 2003;120:1351–1383. doi: 10.1016/j.mod.2003.06.005. [DOI] [PubMed] [Google Scholar]
- Sun D, Baur S, Hay ED. Epithelial-mesenchymal transformation is the mechanism for fusion of the craniofacial primordia involved in morphogenesis of the chicken lip. Dev Biol. 2000;228:337–349. doi: 10.1006/dbio.2000.9946. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Kumanogoh A, Kikutani H. Semaphorins and their receptors in immune cell interactions. Nat Immunol. 2008;9:17–23. doi: 10.1038/ni1553. [DOI] [PubMed] [Google Scholar]
- Tarcic G, et al. EGR1 and the ERK-ERF axis drive mammary cell migration in response to EGF. FASEB J. 2012;26:1582–1592. doi: 10.1096/fj.11-194654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Trimboli AJ, et al. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res. 2008;68:937–945. doi: 10.1158/0008-5472.CAN-07-2148. [DOI] [PubMed] [Google Scholar]
- Vernon AE, LaBonne C. Tumor metastasis: a new twist on epithelial-mesenchymal transitions. Curr Biol. 2004;14:R719–R721. doi: 10.1016/j.cub.2004.08.048. [DOI] [PubMed] [Google Scholar]
- Verykokakis M, Papadaki C, Vorgia E, Le Gallic L, Mavrothalassitis G. The Ras-dependent Erf control of cell proliferation and differentiation is mediated by c-Myc repression. J Biol Chem. 2007;282:30285–30294. doi: 10.1074/jbc.M704428200. [DOI] [PubMed] [Google Scholar]
- Waerner T, Alacakaptan M, Tamir I, Oberauer R, Gal A, Brabletz T, Schreiber M, Jechlinger M, Beug H. ILEI: a cytokine essential for EMT, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell. 2006;10:227–239. doi: 10.1016/j.ccr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Walker MM, Ellis SM, Auza MJ, Patel A, Clark P. The intercellular adhesion molecule, cadherin-10, is a marker for human prostate luminal epithelial cells that is not expressed in prostate cancer. Mod Pathol. 2008;21:85–95. doi: 10.1038/modpathol.3800988. [DOI] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Gunput RA, Pasterkamp RJ. Semaphorin signaling: progress made and promises ahead. Trends Biochem Sci. 2008;33:161–170. doi: 10.1016/j.tibs.2008.01.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.