

Abstract
Integrins link the cell's cytoskeleton to the extracellular matrix, as well as to receptors on other cells. These links occur not only at focal contacts but also at smaller integrin-containing protein complexes outside of focal contacts. We previously demonstrated the importance of focal contact-independent integrin–cytoskeleton interactions of β2 integrins: activation of adhesion resulted from a release of integrins from cytoskeletal constraints. To determine whether changes in integrin–cytoskeleton interactions were related to activation of the integrin, we used single particle tracking to examine focal contact-independent cytoskeletal associations of αIIbβ3-integrin, in which activation results in a large conformational change. Direct activation of αIIbβ3 by mutation did not mimic activation of lymphocytes with phorbol ester, because it enhanced integrin–cytoskeleton interactions, whereas activation of lymphocytes decreased them. Using additional integrin mutants, we found that both α- and β-cytoplasmic domains were required for these links. This suggests that 1) both β2- and β3-integrins interact with the cytoskeleton outside of focal contacts; 2) activation of a cell and activation of an integrin are distinct processes, and both can affect integrin–cytoskeleton interactions; and 3) the role of the α-subunit in integrin–cytoskeleton interactions in at least some circumstances is more direct than generally supposed.
INTRODUCTION
Integrins are a large family of heterodimeric adhesion molecules, present on nearly every metazoan cell type. For the cell to adhere, spread, or locomote, integrins must link the force-generating elements of the cytoskeleton to extracellular structures. This link consists of two components: the integrin–ligand bond and the integrin–cytoskeleton bond. (Faull and Ginsberg, 1996; Yamada and Geiger, 1997).
Regulation of the integrin–ligand bond is relatively well characterized. It has been shown that αIIbβ3-integrins, for example, have two distinct activation states: an unactivated, low-affinity conformation and an activated, high-affinity conformation. Thus, regulation of ligand binding can be accomplished by controlling integrin conformation. This regulation is functionally important. For example, when αIIbβ3-integrins are locked in the unactivated state, as in some cases of Glanzmann's thrombasthenia, hemostasis is abnormal (Ginsberg et al., 1986). Unactivatable integrins also mediate spreading and adhesion poorly compared with the high-affinity integrin (Peter and O'Toole, 1995). Similarly, dysregulation of the strength of integrin–ligand interactions by activating mutations can influence the efficiency of cell motility (Huttenlocher et al., 1996).
The question arises whether functional effects of integrin activation are due solely to the effect on ligand binding or whether integrin–cytoskeleton interactions are affected by integrin activation as well. Within focal contacts, integrin–cytoskeleton interactions are regulated. For example, integrins form connections to the cytoskeleton in focal contacts in response to activation of Rho (Ridley and Hall, 1992; Zhong et al., 1997); in response to epidermal growth factor, these contacts dissolve and the integrins disperse (Xie et al., 1997). Less is known about regulation of integrin–cytoskeleton interactions that occur outside focal contacts. These interactions tend to be weaker and more transient, making them more difficult to detect and study, but they are likely to be very important in initiation of cell adhesion and in cell motility. It is reasonable that the molecular basis for these interactions might differ from that in the focal contact, because the protein complexes are smaller.
We previously demonstrated that focal contact-independent interactions of β2-integrins with the lymphocyte cytoskeleton are an important component of phorbol 12-myristate 13-acetate-induced activation of lymphocyte adhesion (Kucik et al., 1996). We did this using the biophysical technique of single-particle tracking (SPT), which detects integrin–cytoskeleton interactions, in real time, on living cells, by measuring the thermal motion of the integrins. The basis of the SPT technique is that, when a small bead coated with antibody is placed on a cell and allowed to bind specifically to a membrane protein, displacements of the bead reflect the motion of the membrane protein to which the bead is attached (Sheetz et al., 1989; De Brabander et al., 1991; Qian et al., 1991). When integrins are associated with the cytoskeleton, either in focal contacts (Jacobson et al., 1987) or by weaker or more transient interactions (Kucik et al., 1996), their thermal motion is dramatically lower than when the integrins are free to diffuse (Sheetz et al., 1989; Kucik et al., 1990; Schmidt et al., 1993). Thermal motion of beads attached to membrane proteins can therefore be used to determine and quantify the cytoskeletal interactions of membrane proteins.
The aim of the current study was to begin to understand the role of activation-related integrin conformational changes in integrin–cytoskeleton interactions. Activation of integrins often results from activation of various cell-signaling pathways, e.g., by phorbol ester or other, more physiological signals. Such treatments have widespread effects, however, and a resulting change in integrin–cytoskeleton interactions could not be interpreted unambiguously, because there might be other effects in addition to conformational changes in the integrin. To separate integrin activation from cell activation, we used a mutational approach, with wild-type and mutant αIIbβ3-integrins expressed in Chinese hamster ovary (CHO) cells. αIIbβ3 is a platelet integrin that is well characterized with respect to ligand binding, activation of adhesion, and signaling (Du and Ginsberg, 1997). An advantage of β3-integrins is that, unlike β2 receptors, they undergo large, easily detectable conformational changes in response to activation. Therefore, the activation states of αIIbβ3 are unambiguous. Several mutants of αIIbβ3 have been developed that are locked in either the high- or low-affinity state, independently of the state of activation of the cell (Kurzinger et al., 1982; Hughes et al., 1995; Peter and O'Toole, 1995). Because CHO cells do not normally express αIIbβ3-integrins, transfectants can be developed in which there is no background of wild-type integrins to confuse assays of mutant integrin function. Even though αIIbβ3 is not normally present in CHO cells, the transfected integrin can mediate adhesion, spreading, and locomotion (Polte et al., 1991; O'Toole et al., 1994a; Huttenlocher et al., 1996), all of which are functions of normally expressed integrins. Therefore, the αIIbβ3 CHO transfectants have been used extensively for the study of integrin function and represent an especially useful model for understanding integrin structure-function relationships.
Using SPT to track the thermal motion of αIIbβ3-integrins in CHO cells, we found that a fraction of wild-type β3-integrins interact with the cytoskeleton outside of focal contacts at any given time. A mutation that locks integrins in the high-affinity conformation also led to increased interaction with the cytoskeleton, as shown by decreased integrin diffusion. This restriction of diffusion of αIIbβ3 outside of focal contacts required both the α- and the β-cytoplasmic domains, in contrast to integrin–cytoskeleton interactions in focal contacts, where the β-cytoplasmic domain is necessary and sufficient. Thus, regulation of focal contact-independent integrin functions is distinct from the better studied adhesion plaques. In addition, these findings suggest a molecular mechanism by which cytoskeletal proteins that bind integrin α-subunits can regulate cell motility (Liu et al., 1999).
MATERIALS AND METHODS
Integrin Constructs
Mutations in the cytoplasmic domains of the integrin constructs used in this study are shown in Figure 1. All constructs had the wild-type αIIbβ3-transmembrane and extracellular domains and were recognized by the same monoclonal antibody (D57). Modifications were made to the cytoplasmic tails of these integrins. These included truncation of either the α- or the β-cytoplasmic domains, deletion of the membrane-proximal GFFKR sequence common to all integrin α-subunits, and generation of a chimeric integrin with an activated αL-cytoplasmic domain exchanged for that of αIIb.
Figure 1.

Amino acid sequence of wild-type and variant integrin cytoplasmic domains of the constructs used in this study. Single-letter amino acid codes are used. The positions of stop codons producing cytoplasmic truncations (i.e., α991, α996, and β724) are noted by triangles. The residues deleted in the activated constructs (to make αIIbΔ and αLΔ) are underlined.
Antibody Coating of Beads
Protein A was covalently coupled to aminated latex beads (Polysciences, Warrington, PA), as follows. One milliliter of a 2.5% suspension was washed with 1.5 ml of phosphate-buffered saline (PBS) three times. The pellet was resuspended in 8% glutaraldehyde and incubated overnight at 25°C. After washing three times with PBS, 400 μg of protein A (Sigma, St. Louis, MO) was added, and the solution was mixed gently at 25°C for 5 h. The beads were pelleted and resuspended in 0.5 M ethanolamine for 30 min at 25°C and then pelleted and resuspended in 10 mg/ml bovine serum albumin (BSA) in PBS. After a 30-min 25°C incubation, the beads were washed with BSA/PBS and resuspended in BSA/PBS with 0.1% NaN3 and 5% glycerol. On the day of the experiment, 20 μl of the beads were washed in 100 mM Tris, pH 8.0, and resuspended in the same buffer, and 20 μg of D57 antibody were added. After 2 h at 4°C with gentle mixing, the beads were pelleted and resuspended in the stage media, serum-free Iscove's medium without phenol red (Mediatech, Herndon, VA).
Specific Binding of Antibody-coated Latex Beads to Transfected Integrins
Beads were held against the surface of a cell for 5 s with the laser tweezers. Laser intensity was held at a constant value empirically determined to result in specific bead binding. Beads were then released from the optical trap and scored as to whether they adhered to the cell or not. Because of the large amount of thermal motion of these small beads, those that did not bind to membrane proteins diffused away into the medium, rather than remaining on the cell. On transfected cells, the beads coated with specific antibody (D57) typically adhered on ∼60–80% of attempts; beads coated with rabbit immunoglobulin G typically adhered 0–20% of the time. (D57 is specific for the αIIbβ3 complex. It is not ligand substituting.)
Imaging of Cells and Beads
Cells were plated onto glass coverslips (22 × 22 mm, no. 0, Thomas Scientific, Swedesboro, NJ) and allowed to adhere overnight. Before the experiment, the coverslip was placed in a custom-designed cell chamber. Beads were perfused into the chamber at a concentration that was empirically determined to facilitate easy capture of beads from the medium with laser tweezers. Cells to analyze were chosen at random. The cells were viewed on an Axiovert TV100 inverted microscope (Zeiss, Oberkochen, Germany) equipped with differential interference contrast optics. Images were collected with a model NC-70 (Dage-MTI, Michigan City, IN) or a C2400–07 (Hamamatsu Photonic Systems, Bridgewater, NJ) video camera equipped with a newvicon tube and recorded onto sVHS videotape. Sequences showing bead binding were selected, and these were digitized onto a hard disk in a 2000 GP260 computer (Gateway, North Sioux City, SD) using a Perception PVR-2500 digital recording system (Digital Processing Systems, Markham, Ontario, Canada). Particle positions were determined using Metamorph software (Universal Imaging, West Chester, PA) and converted from pixel to nanometer coordinates by comparison with a known standard. Beads were tracked from the point at which they were released from the laser tweezers, and tracking continued for 15 s. This resulted in a particle track containing 450 data points (at 30 video frames/s), sufficient data to accurately determine a diffusion coefficient, D. Particle tracks were then analyzed using software developed for this purpose (Gelles et al., 1988), as follows. SPT measurements permit the separation of the random and systematic contributions to the motion of an individual bead as previously explained (Sheetz et al., 1989; Qian et al., 1991). Briefly, purely random motion results in a linear increase in mean squared displacement (msd) with elapsed time, t, i.e.,
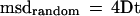 |
1 |
For a constant velocity component of directed motion (such as that contributed by movement of the cell), d = Vt and
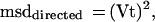 |
2 |
where d = displacement and V = velocity. Thus, for a diffusing particle on the surface of a cell moving at constant velocity,
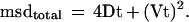 |
3 |
By fitting this quadratic equation to the data, the random diffusion component of motion can be separated from the directed movement of the cell, and a diffusion coefficient can be determined (Kucik et al., 1989; Qian et al., 1991).
Statistical Analysis
Statistical significance of the difference in mobile fractions of the various constructs was tested in all cases by Fisher's exact test, two sided.
RESULTS
Cytoskeletally Restricted and Mobile Populations of αIIbβ3
The degree to which the integrins are constrained by the cytoskeleton can be determined by quantifying the thermal motion of antibody-coated beads specifically bound to them (Qian et al., 1991; Kucik et al., 1996). The measure of this is the diffusion coefficient, D. Because even cytoskeleton-associated integrins have a measurable amount of thermal motion, this quantity can be calculated for all integrins, whether diffusing or not. We first measured the thermal motion of the wild-type integrin, αIIbβ3, using 0.5-μm latex beads coated with D57, a monoclonal antibody that is specific for the αIIbβ3-extracellular domain. These beads, specifically bound to integrins on the dorsal surface of the cell, provide a measure of the cytoskeletal interactions of integrins outside of focal contacts. Computer tracking of the bead trajectories, as described in MATERIALS AND METHODS, results in particle tracks such as those in Figure 2, where it can be readily seen that, although integrins restricted by the cytoskeleton have measurable thermal motion, they are clearly distinguishable from diffusing proteins. Figure 2 also shows mean square displacement (MSD) plots obtained from these particle tracks. By fitting these MSD plots, diffusion coefficients are obtained, as explained in MATERIALS AND METHODS. These diffusion coefficients then provide a quantitative readout of integrin–cytoskeleton interactions.
Figure 2.

Diffusing versus restricted motion. Top, particle tracks of typical diffusing (a and b; mean D = 4 × 10−10 cm2/s) and restricted (c and d; D = 4 × 10−10 cm2/s) integrins. Examples are chosen to represent the average values of each group. Bottom, MSD plots generated from these particle tracks. MSD plots are used to calculate D and are helpful in assessing randomness of motion.
A histogram of diffusion coefficients obtained has a bimodal distribution of measurements (Figure 3a). A cutoff of D ≅ 1 × 10−10 cm2/s is often used to distinguish membrane proteins that are freely diffusing from those constrained by integrin–cytoskeleton interactions (Sako and Kusumi, 1995; Kucik et al., 1996). Therefore, this bimodal distribution likely represents two populations: one diffusing freely in the membrane and one constrained by integrin–cytoskeleton interactions.
Figure 3.

GFFKR loopout mutations lead to increased integrin–cytoskeleton interactions. (a) Wild-type αIIbβ3 has a bimodal distribution of D values, suggesting two populations of integrins, one relatively mobile with respect to the other. For each integrin construct, in three or more experiments on three or more separate days, measurements of integrin mobility were made using SPT. These data were pooled to generate the histograms in this and other figures. In Figure 3a, 40% of the measurements resulted in values greater than D = 1 × 10−10 cm2/s (70 total measurements). The wide distribution of D values is typical of freely diffusing membrane proteins (Kucik et al., 1999). The remaining 60% of measurements yielded a tight distribution of values less than D = 1 × 10−10 cm2/s, consistent with restriction due to interactions with the cytoskeleton. This bimodal distribution is typical of many membrane proteins that are capable of cytoskeletal interactions and has been seen with integrins using fluorescence photobleaching measurements (see text). (b) Loopout of the highly conserved GFFKR sequence in the α-cytoplasmic domain near the membrane is known to activate high-affinity ligand binding. This mutation also results in a shift of integrins from the mobile, diffusing population to the population interacting with the cytoskeleton. (7% mobile, of 66 total measurements). (c) The GFFKR loopout mutation again affects cytoskeletal interactions of a chimeric integrin. When the cytoplasmic domain of αIIbβ3 is replaced by that of αL, a GFFKR loopout mutation also induces a shift to the “immobile,” cytoskeleton-associated population of integrins (19% mobile, 54 measurements). This is not unexpected, because this mutation in this chimeric integrin also has effects on ligand-binding affinity similar to those with the wild type. (d) The data from a to c are plotted as percentages mobile, using a cutoff of D = 1 × 10−10 cm2/s. This allows the shift in populations to be represented by a single number (see text). All three mobile fractions in this figure are significantly different from each other (p < 0.05, Fisher's exact test, two sided).
Two Mutations That Activate Ligand Binding Enhance Cytoskeletal Interactions
There is a highly conserved sequence of five amino acids, GFFKR, in integrin α-chains near the plasma membrane on the cytoplasmic side (Ginsberg, 1995; Marcantonio and David, 1997). This sequence is involved in affinity regulation, because mutations that delete the GFFKR motif lock the αIIbβ3-integrin in a high-affinity state, even if the rest of the α-cytoplasmic domain is intact (O'Toole et al., 1994b). This change in affinity is clearly due to a conformational change, because the monoclonal antibody PAC1 recognizes only the high-affinity state (O'Toole et al., 1990). This mutation also affects integrin function, enhancing spreading and inhibiting cell motility (Huttenlocher et al., 1996). Because these effects on cell function could be consistent not only with increased ligand affinity but also with enhanced interaction of this mutant with the cytoskeleton, we examined the effect of a GFFKR loopout mutation on the cytoskeletal interactions of αIIbβ3. To do this, the thermal motion of D57-coated beads was measured on cells transfected with a mutant integrin missing the GFFKR sequence (αIIbΔβ3; see Figure 1). Figure 3b shows the distribution of diffusion coefficients of this “activated,” high-affinity integrin. Compared with wild-type integrin (Figure 3a), the mobile fraction of the high-affinity form is dramatically reduced, with almost no observed diffusion rates >1 × 10−10 cm2/s. This is consistent with restriction of movement of most of these integrins by the cytoskeleton. Thus, the GFFKR loopout mutation, in addition to activation of ligand binding, induces integrin–cytoskeleton interactions outside of focal contacts that restrict the mobility of this chimeric integrin.
To determine whether the increased cytoskeletal association was specific for the αIIb-cytoplasmic tail or was also a property of other integrins in the high-affinity conformation, we examined αIIbαLΔβ3. This chimeric integrin, with the αL-cytoplasmic domain replacing that of the native αIIb, also is activated to bind ligand by the GFFKR loopout mutation (O'Toole et al., 1994b). Quantitation of integrin–cytoskeleton interactions for this activated construct, αIIbαLΔβ3 (Figure 3c), resulted in a larger immobile fraction than wild-type αIIbβ3 (p < 0.05). Therefore, an activating mutation in two different integrin α-subunit cytoplasmic tails greatly enhanced interactions between the integrin and the cytoskeleton.
To compare the diffusion of wild-type and activated αIIbβ3, the mobile fraction of each integrin was determined using a cutoff of 1 × 10−10 cm2/s to distinguish diffusing membrane proteins from those interacting with the cytoskeleton (Figure 3d). The histograms demonstrate that these changes in distribution of membrane protein mobility measurements can best be characterized not as a shift in the mean of a single population but as a shift from one population to another. Therefore, calculation of mobile fractions is a more appropriate way to characterize the difference induced by a particular mutation than calculation of the mean. Molecularly, the proportion of nondiffusing integrins reflects the probability of a particular form of αIIbβ3 interacting with the cytoskeleton.
α-Cytoplasmic Tail Truncation Does Not Inhibit αIIbβ3 Diffusion
A variety of evidence shows the critical importance of the β-chain cytoplasmic tail for integrin–cytoskeleton interactions. For example, mutant integrins with deleted α-chain cytoplasmic domains can target to focal contacts by interaction of the β-chain cytoplasmic domain with cytoplasmic proteins (Briesewitz et al., 1993; Ylanne et al., 1993). Even a chimera consisting of the β3-cytoplasmic domain and an interleukin 2 receptor transmembrane and extracellular domain will target to focal contacts (LaFlamme et al., 1992). This is strong evidence that integrin–cytoskeleton interactions in focal contacts could be attributed completely to exposure of cytoskeletal interaction sites on the β-cytoplasmic domain.
Because mutations that activate ligand binding also lead to increased integrin–cytoskeleton interactions, it is likely that the affinity-enhancing conformational change alters the cytoplasmic domain interaction sites for cytoskeleton as well. For example, deletion of GFFKR might cause a conformational change in the α-cytoplasmic domain, exposing a pre-existing binding site on the β-cytoplasmic domain. This would allow an as yet unidentified cytoskeletal linker protein to bind to the β-cytoplasmic tail (Figure 4a) and would be consistent with the known dominant role of β-cytoplasmic domains for interaction with cytoskeleton within focal contacts.
Figure 4.

Truncation of the α-cytoplasmic domain does not mimic the activating mutation. (a) Schematic representation of the model tested. The black, filled shape represents a cytoplasmic protein that forms a link between the integrin and the cytoskeleton. Without identifying this protein, it is possible to discriminate among models of how the link to the integrin is formed. In this, the first model, the GFFKR mutation in the α-cytoplasmic domain swings the α-tail away from that of the β-subunit, allowing a cytoplasmic protein to interact with a pre-existing binding site on the β. (b) Inset, schematic representation of the integrin construct used to test the model shown in a. The α-cytoplasmic domain is truncated at amino acid 996, leaving only five amino acids (GFFKR) on the cytoplasmic side of the membrane. This exposes virtually all of the β-cytoplasmic domain. This mutation does not, however, cause a conformational change to activate ligand binding. The frequency histogram shows that a substantial fraction (27%; n = 82) of the αIibΔ996β3 are freely diffusing. This mobile fraction is significantly different from that of the activated construct, αIIbΔβ3 (7% mobile; p < 0.05). (c) The histogram in b is represented as a mobile fraction (percentage with D > 1 × 10−10 cm2/s and is compared with αIIbΔβ3 (which has the activating mutation and both cytoplasmic domains intact) and with wild-type data from Figure 3.
To test the possibility that β-cytoplasmic domain sites are sufficient for integrin interaction with cytoskeleton outside of focal contacts, we measured the thermal motion of the α-chain truncation mutant αIIbΔ996β3, in which the α-chain cytoplasmic domain terminates immediately distal to the GFFKR sequence (Figure 1). This mutation exposes the β-cytoplasmic domain, but does not induce the high-affinity conformation, and is depicted in cartoon form in the inset of Figure 4b. αIIbΔ996β3 had a larger mobile population than did αIIbΔβ3 (Figure 4, b and c), suggesting that merely unmasking sequences on the β-subunit is not sufficient to induce the enhanced integrin–cytoskeleton interactions typical of the GFFKR loopout mutations.
A second possible model is that a combination of unmasking of the β-cytoplasmic domain and the high-affinity conformation of the integrin are required for the observed enhancement of cytoskeletal interaction with the GFFKR loopout (Figure 5a). To test this model, we used another α-truncation integrin construct, αIIbΔ991β3, that differs from the αIIbΔ996β3 in that it is truncated five amino acids closer to the cell membrane, removing the GFFKR sequence (Figure 1). This truncation induces the high-affinity conformation of αIIbβ3 (O'Toole et al., 1994b) as well as unmasking or activating cytoskeleton-binding sites on the β-cytoplasmic domain. Integrin mobility measurements, however, showed that this construct also has a large mobile fraction (Figure 5, b and c). This demonstrates that, surprisingly, outside of focal contacts, exposure of sequences on the β-cytoplasmic domain is not sufficient to induce integrin–cytoskeleton interactions, even in the high-affinity (activated) integrin conformation.
Figure 5.

Truncation of the α-cytoplasmic domain, even with a conformational change inducing high-affinity ligand binding, still does not mimic the activating mutation. (a) Schematic representation of the model tested. This model is distinct from that represented in Figure 4 in that the GFFKR loopout in the α-cytoplasmic domain not only exposes the β-cytoplasmic domain but also induces an “activating” conformational change that results in a new binding site on the β. (b) Inset, schematic representation of the integrin construct to test the model shown in a. The α-cytoplasmic domain is truncated at amino acid 991, deleting virtually all of the α-cytoplasmic domain, including the GFFKR sequence. This mutation also causes a conformational change in the integrin that results in high-affinity ligand binding and might be expected to alter cytoskeletal interactions. Frequency histogram. A large fraction (35%; n = 80) of the measurements are consistent with freely diffusing integrins, compared with 7% with the GFFKR loopout mutation (αIIbΔβ3). This difference is significant (p < 0.05). Therefore, this construct, with an α-tail truncation that also causes a ligand-binding–activating conformational change, does not mimic the activating mutation with respect to cytoskeletal interactions. (c) The histogram in b is represented as a mobile fraction (percentage with D ≥ 1 × 10−10 cm2/s) and compared with wild-type and GFFKR loopout data from Figure 3.
The β-Cytoplasmic Domain Also Plays a Positive Role in Integrin–Cytoskeleton Interactions Outside of Focal Contacts
The above data clearly demonstrate that sequences in the α-chain cytoplasmic tail distal to GFFKR are necessary for activation-induced enhancement of integrin–cytoskeleton interactions. The relevant cytoskeletal interactions are present in both αL- and αIIb-cytoplasmic domains. This raises the question of whether the α-chain alone is sufficient to mediate focal contact-independent integrin–cytoskeleton interactions, e.g., by binding directly to a cytoskeletal linker protein (Figure 6a). In this model, enhanced integrin–cytoskeleton interactions would not require the β-cytoplasmic domain. To test this model, we measured the mobility of αIIbαLΔβ3Δ724, a construct that combines a β-cytoplasmic domain truncation with an activating mutation in the α-cytoplasmic domain. As shown in Figure 6, b and c, this construct has a large mobile fraction. Therefore, truncation of the β-cytoplasmic domain abrogates the increased integrin–cytoskeleton interactions induced by the GFFKR loopout mutation, showing that the β-subunit, as well as the α, is required for the increased integrin–cytoskeleton interactions induced by the activating mutation.
Figure 6.

GFFKR loopout in the α-cytoplasmic domain, combined with truncation of the β-cytoplasmic domain, does not mimic the activating mutation. (a) Schematic representation of the model tested. In this model, the GFFKR loopout mutation on the α-cytoplasmic domain affects only the α-subunit with respect to cytoskeletal interactions. This mutation is depicted as inducing a binding site on the α-cytoplasmic domain that allows it to interact with a cytoplasmic protein that forms a link to the cytoskeleton. In this model, the β-cytoplasmic domain would be unnecessary. (b) Inset, schematic representation of the integrin construct to test the model shown in a. The β-cytoplasmic domain is truncated at amino acid 724, deleting virtually all of the β-cytoplasmic domain. This β-domain truncation does not, by itself, cause a conformational change to activate ligand binding. The α-cytoplasmic domain in this construct, however, is an activated form, with a GFFKR loopout mutation. Therefore, this construct is in an activated conformation with respect to ligand binding. This construct tests the hypothesis that this conformation of the α-cytoplasmic domain is sufficient to enhance cytoskeletal interactions. Frequency histogram. A substantial fraction of the measurements are consistent with freely diffusing integrins. (c) The data are plotted in terms of mobile fractions. Of 56 total measurements, 66% resulted in a D < 1 × 10−10 cm2/s with the β-cytoplasmic domain truncation mutant, compared with 19% with the GFFKR loopout construct (αIIbαLΔβ3). This difference is significant (p < 0.05). Therefore, this construct, with a GFFKR loopout in the α-tail, but a β-tail truncation, does not mimic the activating mutation.
DISCUSSION
Much remains to be learned about integrin–cytoskeleton interactions outside of focal contacts. To examine how activation of an integrin affects these interactions, we used a system of genetically modified αIIbβ3-integrins that are well characterized with respect to activating mutations that enhance ligand binding. This mutational approach enabled us to activate the integrin directly to study the effect of integrin activation without the need to activate cell-signaling pathways, which might complicate interpretation of results. By combining molecular biology with the biophysical technique of SPT, we gained important information about which parts of the integrin are necessary and/or sufficient to mediate focal contact-independent interactions, even without identifying the cytoplasmic proteins involved.
Our measurements demonstrated that the wild-type αIIbβ3 is present in two populations when transfected into CHO cells: one freely mobile, diffusing population and another with restricted motion. This was not surprising. The existence of two such populations is common in protein mobility measurements, especially for proteins known to interact with the cytoskeleton (Jacobson et al., 1987). For example, the mobile fraction of β1-integrins on locomoting chick embryo heart fibroblasts (which did not have well established focal contacts) was 73%, as measured by fluorescence photobleaching (Duband et al., 1988); thermal motion of the remaining fraction was restricted by the cytoskeleton. In our system, for the wild-type αIIbβ3-integrin, the mobile and immobile populations were of approximately equal size (Figure 3). This implies that, as in many biological systems, integrin binding to the cytoskeleton is not an all-or-nothing phenomenon but can increase or decrease in response to signals.
The highly conserved GFFKR sequence in the α-cytoplasmic domain is involved in regulation of integrin function, and, for αIIbβ3 in particular, the GFFKR loopout mutation activates ligand binding. This mutation also has effects on integrin-mediated cell functions such as adhesion, spreading, and locomotion. Our measurements demonstrated that the GFFKR loopout increases interactions of the integrin with the cytoskeleton outside of focal contacts, shifting most integrins into the immobile (cytoskeleton-associated) population. This has important implications for interpretation of the effects of these mutations on cell functions. That is, although activation of integrins certainly increases ligand-binding affinity, the effect of integrin activation on integrin–cytoskeleton interactions outside of focal contacts is to decrease integrin diffusion, which likely also will have significant functional consequences.
The cause and effect relationship between increased ligand-binding affinity and increased integrin–cytoskeleton interactions remains to be determined. It may be that, when the integrins assume the high-affinity conformation, this increases their interactions with the cytoskeleton as well. Alternatively, it may be that binding to the cytoskeleton results in high-affinity conformation. Our data do not address this issue.
A related issue is whether ligand binding itself might affect integrin–cytoskeleton interactions. In the β1-integrin system, this is the case. In SPT experiments, Felsenfeld et al. (1996) found that addition of RGD peptide caused attachment of integrins to the rearward-moving cytoskeleton. In the same system, Choquet et al. (1997) found that ligand occupancy led to strengthening of integrin–cytoskeleton linkages. We were unable to demonstrate an effect of RGDS peptide binding in our system (Kucik and Busettini, unpublished results). This may be due to a difference in the effect on cytoskeletal interactions of ligand binding, or a difference in affinity for soluble ligand, in the two integrins systems.
Felsenfeld et al. (1996) found that, in their system, when the β1-integrins attached to the cytoskeleton in response to ligand binding, they were transported toward the center of the cell by the movement of the actin cytoskeleton. Therefore, we analyzed our data for a component of directed motion (independent of the changes in thermal motion). This can be done by a statistical method that determines the likelihood of a given particle track having arisen from random movements alone (as described in MATERIALS AND METHODS). We found that few integrins, either wild-type (2/40) or the activated αIIbΔβ3-construct (13/66) had a significant component of directed motion. Although this difference is significant, it does not demonstrate a clear relationship between cytoskeletal attachment and directed motion. This may be due to the fact that our CHO cells are less motile, with less pronounced cytoskeletal motion, than the fibroblasts used in the study of Felsenfeld et al. (1996), making directed motion less detectable on the time course of our measurements (15 s).
An important question is whether αIIbβ3-integrin activation increases integrin interaction with the cytoskeleton via the α- or the β-integrin subunit. The β-cytoplasmic domain is sufficient for localization of integrins, and even chimeric molecules, to focal contacts (LaFlamme et al., 1992). Therefore, although several cytoplasmic proteins have been shown to bind to integrin α-subunits (Shattil and Ginsberg, 1997; Yamada and Geiger, 1997; Liu et al., 1999), the significance of binding of cytoskeletal proteins to the α-cytoplasmic domain has not always been obvious. A key observation is that, whereas the α-cytoplasmic domain seems to play a passive role in localization of integrins to focal contacts, it plays a more active role in some integrin-mediated cell functions, such as cell motility and collagen fibril formation (Kassner and Hemler, 1993; Kawaguchi and Hemler, 1993; Shaw and Mercurio, 1993; Filardo and Cheresh, 1994; Kassner et al., 1994). It was recently demonstrated that the integrin α4-chain binds specifically to paxillin and that this binding correlates with increased cell migration, decreased spreading, and stress fiber and focal contact formation (Liu et al., 1999). Therefore, integrin–cytoskeleton interactions that require a contribution by the α-subunit may be more important for functions like cell motility that make use of integrins outside of focal contacts. This is consistent with the finding that, although CHO cells expressing an αIIbβ3-construct with a β-cytoplasmic domain truncation do not localize to focal contacts (Ylanne et al., 1993), they are capable of locomotion, a function that requires integrin–cytoskeleton interactions (Huttenlocher et al., 1996). Our data indicate that enhancement of integrin–cytoskeleton interactions outside of focal contacts by integrin activation depends on the presence of both the α- and the β-cytoplasmic domain, because deletion of either the α- or the β-cytoplasmic domain abrogated the effect of integrin activation on integrin mobility. Because neither the α- nor β-cytoplasmic domain alone is sufficient to mediate this interaction, the enhanced cytoskeletal interactions associated with activation must involve the cytoplasmic tails of both subunits.
Experiments with the αL-chimera address the generality of the α-subunit interaction. A GFFKR loopout mutation in this construct also increases interactions with the cytoskeleton compared with the wild-type but not as completely as the same mutation in the αIIb-cytoplasmic domain. Two possible explanations for this difference should be considered. The first is that interactions of the αL-cytoplasmic domain with the cytoskeleton are not as strong as those of the activated αIIb. Given that the mobile fraction of the αL-construct is similar to that of the Δ996 α-tail truncation, however, a second possibility is that the αL might have no direct interaction with the cytoskeleton, but, in contrast to the αIIb, might simply expose a site for cytoskeleton interaction on the β-cytoplasmic domain. We favor the former explanation, for two reasons. First, the complete removal of the α-chain cytoplasmic tail (αIIbΔ991), where the β-chain cytoplasmic tail is potentially most exposed, results in little restriction of diffusion compared with wild-type αIIbβ3. This suggests that the exposed β on its own does not result in significant restriction of diffusion. Second, The combination of a β-cytoplasmic domain truncation with an activated αL-tail results in a high mobile fraction, higher even than the wild type. However, there is still a substantial fraction of these integrins restricted by the cytoskeleton. It is, of course, possible that the activating mutation exposes a cryptic binding site on the α-cytoplasmic domain that does not normally bind cytoskeletal proteins; such possibilities are inherent in the mutational approach. Although we cannot rule this out, nor can we rule out interactions of the extracellular domains with immobile membrane proteins, the simplest explanation is that this restriction of integrin mobility is due to interactions of the cytoskeleton with a physiological binding site on the α-subunit.
We found earlier (Kucik et al., 1996) that activation of lymphocytes by phorbol ester involves a release of integrins from cytoskeletal constraints. The current study demonstrates that direct activation of integrins has the opposite effect. This implies that the release of cytoskeletal constraints in response to phorbol ester operates by a mechanism other than integrin activation. What, then, is the role of integrin activation in adhesion-related integrin–cytoskeleton interactions? We have speculated that, although release of integrins from cytoskeletal constraints is an early event in activation of adhesion, later events probably require a reassociation (Kucik et al., 1996). Indeed, strong adhesion does require integrin binding to cytoskeletal proteins. A variety of studies have demonstrated that both β-cytoplasmic domain truncation and disruption of the cytoskeleton with cytochalasin D, although not interfering with integrin-ligand binding, do prevent the development of strong cell adhesion (Hibbs et al., 1991; Peter and O'Toole, 1995). Because integrin activation involves a conformational change in the integrin, it is reasonable that it might trigger both ligand binding and connections to the cytoskeleton. Thus, phorbol esters trigger an early step in activation of lymphocyte adhesion by inducing release of cytoskeletal constraints, allowing the integrin to diffuse to ligand. Ligand binding will be simultaneous with or followed by integrin activation, which would stabilize the integrin-ligand bond and, as demonstrated in this study, induce reassociation of the integrin with the cytoskeleton. Indeed, integrin binding to ligand can itself cause association with the cytoskeleton (Felsenfeld et al., 1996). All these steps might occur before integrin aggregation consequent to focal contact formation, where interactions with cytoskeleton apparently become independent of the α-chain cytoplasmic domain.
Much remains to be learned about the molecular basis of integrin–cytoskeleton interactions outside of focal contacts. At this point, we cannot distinguish whether the conformational change associated with activation induces a new binding site, requiring both cytoplasmic domains, or brings two halves of a pre-existing binding site into a new alignment such that they can both participate in an interaction. Importantly, however, the requirement for the α-cytoplasmic domain of integrins is primarily important for focal contact-independent cytoskeletal interactions.
Our model builds on a previously published model of affinity modulation by GFFKR loopout mutations (O'Toole et al., 1994b). In that model, the GFFKR sequence constituted a hinge region, important in transmitting a conformational change in the integrin in response to a force supplied by an integrin activation complex (IAC). Deletion of the GFFKR sequence mimicked IAC action by artificially breaking the hinge to induce the activated conformation. Our data are consistent with membrane distal cytoskeleton interaction sites on both α and β exposed by the hinge mutation and presumably by the IAC. The fact that the molecular basis of integrin–cytoskeleton interactions differs between focal contacts and other parts of the cell has implications for how the cell regulates which function its integrins will perform.
We speculate that firm adhesion (mediated by focal contacts) may involve an integrin linked to one or more cytoplasmic proteins via its β-cytoplasmic domain; other functions, such as cell motility, or activation of leukocyte adhesion, may involve links that require both subunits. This is illustrated in Figure 7, which, although it depicts development of leukocyte adhesion, is meant to illustrate the concept of multiple modes of integrin interaction with the cytoskeleton, which is applicable to other integrin-mediated cell functions as well. This model suggests that it is important to begin to think of regulation of integrin–cytoskeleton interactions not simply as whether or not integrins are linked to the cytoskeleton, but how they are linked. The possibility of multiple forms of mechanical links between integrins and the cytoskeleton, dependent on the activation state and location (in or out of focal contacts) of the integrin suggests an additional level of cellular control over integrin function.
Figure 7.

A model of integrin function determined by alternative connections to the cytoskeleton. In this example, activation of leukocyte adhesion is depicted, but the concept is applicable for other integrin-mediated cell functions as well. In a, neither the integrin nor the cell are activated. The integrin's mobility in the membrane is restricted by a link to the cytoskeleton. In b, the cell is activated, e.g., by phorbol ester, and the integrin becomes free to diffuse and bind ligand. In c, several integrins have bound ligand, which either induces or is closely followed by integrin activation, and now a new link to the cytoskeleton is formed, requiring both integrin cytoplasmic domains. In d, integrins have clustered, and firm adhesion has developed, and now the β-cytoplasmic domain is sufficient for cytoskeletal association.
In summary, this work represents a first step in an approach combining biophysical measurements with molecular biology to investigate the regulation of integrin–cytoskeleton interactions in living cells. We have focused on interactions outside of focal contacts and have shown that an activating mutation that enhances ligand binding and has effects on cell spreading and motility also affects integrin–cytoskeleton interactions. We have determined that relatively intact cytoplasmic domains of both subunits are required for this effect. This finding has important implications for integrin function and its regulation.
ACKNOWLEDGMENTS
D.K. is supported by a Career Development Award from the Department of Veterans Affairs.
Abbreviations used:
- BSA
bovine serum albumin
- CHO
Chinese hamster ovary
- IAC
integrin activation complex
- MSD
mean square displacement
- PBS
phosphate-buffered saline
- SPT
single-particle tracking
REFERENCES
- Briesewitz R, Kern A, Marcantonio EE. Ligand-dependent and -independent integrin focal contact localization: the role of the alpha chain cytoplasmic domain. Mol Biol Cell. 1993;4:593–604. doi: 10.1091/mbc.4.6.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquet D, Felsenfeld DP, Sheetz MP. Extracellular matrix rigidity causes strengthening of integrin–cytoskeleton linkages. Cell. 1997;88:39–48. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- De Brabander M, Nuydens R, Ishihara A, Holifield B, Jacobson K, Geerts H. Lateral diffusion and retrograde movements of individual cell surface components on single motile cells observed with Nanovid microscopy. J Cell Biol. 1991;112:111–124. doi: 10.1083/jcb.112.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Ginsberg MH. Integrin αIIbβ3 and platelet function. Thromb Haemostasis. 1997;78:96–100. [PubMed] [Google Scholar]
- Duband JL, Nuckolls GH, Ishihara A, Hasegawa T, Yamada KM, Thiery JP, Jacobson K. Fibronectin receptor exhibits high lateral mobility in embryonic locomoting cells but is immobile in focal contacts and fibrillar streaks in stationary cells. J Cell Biol. 1988;107:1385–1396. doi: 10.1083/jcb.107.4.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faull RJ, Ginsberg MH. Inside-out signaling through integrins. J Am S Nephrol. 1996;7:1091–1097. doi: 10.1681/ASN.V781091. [DOI] [PubMed] [Google Scholar]
- Felsenfeld DP, Choquet D, Sheetz MP. Ligand binding regulates the directed movement of β1 integrins on fibroblasts. Nature. 1996;383:438–440. doi: 10.1038/383438a0. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Cheresh DA. A β turn in the cytoplasmic tail of the integrin αv subunit influences conformation and ligand binding of αvβ3. J Biol Chem. 1994;269:4641–4647. [PubMed] [Google Scholar]
- Gelles J, Schnapp BJ, Sheetz MP. Tracking kinesin-driven movements with nanometre-scale precision. Nature. 1988;331:450–453. doi: 10.1038/331450a0. [DOI] [PubMed] [Google Scholar]
- Ginsberg MH. Integrins: dynamic regulation of ligand binding. Biochem Soc Trans. 1995;23:439–446. doi: 10.1042/bst0230439. [DOI] [PubMed] [Google Scholar]
- Ginsberg MH, Lightsey A, Kunicki TJ, Kaufmann A, Marguerie G, Plow EF. Divalent cation regulation of the surface orientation of platelet membrane glycoprotein IIb: correlation with fibrinogen binding function and definition of a novel variant of Glanzmann's thrombasthenia. J Clin Invest. 1986;78:1103–1111. doi: 10.1172/JCI112667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs ML, Xu H, Stacker SA, Springer TA. Regulation of adhesion to ICAM-1 by the cytoplasmic domain of LFA-1 integrin β subunit. Science. 1991;251:1611–1613. doi: 10.1126/science.1672776. [DOI] [PubMed] [Google Scholar]
- Hughes PE, O'Toole TE, Ylanne J, Shattil SJ, Ginsberg MH. The conserved membrane-proximal region of an integrin cytoplasmic domain specifies ligand binding affinity. J Biol Chem. 1995;270:12411–12417. doi: 10.1074/jbc.270.21.12411. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A, Ginsberg MH, Horwitz AF. Modulation of cell migration by integrin-mediated cytoskeletal linkages and ligand-binding affinity. J Cell Biol. 1996;134:1551–1562. doi: 10.1083/jcb.134.6.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K, Ishihara A, Inman R. Lateral diffusion of proteins in membranes. Annu Rev Physiol. 1987;49:163–175. doi: 10.1146/annurev.ph.49.030187.001115. [DOI] [PubMed] [Google Scholar]
- Kassner PD, Hemler ME. Interchangeable alpha chain cytoplasmic domains play a positive role in control of cell adhesion mediated by VLA-4, a beta 1 integrin. J Exp Med. 1993;178:649–660. doi: 10.1084/jem.178.2.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassner PD, Kawaguchi S, Hemler ME. Minimum α chain cytoplasmic tail sequence needed to support integrin-mediated adhesion. J Biol Chem. 1994;269:19859–19867. [PubMed] [Google Scholar]
- Kawaguchi S, Hemler ME. Role of the α subunit cytoplasmic domain in regulation of adhesive activity mediated by the integrin VLA-2. J Biol Chem. 1993;268:16279–16285. [PubMed] [Google Scholar]
- Kucik DF, Dustin ML, Miller JM, Brown EJ. Adhesion activating phorbol ester increases the mobility of leukocyte integrin LFA-1 in cultured lymphocytes. J Clin Invest. 1996;97:2139–2144. doi: 10.1172/JCI118651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucik DF, Elson EL, Sheetz MP. Forward transport of glycoproteins on leading lamellipodia in locomoting cells [see comments] Nature. 1989;340:315–317. doi: 10.1038/340315a0. [DOI] [PubMed] [Google Scholar]
- Kucik DF, Elson EL, Sheetz MP. Cell migration does not produce membrane flow. J Cell Biol. 1990;111:1617–1622. doi: 10.1083/jcb.111.4.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucik DF, Elson EL, Sheetz MP. Weak dependence of mobility of membrane protein aggregates on aggregate size supports a viscous model of retardation of diffusion. Biophys J. 1999;76:314–322. doi: 10.1016/S0006-3495(99)77198-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzinger K, Ho M-K, Springer TA. Structural homology of a macrophage differentiation antigen and an antigen involved in T-cell-mediated killing. Nature. 1982;296:668–670. doi: 10.1038/296668a0. [DOI] [PubMed] [Google Scholar]
- LaFlamme SE, Akiyama SK, Yamada KM. Regulation of fibronectin receptor distribution. J Cell Biol. 1992;117:437–447. doi: 10.1083/jcb.117.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Thomas SM, Woodside DG, Rose DM, Kiosses WB, Pfaff M, Ginsberg MH. Binding of paxillin to α4 integrins modifies integrin-dependent biological responses. Nature. 1999;402:676–681. doi: 10.1038/45264. [DOI] [PubMed] [Google Scholar]
- Marcantonio EE, David FS. Integrin receptor signaling: the propagation of an alpha-helix model. Matrix Biol. 1997;16:179–184. doi: 10.1016/s0945-053x(97)90006-8. [DOI] [PubMed] [Google Scholar]
- O'Toole TE, Katagiri Y, Faull RJ, Peter K, Tamura R, Quaranta V, Loftus JC, Shattil SJ, Ginsberg MH. Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol. 1994a;124:1047–1059. doi: 10.1083/jcb.124.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole TE, Katagiri Y, Faull RJ, Peter K, Tamura R, Quaranta V, Loftus JC, Shattil SJ, Ginsberg MH. Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol. 1994b;124:1047–1059. doi: 10.1083/jcb.124.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole TE, Loftus JC, Du XP, Glass AA, Ruggeri ZM, Shattil SJ, Plow EF, Ginsberg MH. Affinity modulation of the αIIbβ3 integrin (platelet GPIIb-IIIa) is an intrinsic property of the receptor. Cell Regul. 1990;1:883–893. doi: 10.1091/mbc.1.12.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter K, O'Toole TE. Modulation of cell adhesion by changes in αLβ2 (LFA-1, CD11a/CD18) cytoplasmic domain/cytoskeleton interaction. J Exp Med. 1995;181:315–326. doi: 10.1084/jem.181.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polte T, Newman W, Raghunathan G, Venkat Gopal T. Structural and functional studies of full-length vascular cell adhesion molecule-1: internal duplication and homology to several adhesion proteins. DNA Cell Biol. 1991;10:349–357. doi: 10.1089/dna.1991.10.349. [DOI] [PubMed] [Google Scholar]
- Qian H, Sheetz MP, Elson EL. Single particle tracking: analysis of diffusion and flow in two-dimensional systems. Biophys J. 1991;60:910–921. doi: 10.1016/S0006-3495(91)82125-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Sako Y, Kusumi A. Barriers for lateral diffusion of transferrin receptor in the plasma membrane as characterized by receptor dragging by laser tweezers: fence versus tether. J Cell Biol. 1995;129:1559–1574. doi: 10.1083/jcb.129.6.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CE, Horwitz AF, Lauffenburger DA, Sheetz MP. Integrin-cytoskeletal interactions in migrating fibroblasts are dynamic, asymmetric, and regulated. J Cell Biol. 1993;123:977–991. doi: 10.1083/jcb.123.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattil SJ, Ginsberg MH. Integrin signaling in vascular biology. J Clin Invest. 1997;100(suppl):S91–S95. [PubMed] [Google Scholar]
- Shaw LM, Mercurio AM. Regulation of α6β1 integrin laminin receptor function by the cytoplasmic domain of the α6 subunit. J Cell Biol. 1993;123:1017–1025. doi: 10.1083/jcb.123.4.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheetz MP, Turney S, Qian H, Elson EL. Nanometre-level analysis demonstrates that lipid flow does not drive membrane glycoprotein movements. Nature. 1989;340:284–288. doi: 10.1038/340284a0. [DOI] [PubMed] [Google Scholar]
- Xie H, Pallero MA, Gupta K, Chang P, Ware MF, Witke W, Kwiatkowski DJ, Lauffenburger DA, Murphy-Ullrich JE, Wells A. EGF receptor regulation of cell motility: EGF induces disassembly of focal adhesions independently of the motility-associated PLCgamma signaling pathway. J Cell Sci. 1998;111:615–624. doi: 10.1242/jcs.111.5.615. [DOI] [PubMed] [Google Scholar]
- Yamada KM, Geiger B. Molecular interactions in cell adhesion complexes. Curr Opin Cell Biol. 1997;9:76–85. doi: 10.1016/s0955-0674(97)80155-x. [DOI] [PubMed] [Google Scholar]
- Ylanne J, Chen Y, O'Toole TE, Loftus JC, Takada Y, Ginsberg MH. Distinct functions of integrin alpha and beta subunit cytoplasmic domains in cell spreading and formation of focal adhesions. J Cell Biol. 1993;122:223–233. doi: 10.1083/jcb.122.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong C, Kinch MS, Burridge K. Rho-stimulated contractility contributes to the fibroblastic phenotype of Ras-transformed epithelial cells. Mol Biol Cell. 1997;8:2329–2344. doi: 10.1091/mbc.8.11.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]