

Abstract
The KIR complex appears to be evolving rapidly in humans, and more than 50 different haplotypes have been described, ranging from four to 14 KIR loci. Previously it has been suggested that most KIR haplotypes consist of framework genes, present in all individuals, which bracket a variable number of other genes. We used a new technique to type 793 families from the United Kingdom and United States for both the presence/absence of all individual KIR genes as well as copy number and found that KIR haplotypes are even more complex. It is striking that all KIR loci are subject to copy number variation (CNV), including the so-called framework genes, but CNV is much more frequent in KIR B haplotypes than KIR A haplotypes. These two basic KIR haplotype groups, A and B, appear to be following different evolutionary trajectories. Despite the great diversity, there are 11 common haplotypes, derived by reciprocal recombination near KIR2DL4, which collectively account for 94% of KIR haplotypes determined in Caucasian samples. These haplotypes could be derived from combinations of just three centromeic and two telomeric motifs, simplifying disease analysis for these haplotypes. The remaining 6% of haplotypes displayed novel examples of expansion and contraction of numbers of loci. Conventional KIR typing misses much of this additional complexity, with important implications for studying the genetics of disease association with KIR that can now be explored by CNV analysis.
The killer immunoglobulin-like receptor (KIR) genes are part of the leukocyte receptor complex (LRC), on chromosome 19q13.4. These genes are highly polymorphic and occupy one of the most rapidly evolving regions of the human genome. A KIR complex is missing in the mouse genome, and even closely related primate species show substantial differences in organization (Parham et al. 2012).
KIR molecules modulate the development and activity of natural killer (NK) and some T-cells through interaction with major histocompatibility complex (MHC) class I receptors. Genetic analysis comparing disease with control samples indicates that KIR variation, in conjunction with polymorphic MHC class I molecules, plays a key role both in human reproduction as well as in immune defense (Khakoo and Carrington 2006).
Currently, 15 different KIR loci (KIR2DL1, KIR2DL2/KIR2DL3 [2DL2L3], KIR2DL4, KIR2DL5A, KIR2DL5B, KIR2DS1–5, KIR3DL1/KIR3DS1 [3DL1S1], KIR3DL2–3, and two pseudogenes, KIR2DP1 and KIR3DP1; all gene names abbreviated in subsequent text, without “KIR”) have been identified (http://www.ebi.ac.uk/ipd/kir/). Selected combinations of these genes are encoded on haplotypes within a 100- to 200-kb region of the LRC (Martin et al. 2000; Wilson et al. 2000). Most KIR haplotypes contain between seven and 12 genes plus the two pseudogenes. Based on this variation in gene content alone, more than 50 distinct KIR haplotypes have been identified (Hsu et al. 2002; Khakoo and Carrington 2006). Further complexity is introduced by extensive allelic variation, with more than 50 different alleles determined for some loci (Robinson et al. 2010).
Previous analyses of these haplotypes suggested that they are subdivided into two distinct groups. Subsequently these two groups were shown to have differential associations with disease and reproductive success (Khakoo and Carrington 2006). Group A haplotypes comprise seven genes and two pseudogenes. Six of the seven KIRs are inhibitory in nature, because they have immunoreceptor tyrosine-based motifs (ITIMs) in their cytoplasmic tails. The seventh gene, KIR2DS4, is potentially activating but is disabled by a 22-bp frameshift deletion on ∼75% of A haplotypes and is only therefore functional in a minority of individuals (Hsu et al. 2002). Activating KIR genes do not contain ITIM motifs and instead are coupled to adaptor proteins, such as DAP12, which contain immunoreceptor tyrosine-based activating motifs (ITAMs).
In contrast, Group B haplotypes are composed of variable numbers of KIR genes, and one or more of these are activating. This segregation into distinct and evolutionarily divergent haplotype groups (A and B) is unique to the human species (Parham et al. 2012). Furthermore, both haplotypic groups have been found in all human populations studied to date and are maintained by balancing selection (Gendzekhadze et al. 2009).
The recent evolutionary plasticity of the KIRs and human-specific high diversity, in addition to their biomedical relevance, makes it important to study their organization. There is mounting evidence that receptor–ligand specificity between polymorphic KIR and polymorphic MHC class I influences susceptibility to infections, such as HIV and hepatitis C, in addition to cancer, autoimmune diseases, and disorders of pregnancy, as well as outcomes after hematological and solid organ transplantation (Parham 2005).
The genomic organization of KIR haplotypes is structured relative to the fixed position of the so-called framework genes, 3DL3, 2DL4, and 3DL2 (Robinson et al. 2010). This gene order has been determined from the mapping and sequencing of several prototypic KIR haplotypes (Pyo et al. 2010). 3DL3 and 3DL2 mark the centromeric and telomeric boundaries of the cluster, respectively. 3DP1 and 2DL4 are placed centrally. Between 3DP1 and 2DL4 is a recombination hotspot. Annotating the haplotype motif structures centromeric (Cen) and telomeric (Tel) of this hotspot based on gene content (Pyo et al. 2010) has simplified and strengthened analysis in disease association studies (Hiby et al. 2010).
It has been proposed that the arrangement of KIR genes in close head-to-tail orientation and their high sequence similarity facilitates gene gain and loss or copy number variation (CNV) by unidirectional alignment and sequential non-allelic homologous recombination (NAHR) (Martin et al. 2003). Consistent with this, unusual KIR haplotypes that possess aberrant gene content and fusion genes have been identified (Norman et al. 2002, 2007, 2009; Martin et al. 2008; Traherne et al. 2010).
Given this background, we set out to explore variation in KIR haplotypes that may not be detected by conventional typing methods. To do this, we measured copy number as well as KIR presence or absence, using DNA from family-based cohorts in order to ensure the provenance of each haplotype.
Results
KIR haplotypes are highly diversified by copy number variability
KIR content and copy number were determined by typing 3466 samples (Table 1). Variation between sample replicates was typically <0.17 SD allowing accurate copy number calling for 99.6% of typed subjects. Copy number variability was observed, with variable frequency, for all KIR loci (Table 2). Segregation of gene copies in families resolved 2999 parental haplotypes. Of these, 71 unique haplotypes were identified, defined by gene content. Fifty-eight of the 71 haplotypes represented rare gene configurations, reflecting considerable CNV. The longest haplotype identified contained 20 loci, and the shortest carried four loci.
Table 1.
Composition of the HBDI and DBN sets
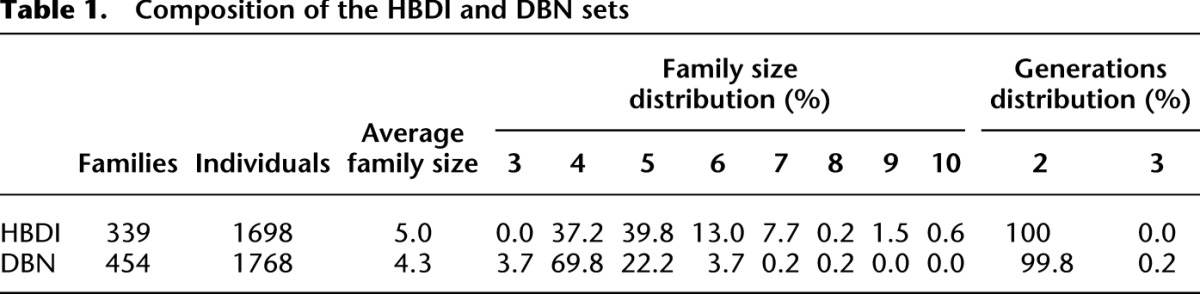
Table 2.
KIR gene and allele copy number frequencies (n = 2999)

KIR haplotype frequencies are nonrandom
Figure 1 shows the frequency distribution of the haplotypes and the centromeric (Cen) and telomeric (Tel) haplotype motifs. Consistent with previous studies of samples of the same ethnicity, the standard A haplotype was present at 55% frequency (Martin et al. 2008). KIR A haplotypes, 01 and 02, had identical gene content but differed according to their 2DS4 allele type. Haplotype 01 contained the 22-bp frameshift deletion variant of the 2DS4 gene, whereas haplotype 02 contained the full-length form. KIR B haplotypes with conventional gene configurations were present at 39% frequency. Dividing the KIR haplotypes into centromeric and telomeric regions separated by the recombination hotspot between 3DP1 and 2DL4 showed that the frequencies of the previously defined centromeric and telomeric motifs were mostly consistent with previous studies (Pyo et al. 2010; Hou et al. 2012). Exceptions were ∼6% of haplotypes that, although still definable as altered A or B haplotypes, were unclassifiable using current KIR haplotype nomenclature because their composition did not conform to the established centromeric or telomeric gene content motifs. These unusual cases represent extended or contracted haplotypes where the standard motifs are disrupted (see below). DNA sequencing of these haplotypes will be necessary to elucidate their precise structures.
Figure 1.

Frequency distributions of KIR haplotypes and motifs. Seventy-one unique haplotypes based on gene content were identified in 793 nuclear families. Eleven haplotypes with frequencies >1% (shaded) account for 94% of the total 2999 parental haplotypes. Haplotypes 01 and 02 (both Cen-A:Tel-A) are the two basic A haplotypes that differ by the type of 2DS4 present; 01 carries the 22-bp frameshift deletion variant and 02 carries the full-length form. Pie charts show the frequencies of unique KIR haplotypes, A (Cen-A:Tel-A) and B (Cen-A:Tel-B, Cen-B:Tel-A, Cen-B:Tel-B) haplotype groups, individual structural motifs, and motif combinations. KIR A haplotypes contain a Cen-A motif (defined by presence of 2DL3) and a Tel-A motif (2DS4 present). B haplotypes can be defined by the presence of a Cen-B (2DS2 present) and/or a Tel-B (2DS1 present); Cen-A motif 3DL3–2DL3–2DP1–2DL1-3DP1, Cen-B motif 3DL3–2DS2–2DL2–(2DL5C–2DS3S5C-2DP1–2DL1)–3DP1, Tel-A motif 2DL4–3DL1–2DS4 full-length or deletion variant–3DL2, Tel-B motif 2DL4–3DS1–2DL5T–2DS3S5T–2DS1–3DL2. Sixty haplotypes, together accounting for 6% of the 2999 resolvable haplotypes, have frequencies <1% and typically carry unconventional gene content. Haplotypes that did not strictly conform to standard A/B haplotypes or motifs because of altered structural composition are labeled “U.”
Common KIR haplotypes are created by reciprocal recombination
Eleven haplotypes, each with frequencies >1%, accounted for 94% of the total 2999 parental haplotypes (Fig. 2). A parsimonious mechanism to account for the derivation of all of these common haplotypes invokes homologous recombination between common precursor haplotypes at the known recombination hotspot upstream of 2DL4, as illustrated in Figure 3. All possible combinations of centromeric (cA01, cB01, cB02) and telomeric (tA01, tB01) haplotype structures are represented in these 11 common haplotypes (Fig. 2).
Figure 2.

Common KIR haplotypes. Depicted are the 11 common haplotypes that encompass 94% of Caucasian haplotypes studied. (Right) Haplotype frequencies, A/B group classification, and constituent motifs; for example, cA01 and tA01 denote centromeric-A haplotype 1 and telomeric-A haplotype 1, respectively. Gene and allele content were determined by segregation analysis. (Black spot) Site of the recombination hotspot between 3DP1 and 2DL4. The gene order is based on published sequences of KIR haplotypes that were sequenced in their entirety. Locations of 2DS3S5 may vary on unsequenced haplotypes. The boxed key identifies alleles, variants, and which genes are considered framework. 2DS4v represents the 22-bp frameshift deletion variant, and 2DS4f is the full-length form. 2DL2 and 2DL3 are allelic variants of 2DL2L3, 3DL1 and 3DS1 are variants of 3DL1S1, and 2DS3 and 2DS5 are variants of 2DS3S5. The motif annotation is at the resolution of gene content and does not distinguish allelic variation of individual genes. For example, the haplotype motif tA01 may have either form of 2DS4 (2DS4f or 2DS4v), and tB01 may have either variant of 2DS3S5 (2DS3 or 2DS5). T and C suffixes for 2DL5 (2DL5A or 2DL5B) and 2DS3S5 indicate the telomeric and centromeric versions of the genes, respectively (Gomez-Lozano et al. 2002; Ordonez et al. 2008; Pyo et al. 2010). For simplicity, 2DS1 and 2DS4 are shown at the same site because they showed a mutually exclusive relationship.
Figure 3.

The 11 common haplotypes are derived from recombination of a small number of haplotypes. The figure shows the putative mechanism for formation of common recombinant haplotypes from parental haplotypes by reciprocal recombination. All 11 haplotypes can be accounted for by recombination at the recombination hotspot near 2DL4. The mechanism effectively swaps the centromeric and telomeric motifs (identification in key above) between haplotypes by reciprocal recombination. Genes are not duplicated or deleted by this process, but haplotype motifs are interchanged. Some recombinations generate equivalent recombinant haplotypes to the others, in terms of gene content and not considering alleles. (Right) Constituent haplotype motifs based on gene content; (f) frequency. The numbers in the parentheses refer to haplotypes defined in Figure 1. Where there are two haplotypes in parentheses, e.g., (01/02), then these haplotypes only differ by 2DS4 type (2DS4f or 2DS4v; see below). (Gray) Cen-B and Tel-B motif genes; (white) Cen-A and Tel-A motif genes. cA01 and tA01 denote Cen-A haplotype motif 1 and Tel-A haplotype motif 1, respectively. tA01 may have either form of 2DS4 (2DS4f or 2DS4v). tB01 may have either variant of 2DS3S5 (2DS3 or 2DS5). The arrows point both up and down because the precursor and recombinant haplotypes are not known and the process could occur in either direction, exchanging parental and progeny haplotypes.
Many KIR haplotypes exhibit novel structural variation
Sixty rare haplotypes, each with frequencies <1%, exhibited unconventional gene copy number or gene arrangement. These haplotypes, which often had framework genes duplicated or deleted, accounted for 6% of the 2999 parental haplotypes (Fig. 4). For example, 10 of these 61 haplotypes lack either one or both of the so-called framework genes 3DP1 and 2DL4. Altogether 22 expanded KIR haplotypes and 38 contracted haplotypes were identified. Less than ∼5% of these novel haplotypes would be distinguishable using published KIR typing methods such as SSP and Luminex-based SSO technology, even if their configurations were provided as templates. The reason for this is that without determining copy number or defining alleles, ascertainment of the gene arrangements on these haplotypes can be obscured by variation in KIR gene content of the partnering chromosome, even with pedigrees to track gene transmission (Martin et al. 2008). Six of the uncommon gene combinations (haplotypes 12, 13, 14, 15, 18, and 33) appeared to match recombinant haplotypes reported previously by us and others (Gomez-Lozano et al. 2003, 2005; Martin et al. 2003; Ordonez et al. 2008, 2011; Traherne et al. 2010). Using published methods for detecting hybrid genes (e.g., 2DL5/3DP1 [3DP1*004], 2DS2/2DS3 [2DS2*005], and 2DL3/2DP1) and specific alleles associated with these recombinant haplotypes, we were able to confirm the identity of these haplotypes (data not shown). The identification of these recombinant haplotypes further corroborates the copy number typing method used in this study for detecting novel haplotype rearrangements.
Figure 4.

Unconventional KIR haplotypes and their frequencies. Fifteen altered A haplotypes and 56 B haplotypes all with frequencies <1%. Duplicated genes are not depicted in order, but for simplicity, the paralog has been arbitrarily placed adjacent to the ancestral gene. In some cases, the order of all of the genes has not been determined. Genes suffixed with “v” have parts altered or deleted including fused genes resulting from NAHR or a novel allele. 2DS4v represents the 22-bp frameshift deletion variant, and 2DS4f is the full-length form. 3DL1v on haplotypes 63 and 48 tested negative for exon 4 and on haplotypes 59, 61, 71, and 43 negative for exon 9. 3DL2v on haplotypes 64 and 26 were negative for exon 4 and on haplotype 32 negative for exon 9. Nineteen haplotypes, marked with a black dot, were missing a copy or part of a single locus but otherwise could be the same as standard haplotypes (common arrangements of genes). Apart from haplotypes 15 and 21, which carried a hybrid gene (2DS2/2DS3; 2DS2*005), these marked haplotypes may not reflect true copy number variation because in these cases, we could not rule out the possibility that the gene was present but was erroneously scored as missing due to sequence variation that did not complement our generic primers (see Methods; Supplemental Material). To help resolve novel KIR haplotypes from unphased genotypes of unrelated individuals, we developed a tool for imputing haplotype pairs given observed copy number for each KIR loci. This tool is provided at http://www.bioinformatics.cimr.cam.ac.uk/haplotypes/.
Even though the primers and probes were designed specifically to avoid sequence polymorphism known to date within each gene (current IPD–KIR Database Release 2.4.0), it is possible that some of the novel deletion and extended haplotypes could rather be common arrangements of genes carrying allelic polymorphisms not detected by our PCR primers or probes (see the Supplemental Material). In the few instances in which a rare SNP lies within an annealing site for a primer or probe and may therefore disrupt binding, the corresponding allele designation is given in the Supplemental Material. The majority of the haplotypes involve loss or duplication of several genes (as evaluated by several independent assays). It is unlikely that these haplotypes correspond to rare allelic polymorphism in multiple genes. In two deletion haplotypes missing only a single gene (haplotypes 15 and 21), a hybrid gene is present, confirming haplotype rearrangement. In the remaining 17 deletion haplotypes that are missing a single gene (marked by a black dot in Fig. 4), we do not rule out the possibility that these may relate to allelic polymorphism.
Alternative methods such as SSP-PCR KIR genotyping have been carried out for the families carrying unusual genotypes. In some cases, this has confirmed the CNV, although in many cases, and for all framework genes, it has not been possible to verify gene absence using this approach. This is because without determining copy number, it cannot be seen whether the gene in question is deleted since it is hidden by a copy of the same gene on the partnering chromosome.
KIR copy number variation is spatially biased
The data indicate that some genes are rarely subject to CNV (Fig. 5). For example, the two genes defining the ends of the KIR region, 3DL2 and 3DL3, only deviated from one copy on four of the 71 haplotypes (3DL3: 0, 1, 2 copy frequencies 0.07, 99.9, and 0.03, respectively, and 3DL2: 0, 1 copy frequencies 0.3, 99.7). Variation may be confined between the flanking framework genes because these genes represent the limit of complementary sequence for misalignment and repair. There were no haplotypes with 2DS2 duplicated and only two haplotypes with 2DS1 or 2DS4 duplicated, although these genes were missing on certain haplotypes and thus are subject to CNV. This raises the possibility that duplication of these activating genes may be selectively disadvantageous in this population or that it is restricted by their position within the locus. On the other hand, some genes were more frequently subject to CNV. For example, 3DL1S1 deviated from one copy on 25 of the 71 haplotypes.
Figure 5.

Gene duplication:absence ratios in the 71 unique haplotypes. CNV is spatially biased within the KIR locus, as illustrated by this plot showing the proportion of the 71 haplotypes showing duplication or absence for each respective gene.
Novel fusion genes may be older than many extant haplotypes
Gene fusions that have previously been described (2DL5/3DP1 [now designated 3DP1*004], 2DL1/2DS1, 2DL3/2DP1) were identified in a small number of samples (Martin et al. 2003; Traherne et al. 2010). Certain fusion genes, with identical chromosome breakpoints, were present on different haplotypes. These may have arisen by recombination with other haplotypes that took place after the hybrid genes were derived. For example, the same 2DL5/3DP1 (3DP1*004) fusion gene was present in haplotypes 13, 16, 20, and 39, suggesting the occurrence of at least three recombination events since the generation of this fusion gene (Fig. 6). In these cases, the hybrids may predate extant haplotypes. Using a range of hotspot recombination rates from 2 × 10−3 to 1 × 10−4 per generation accounting for normal hotspot widths of 1.0–2.0 kb (Jeffreys et al. 2001; Jeffreys and Neumann 2002), and taking into account three recombination events over two recombination hotspots, we estimate that the 2DL5/3DP1 (3DP1*004) fusion gene was formed 750–15,000 generations ago. Very few 2DL4-3DL1S1 duplication haplotypes have been found in Africa compared with other populations (Norman et al. 2009), which may fit with the more recent end of the age estimation. Haplotypes that have expanded the central region of the KIR locus must have duplicated the recombination hotspot upstream of 2DL4 and may therefore be more prone to reciprocal recombination than standard KIR haplotypes. In addition, hybrid genes with differently sited breakpoints were identified, consistent with recurrent recombination within KIR (Traherne et al. 2010).
Figure 6.

The fusion gene 2DL5/3DP1 is carried on four different KIR haplotypes. The precise chromosome breakage in 2DL5/3DP1 (3DP1*004) is shared by these four haplotypes, suggesting a common ancestry rather than independent formation of the fusion gene by NAHR between different haplotypes. Instead, the haplotype structures are consistent with recombination occurring with different haplotypes, i.e., motif interchange, at the recombination hotspots sited between 3DP1 and 2DL4, after the fusion gene was created. Constituent haplotype motifs based on gene content are boxed. (f) Frequency. The numbers in the parentheses refer to the haplotypes defined in Figure 1.
Only KIR B haplotypes show rich CNV haplotype diversity
Interestingly, more than half of the 2999 resolvable haplotypes in the two sets of samples were A haplotypes, yet <1% of A haplotypes were affected by copy number variation (i.e., products of NAHR between two A haplotypes, so comprising no B haplotype-specific genes, were very rare), compared with >80% of the B haplotypes. Most of these A haplotypes appeared to be affected in a single gene. These could have been due to deletions or alternatively to novel/rare sequence variants that resulted in lack of priming with our oligonucleotides as described above.
Discussion
More KIR haplotypes will exist
We show that KIR variation is even more extensive than previously suspected. Out of 1605 founders, we obtained evidence for 71 haplotypes with different compositions of genes. The KIR locus is thus the most variable protein-encoding site in the human genome in terms of size and haplotype gene content diversity. So far, we have determined several novel KIR haplotypes having new gene arrangements and loci in Caucasian populations. Collectively these novel haplotypes exceed 5% in frequency.
The families were selected for the presence of two common autoimmune diseases so it is possible that risk haplotypes may be present in the study population. Having said this, the unrelated individuals studied contained a high proportion of unaffected individuals, and haplotype frequencies did not differ between unaffected and disease subjects. This is reflected in the frequencies for the 11 common haplotypes that did not differ significantly compared with an earlier family-based study consisting of unaffected Caucasian individuals (Martin et al. 2008).
HBDI (687 founders) contained 20 haplotypes that were not seen in DBN (918 founders), and DBN contributed 24 haplotypes that were not present in HBDI, indicating that more novel haplotypes remain to be described. It can be estimated from our data that there are more than 90 KIR haplotypes in the U.K./U.S. populations of European descent (Supplemental Material). Preliminary assessment of haplotypes from other populations, especially from Africa and Asia, suggests that this number will be greatly exceeded. This considerable reservoir of variation and its broad distribution in frequency provide the substrate on which natural selection acts at the population and individual levels.
The expanded KIR gene cluster is unique to primates, and typically there are only a few KIR genes in other vertebrate species (Parham et al. 2012). In humans, the KIRs are highly variable and in a state of evolutionary flux, and, curiously, humans are the only species that have distinctive A and B haplotypes (Parham et al. 2012). The reasons for these phenomena have not been established, but they are indicative of balancing selection, potentially driven by immunity and reproduction. The physical nature of the locus may facilitate gene rearrangements. In regard to selection, KIR functions are tied in with those of polymorphic HLA class I molecules. Combinatorial KIR and HLA diversity leads to an extreme degree of heterogeneity between different individuals, and these combinations of ligand and receptor affect susceptibility to a broad spectrum of disease.
Disease analyses can be simplified, but current KIR typing is inadequate
With only 11 common haplotypes accounting for 94% in the population, classifiable into Cen-A and Tel-B, analysis in disease studies can be simplified if unusual haplotypes can be detected easily. An improved strategy might therefore exclude the 6% with unusual CNV in large cohorts and analyze these separately from the other 94%.
However, our findings also signal some caution in the interpretation of KIR typing, because current routine methods may be blind to the existence of novel hybrid genes and many of the genomic rearrangements we describe. Our data can be used to help identify new hybrid genes and develop methods to detect them. Methods for detecting some hybrid genes such as 2DL5/3DP1 (3DP1*004), 2DS2/2DS3 (2DS2*005), and 3DL1/3DL2 have already been described (Gomez-Lozano et al. 2005; Norman et al. 2009; Traherne et al. 2010). As with the MHC, novel refinements of typing methods will need to be introduced, based on imputation from SNP data and next-generation sequencing techniques (Leslie et al. 2008; Holcomb et al. 2011).
As mentioned above, many studies have indicated a relationship between MHC class I, KIR, and disease. We propose that these data are in need of further refinement, once copy number has been taken into account. Furthermore, there is evidence that copy number, which leads to expression differences (Norman et al. 2009), may be important to susceptibility to some diseases. For example, CNV of 3DL1S1 influences HIV control (Pelak et al. 2011) and expression differences of 2DL3, interacting with HLA-C, may have a profound effect on resolution of hepatitis C virus infection with an odds ratio of more than 2 (Khakoo et al. 2004; Li et al. 2008).
B haplotypes are subject to gene loss and gain
In compiling the many KIR arrangements, we noted that A haplotypes are generally stable in terms of copy number when compared with B haplotypes. Out of the 71 haplotypes studied, only one exhibited any duplications in genes specific to A haplotypes. One interpretation of this finding is that individuals carrying a deletion or duplication of genes on A haplotypes would have no selective advantage. In contrast, extreme B haplotypes containing between four and 15 genes are consistent with a survival advantage of variability. We have some examples of homozygotes of these in studies of other populations (Traherne et al. 2010; L Farrell, A Munro, O Chazara, H Laivuori, J Jayaraman, J Traherne, K Kivinen, L Järvinen, S Hiby, and A Moffett, unpubl.). Consequently, B haplotypes may be under stronger diversifying selection than A haplotypes. The nature of this selection is not clear, but it could relate to several factors, including reproductive success; risk of pre-eclampsia or extreme birth-weight; selective pressure on unfavorable genes that, for example, carry a risk of autoimmunity; or to creation or loss of novel functional genes driven by waves of infection. In this case, B haplotypes, or more specifically activating genes, which are mostly confined to B haplotypes, are the driving force.
An alternative explanation, which does not necessarily invoke selection, is that B, but not A, haplotypes are prone to rearrange. One possibility is that NAHR only generates copy number diversity in B haplotypes. What features of B haplotypes could be responsible for such a phenomenon? The obvious difference between A and B is the presence of one or more activating KIR genes on the latter, although an intact 2DS4 gene is present on 24% of A haplotypes. Another possibility is that A haplotypes are restrained from recombination (NAHR) by histone protection or because they are missing sequence elements that facilitate recombination. However A haplotype components appear on many extended and truncated haplotypes through recombination with B haplotypes. In principle, the A haplotype may have spread recently under directional selection without sufficient time for rearrangement, although this may be unlikely given its worldwide distribution.
In conclusion, through our description of a number of novel haplotypes in Caucasian families, we have refined our understanding of the genetic variation of the KIR cluster. We have shown that this variability is far greater than previously thought, reflecting extreme evolutionary pressures that have exerted their effects recently and rapidly. This is consistent with the broad spectrum of disease for which there are reported associations between susceptibility and KIR gene content. The additional variability that we have described at this locus is currently only detectable by careful analysis of gene copy number. This has implications for previous genetic studies of human disease, and it will be imperative to consider this variation in future investigations. Furthermore, given the structural complexities of the cluster, it presents a significant challenge to the use of SNP or sequencing-based typing techniques.
Methods
DNA samples
Two sets of subjects were studied. The first set was from the Human Biological Data Interchange (HBDI) and consisted of 1698 white American subjects within 339 families selected as part of a study of type I diabetes. The second set was from the UK DNA Banking Network (DBN) and consisted of 1768 white British individuals from 454 families, ascertained in a separate study of asthma or atopic dermatitis. All subjects were of initial European ancestry.
Copy number determination by quantitative PCR
Genomic DNAs were typed for copy number of the following KIR genes, pseudogenes, and major alleles: 2DL1–5, 2DS1–3, 2DS4 (separate assays for the gene, full-length variant [f], and deletion variant [v]), 2DS5, 3DL1–3, 3DS1, 2DP1, and 3DP1. KIR genotype and copy number were measured using a quantitative PCR comparative Ct method (Schmittgen and Livak 2008). Our copy number assay for all KIR genes was developed from the previously published detailed protocol for determining genomic copy number for 2DL4 (Martin et al. 2003). The primer and probe sequences for analysis of all KIR loci are given in the Supplemental Material to allow judgment of whether they are appropriate for defining the currently known polymorphisms and variants discovered in the future. The primers and probes were designed specifically to avoid the sequence polymorphism known to date within each gene (current IPD–KIR Database Release 2.4.0, 15-April-2011, http://www.ebi.ac.uk/ipd/kir/). In the few instances in which a rare SNP lies within an annealing site for a primer or probe and may therefore disrupt binding, the corresponding allele designation is given in the Supplemental Material. A set of multiplex qPCR assays was used to ascertain copy number for all 17 KIR genes and their major variants. Reactions were carried out in quadruplicate to ensure accuracy of the copy number scoring. Three control samples of known copy number were included in each run. For 3DL1 and 3DL2 we used two reactions to target different parts of the gene to identify known fusion genes (Norman et al. 2009). To verify the accuracy of the KIR typing, we used independent panels previously typed by others with standard methods (PCR-sequence-specific primers [PCR-SSP] and PCR-sequence-specific oligonucleotide [PCR-SSO] probes), including CEPH pedigrees available from Coriell Cell Repositories (Martin et al. 2008), the POPS cohort (Pasupathy et al. 2008) (n = 50, KIR typed by Ashley Moffett's group, Cambridge, UK), and samples from the International KIR exchange program (http://www.hla.ucla.edu/cellDna.htm; Arlene Locke, Marie Lau, Elaine F. Reed, and Raja Rajalingam, UCLA Immunogenetics Center, Department of Pathology and Laboratory Medicine, David Geffen School of Medicine, University of California, Los Angeles, CA). Complete concordance with previous typing verified the accuracy of the KIR typing. In collaboration with the Addenbrooke's Hospital Tissue Typing laboratory in Cambridge, we are developing plans to establish a KIR typing service using the quantitative KIR automated-typing (qKAT) method.
KIR haplotype determination and statistical analysis
KIR haplotypes were determined by segregation analysis in families. Gene frequencies did not differ between the two populations (χ2 test for independence, P > 0.05), so the data were pooled. For each pedigree, all nonrecombinant haplotypes were identified by the Merlin program (Abecasis et al. 2002). Approximately 8% of the parental (founder) haplotypes were not fully deduced because of phase ambiguity or missing typing data. Standards for KIR haplotype nomenclature are still in development; therefore, we arbitrarily labeled haplotypes numerically based on frequency.
Acknowledgments
This work was supported by grants from the MRC and the Wellcome Trust with partial funding from the National Institute for Health Research Cambridge Biomedical Research Centre, Pathological Society, Newton Trust, and a Wellcome Trust strategic award (079895/Z/06/Z). We acknowledge services and data access provided by BioBanking Solutions administered from the Centre for Integrated Genomic Medical Research (University of Manchester). We are grateful to Professor Jim Kaufman and Professor Ashley Moffett (Department of Pathology, University of Cambridge, UK) for critical reading of this manuscript. We also thank Professor Ashley Moffett for permission to use the data from their KIR typing of the POPS cohort.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.137976.112.
Freely available online through the Genome Research Open Access option.
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR 2002. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30: 97–101 [DOI] [PubMed] [Google Scholar]
- Gendzekhadze K, Norman PJ, Abi-Rached L, Graef T, Moesta AK, Layrisse Z, Parham P 2009. Co-evolution of KIR2DL3 with HLA-C in a human population retaining minimal essential diversity of KIR and HLA class I ligands. Proc Natl Acad Sci 106: 18692–18697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Lozano N, Gardiner CM, Parham P, Vilches C 2002. Some human KIR haplotypes contain two KIR2DL5 genes: KIR2DL5A and KIR2DL5B. Immunogenetics 54: 314–319 [DOI] [PubMed] [Google Scholar]
- Gomez-Lozano N, de Pablo R, Puente S, Vilches C 2003. Recognition of HLA-G by the NK cell receptor KIR2DL4 is not essential for human reproduction. Eur J Immunol 33: 639–644 [DOI] [PubMed] [Google Scholar]
- Gomez-Lozano N, Estefania E, Williams F, Halfpenny I, Middleton D, Solis R, Vilches C 2005. The silent KIR3DP1 gene (CD158c) is transcribed and might encode a secreted receptor in a minority of humans, in whom the KIR3DP1, KIR2DL4 and KIR3DL1/KIR3DS1 genes are duplicated. Eur J Immunol 35: 16–24 [DOI] [PubMed] [Google Scholar]
- Hiby SE, Apps R, Sharkey AM, Farrell LE, Gardner L, Mulder A, Claas FH, Walker JJ, Redman CW, Morgan L, et al. 2010. Maternal activating KIRs protect against human reproductive failure mediated by fetal HLA-C2. J Clin Invest 120: 4102–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb CL, Hoglund B, Anderson MW, Blake LA, Bohme I, Egholm M, Ferriola D, Gabriel C, Gelber SE, Goodridge D, et al. 2011. A multi-site study using high-resolution HLA genotyping by next generation sequencing. Tissue Antigens 77: 206–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Chen M, Ng J, Hurley CK 2012. Conserved KIR allele-level haplotypes are altered by microvariation in individuals with European ancestry. Genes Immun 13: 47–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu KC, Liu XR, Selvakumar A, Mickelson E, O'Reilly RJ, Dupont B 2002. Killer Ig-like receptor haplotype analysis by gene content: Evidence for genomic diversity with a minimum of six basic framework haplotypes, each with multiple subsets. J Immunol 169: 5118–5129 [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Neumann R 2002. Reciprocal crossover asymmetry and meiotic drive in a human recombination hot spot. Nat Genet 31: 267–271 [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Kauppi L, Neumann R 2001. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat Genet 29: 217–222 [DOI] [PubMed] [Google Scholar]
- Khakoo SI, Carrington M 2006. KIR and disease: A model system or system of models? Immunol Rev 214: 186–201 [DOI] [PubMed] [Google Scholar]
- Khakoo SI, Thio CL, Martin MP, Brooks CR, Gao X, Astemborski J, Cheng J, Goedert JJ, Vlahov D, Hilgartner M, et al. 2004. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 305: 872–874 [DOI] [PubMed] [Google Scholar]
- Leslie S, Donnelly P, McVean G 2008. A statistical method for predicting classical HLA alleles from SNP data. Am J Hum Genet 82: 48–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Pascal V, Martin MP, Carrington M, Anderson SK 2008. Genetic control of variegated KIR gene expression: Polymorphisms of the bi-directional KIR3DL1 promoter are associated with distinct frequencies of gene expression. PLoS Genet 4: e1000254 doi: 10.1371/journal.pgen.1000254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AM, Freitas EM, Witt CS, Christiansen FT 2000. The genomic organization and evolution of the natural killer immunoglobulin-like receptor (KIR) gene cluster. Immunogenetics 51: 268–280 [DOI] [PubMed] [Google Scholar]
- Martin MP, Bashirova A, Traherne J, Trowsdale J, Carrington M 2003. Cutting edge: Expansion of the KIR locus by unequal crossing over. J Immunol 171: 2192–2195 [DOI] [PubMed] [Google Scholar]
- Martin MP, Single RM, Wilson MJ, Trowsdale J, Carrington M 2008. KIR haplotypes defined by segregation analysis in 59 Centre d'Etude Polymorphisme Humain (CEPH) families. Immunogenetics 60: 767–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman PJ, Carrington CV, Byng M, Maxwell LD, Curran MD, Stephens HA, Chandanayingyong D, Verity DH, Hameed K, Ramdath DD, et al. 2002. Natural killer cell immunoglobulin-like receptor (KIR) locus profiles in African and South Asian populations. Genes Immun 3: 86–95 [DOI] [PubMed] [Google Scholar]
- Norman PJ, Abi-Rached L, Gendzekhadze K, Korbel D, Gleimer M, Rowley D, Bruno D, Carrington CV, Chandanayingyong D, Chang YH, et al. 2007. Unusual selection on the KIR3DL1/S1 natural killer cell receptor in Africans. Nat Genet 39: 1092–1099 [DOI] [PubMed] [Google Scholar]
- Norman PJ, Abi-Rached L, Gendzekhadze K, Hammond JA, Moesta AK, Sharma D, Graef T, McQueen KL, Guethlein LA, Carrington CV, et al. 2009. Meiotic recombination generates rich diversity in NK cell receptor genes, alleles, and haplotypes. Genome Res 19: 757–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordonez D, Meenagh A, Gomez-Lozano N, Castano J, Middleton D, Vilches C 2008. Duplication, mutation and recombination of the human orphan gene KIR2DS3 contribute to the diversity of KIR haplotypes. Genes Immun 9: 431–437 [DOI] [PubMed] [Google Scholar]
- Ordonez D, Gomez-Lozano N, Rosales L, Vilches C 2011. Molecular characterisation of KIR2DS2*005, a fusion gene associated with a shortened KIR haplotype. Genes Immun 12: 544–551 [DOI] [PubMed] [Google Scholar]
- Parham P 2005. MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol 5: 201–214 [DOI] [PubMed] [Google Scholar]
- Parham P, Norman PJ, Abi-Rached L, Guethlein LA 2012. Human-specific evolution of killer cell immunoglobulin-like receptor recognition of major histocompatibility complex class I molecules. Philos Trans R Soc Lond B Biol Sci 367: 800–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasupathy D, Dacey A, Cook E, Charnock-Jones DS, White IR, Smith GC 2008. Study protocol. A prospective cohort study of unselected primiparous women: The pregnancy outcome prediction study. BMC Pregnancy Childbirth 8: 51 doi: 10.1186/1471-2393-8-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelak K, Need AC, Fellay J, Shianna KV, Feng S, Urban TJ, Ge D, De Luca A, Martinez-Picado J, Wolinsky SM, et al. 2011. Copy number variation of KIR genes influences HIV-1 control. PLoS Biol 9: e1001208 doi: 10.1371/journal.pbio.1001208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyo CW, Guethlein LA, Vu Q, Wang R, Abi-Rached L, Norman PJ, Marsh SG, Miller JS, Parham P, Geraghty DE 2010. Different patterns of evolution in the centromeric and telomeric regions of group A and B haplotypes of the human killer cell Ig-like receptor locus. PLoS ONE 5: e15115 doi: 10.1371/journal.pone.0015115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J, Mistry K, McWilliam H, Lopez R, Marsh SG 2010. IPD—the Immuno Polymorphism Database. Nucleic Acids Res 38: D863–D869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108 [DOI] [PubMed] [Google Scholar]
- Traherne JA, Martin M, Ward R, Ohashi M, Pellett F, Gladman D, Middleton D, Carrington M, Trowsdale J 2010. Mechanisms of copy number variation and hybrid gene formation in the KIR immune gene complex. Hum Mol Genet 19: 737–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MJ, Torkar M, Haude A, Milne S, Jones T, Sheer D, Beck S, Trowsdale J 2000. Plasticity in the organization and sequences of human KIR/ILT gene families. Proc Natl Acad Sci 97: 4778–4783 [DOI] [PMC free article] [PubMed] [Google Scholar]