

Abstract
The recognition potential of most families of DNA-binding domains (DBDs) remains relatively unexplored. Homeodomains (HDs), like many other families of DBDs, display limited diversity in their preferred recognition sequences. To explore the recognition potential of HDs, we utilized a bacterial selection system to isolate HD variants, from a randomized library, that are compatible with each of the 64 possible 3′ triplet sites (i.e., TAANNN). The majority of these selections yielded sets of HDs with overrepresented residues at specific recognition positions, implying the selection of specific binders. The DNA-binding specificity of 151 representative HD variants was subsequently characterized, identifying HDs that preferentially recognize 44 of these target sites. Many of these variants contain novel combinations of specificity determinants that are uncommon or absent in extant HDs. These novel determinants, when grafted into different HD backbones, produce a corresponding alteration in specificity. This information was used to create more explicit HD recognition models, which can inform the prediction of transcriptional regulatory networks for extant HDs or the engineering of HDs with novel DNA-recognition potential. The diversity of recovered HD recognition sequences raises important questions about the fitness barrier that restricts the evolution of alternate recognition modalities in natural systems.
Homeodomains (HDs) play a prominent role in regulating a multitude of biological processes in eukaryotes, ranging from mating type switching in yeast to embryonic patterning in metazoans (Kornberg 1993; Gehring et al. 1994). Emblematic of their central role in gene regulation, HDs are broadly represented across eukaryotic species; in humans, they are the second most common family of DNA-binding domains (Vaquerizas et al. 2009). Consistent with their abundance, HDs display a diverse array of functions in development and cell-type specification, and they can be subdivided into a number of distinct families based on common sequence features and recognition motifs (Burglin 2011). Sequence-specific DNA recognition is central to many aspects of the regulatory function of HDs and as a consequence this characteristic has been extensively studied through genetic, biochemical, and structural analyses (Wolberger et al. 1991; Ades and Sauer 1994, 1995; Ekker et al. 1994; Gehring et al. 1994; Damante et al. 1996; Fraenkel et al. 1998; Grant et al. 2000; Hovde et al. 2001; Joshi et al. 2007; Rohs et al. 2010; Slattery et al. 2011). HDs are typically composed of an ∼60-amino-acid motif that folds into a three-helix bundle preceded by an N-terminal arm. Sequence-specific recognition is mediated by the third (recognition) helix docking in the major groove and the N-terminal arm docking in the minor groove (Fig. 1), where a HD typically specifies a site of 3–8 bp.
Figure 1.

Structure of the Engrailed HD and distribution of HD recognition residues. (A) Structure of the Engrailed HD–DNA complex (Fraenkel et al. 1998), which serves as the framework for library construction. The numbers (white) on the HD recognition helix (yellow) indicate amino acid positions (green side chains) that were randomized, where the primary strand of the core 6-bp binding site is highlighted (green) to emphasize the proximity of these residues to the 3′ end of the recognition sequence. Asn51 (orange), which is highly conserved within the homeodomain family, is shown for reference. (B) Frequency logo displaying the diversity of residues (the residues randomized in the HD library are circled in red) at various positions in the N51-containing HDs in the genomes of humans, mice, Danio rerio, Caenorhabditis elegans, Drosophila melanogaster, and Saccharomyces cerevisiae.
Many specificity determinants central to sequence-specific DNA recognition by HDs have been defined. A subset of these determinants function semi-autonomously, such that the transfer of a single residue between HDs can result in a predictable alteration in specificity. This is demonstrated by seminal studies investigating the role of position 50 in the recognition preference of PRD, BCD, and FTZ (Treisman et al. 1989; Percival-Smith et al. 1990; Hanes and Brent 1991). The critical features determining sequence-specific recognition by the N-terminal arm remain nebulous, and consequently, achieving alterations in specificity typically necessitates the substitution of multiple residues between HDs (Ekker et al. 1994; Damante et al. 1996).
Recent comprehensive analysis of HD specificity in the mouse and fruit fly (194 and 84, respectively) have somewhat clarified the breadth of DNA sequences HDs recognize in natural systems (Berger et al. 2008; Noyes et al. 2008a). While these studies used different approaches for determining DNA-binding specificity, they are in general concordant on the core DNA-binding specificity of homologous HDs. Limited sequence diversity is observed in the residues at the critical recognition helix positions within most eukaryotes (Fig. 1), and there is a corresponding paucity in the diversity of preferred recognition sequences observed for the characterized HD population (Berger et al. 2008; Noyes et al. 2008a). This focused sequence preference is similar to many other families of DNA-binding domains (Deppmann et al. 2006; Wei et al. 2010; De Masi et al. 2011) and could be the result of a general constraint of the domain architecture on its recognition potential. Consistent with this conjecture, previous attempts to select HDs with novel specificity have not succeeded in achieving dramatic alterations in recognition potential (Pomerantz and Sharp 1994; Connolly et al. 1999). These attempts, however, allowed variation at only a modest number of recognition positions. Thus, it remains possible that HDs can recognize a broader range of DNA sequences than is currently observed.
Here we describe radically reengineering the DNA-binding specificity of the Engrailed homeodomain to clarify the general recognition properties of this family. We systematically selected HD variants from a randomized library against all 64 possible combinations of the target site TAANNN. From these selections, we were able to recover HDs that preferentially recognize 44 of the 64 sites, far more than anticipated based on the characterized set of extant HDs. The majority of these HDs harbor distinct combinations of specificity determinants, many of which appear to be uncommon or absent in extant HDs. These determinants expand our understanding of HD recognition, allowing the creation of more explicit recognition models for this family. The potential for this domain to recognize a broader range of DNA sequences raises questions about the fitness barrier that restricts the evolution of more diverse recognition properties for this family in natural systems.
Results
Selection of HDs with novel DNA-binding specificity
To explore the DNA-recognition potential of HDs, we investigated their ability to specify all possible TAANNN sites by selecting compatible HDs from a randomized library. These selections were performed using our bacterial one-hybrid (B1H) system (Noyes et al. 2008a,b), where the HD library is expressed as a fusion to two zinc fingers that position the library over the preferred target site (Supplemental Fig. S1). The Engrailed (EN) HD was chosen as the library backbone because it is amenable to substitutions that change its DNA-binding specificity (Ades and Sauer 1994; Tucker-Kellogg et al. 1997; Noyes et al. 2008a).
Recognition of the 3′ region (bases 4, 5, and 6) of the HD binding site is mediated by specificity determinants within the recognition helix. To select HD variants with altered sequence recognition preferences, residues 43, 46, 47, 50, and 54 were fully randomized (Fig. 1). These positions, which all point toward the major groove in the EN–DNA complex, were chosen based on their potential function as primary or secondary recognition determinants within the 3′ region of the target site. Direct base-specific contacts have been observed between residues 47 and 54 and base 4, as well as between residue 50 and bases 5 and 6 (Wolberger et al. 1991; Tucker-Kellogg et al. 1997; Fraenkel et al. 1998; Passner et al. 1999; Piper et al. 1999; Grant et al. 2000; Joshi et al. 2007), where sequence alteration at these positions has a direct influence on specificity (Treisman et al. 1989; Percival-Smith et al. 1990; Hanes and Brent 1991; Damante et al. 1996; Noyes et al. 2008a). Residues at positions 43 and 46 play a subtler role in recognition (Kissinger et al. 1990; Fraenkel et al. 1998; Mahony et al. 2007; Noyes et al. 2008a). One additional prominent determinant, position 51, is almost exclusively asparagine within the extant HD population, where it specifies adenine at base 3. This position was held constant in our library, in anticipation that our selected HDs could be used to inform a predictive recognition model for extant HDs.
Selections employing the HD library were performed separately against each of the 64 TAANNN sites to recover interacting HDs. We observed variability in the selection stringency required to cull the population down to 1000–2000 surviving clones for each target site (Supplemental Fig. S2). Overall, selections employing the HD library yielded a 20- to 200-fold increase in surviving colonies when compared to a negative control entirely lacking the HD. Sequencing the recovered clones from each target site yielded a catalog of ∼4.4 × 104 HDs (Supplemental Table S3) and revealed striking amino acid preferences at some randomized positions within populations recovered from different target sites (Supplemental Fig. S3). Some of these preferences were anticipated based on prior studies of HD specificity (Wolberger et al. 1991; Ades and Sauer 1994; Passner et al. 1999; Noyes et al. 2008a), but many appear to represent novel determinants.
Analysis of selected HDs
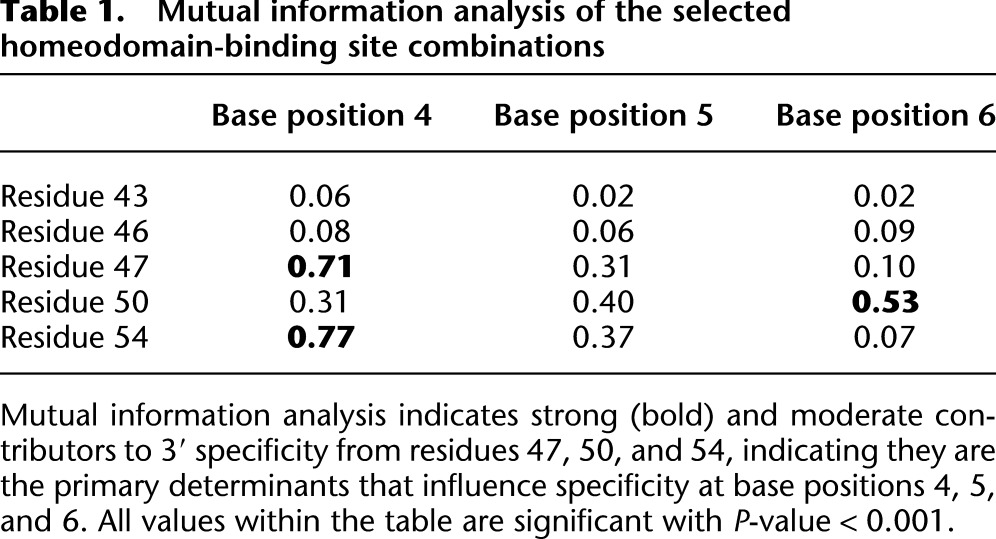
Prominent HD positions influencing base preference were identified by Mutual Information (MI) analysis on the catalog of selected HDs for each target site (Mahony et al. 2007). This analysis identified positions 47, 50, and 54 as strong contributors to 3′ specificity, whereas positions 43 and 46 appeared to have little global influence on the 3′ site preference (Table 1). Significant covariation was observed between residues 47 and 54 and base 4. In addition, a moderate degree of covariation is observed between both of these residue positions and base 5. Moderate covariation is also observed between residue 50 and all of the 3′ base positions but is most pronounced with base 6. The most significant relationships identified between HD position and binding site position are consistent with previously published structural and biochemical data (Treisman et al. 1989; Percival-Smith et al. 1990; Hanes and Brent 1991; Wolberger et al. 1991; Damante et al. 1996; Noyes et al. 2008a).
Table 1.
Mutual information analysis of the selected homeodomain-binding site combinations
Defining the specificity of selected HDs
In an attempt to distinguish selected HD variants that can preferentially bind to each of the 64 TAANNN sites from those that can merely associate favorably with a target site, we determined the DNA-binding specificity for 151 HD variants (Supplemental Table S6; Supplemental Fig. S4). HDs variants were chosen for analysis based on their overlap with the consensus sequence recovered in each selected population or the presence of combinations of recognition residues that were deemed interesting (Supplemental Fig. S3 and Supplemental Table S3). For example, in anticipation of identifying a HD variant that specifies TAACGG, we characterized a clone containing residues R47, E50, and R54 that reflects the predominant consensus sequence recovered for this target site. Preferential DNA-binding specificity for each HD was determined using the B1H system (Noyes et al. 2008a), where the entire population of hundreds to thousands of recovered binding sites was sequenced to construct a recognition motif (Supplemental Fig. S4).
Based on this analysis, we are able to identify HD variants that preferentially bind to or are compatible with 44 out of the 64 target sites (Fig. 2), which represents a sizeable expansion of the 3′ specificities observed in characterized extant HDs (Supplemental Fig. S5). Our analysis of specificities further clarifies the significant association of specificity determinants with certain sequence preferences (Supplemental Table S7) and validates many novel specificity determinants (Fig. 3; Supplemental Table S8). Although this analysis expands the number of primary determinants that can dictate recognition preferences, it is not possible to codify DNA recognition as a set of independent determinants because of the overlapping influence of neighboring determinants. Moreover, specifying some sequence features, such as T at base 6, appears challenging in any sequence context with this HD backbone and randomization scheme.
Figure 2.

Selected HDs with favorable recognition preferences for each target site. A grid illustrating the selected HD variants that preferentially recognize or are compatible with particular 3′ binding site sequences. The amino acids that are present at the randomized recognition positions (43, 46, 47, 50, and 54) are indicated above each motif. Sequences in red indicate those that are present in more than one grid position (i.e., are compatible with two different sites). Empty boxes indicate the absence of quality HD recognizing these sequences.
Figure 3.
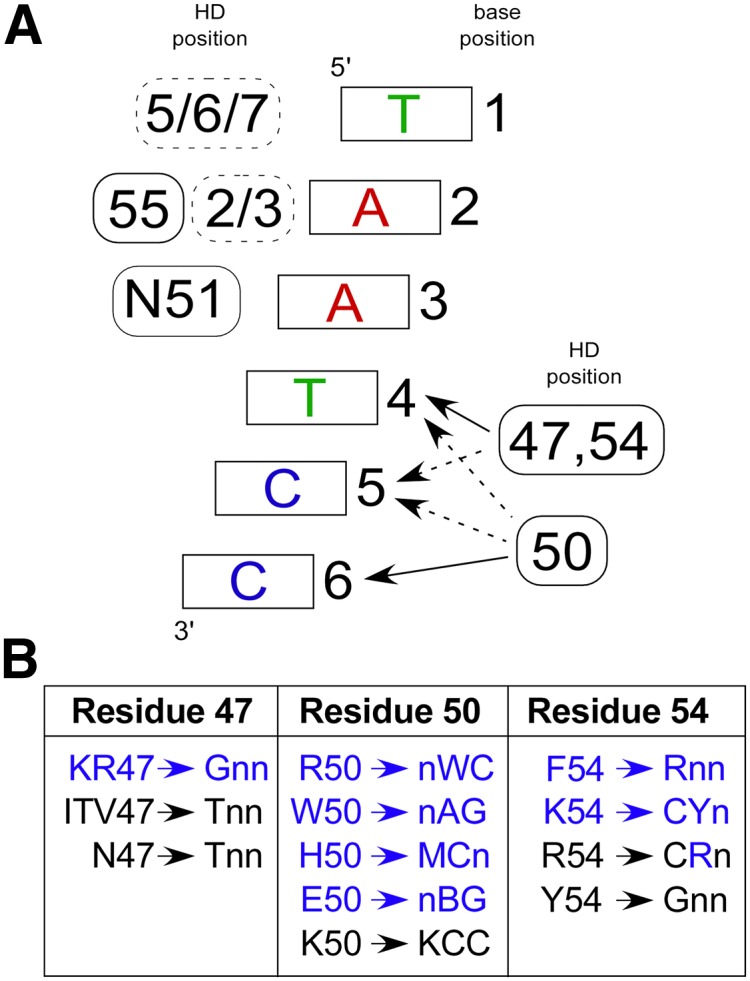
Robust specificity determinants observed in the selected HDs. (A) Canonical recognition pattern for HD–DNA interaction. At the 5′ end of the binding site (bases 1, 2, and 3), positions on the recognition helix (solid boxes) and the N-terminal arm (dashed boxes) contribute to specificity, where the position(s) of the contributing determinants are indicated to the left of the base pair. At the 3′ end of the binding site (bases 4, 5, and 6), homeodomain specificity is primarily defined by positions 47, 50, and 54, where these determinants have overlapping regions of influence. (Solid arrows) Primary positions of interaction; (dotted arrows) secondary influences on specificity. (B) New specificity determinants (blue) and previously described specificity determinants (black) for HDs containing the conserved N51 are broken down by position and trends in base preference within the 3 bp at the 3′ end of the target site. Note that there are exceptions within our characterized HDs to these specificity preferences, likely reflecting the overlapping influence of these determinants.
Sequence discrimination by HD variants
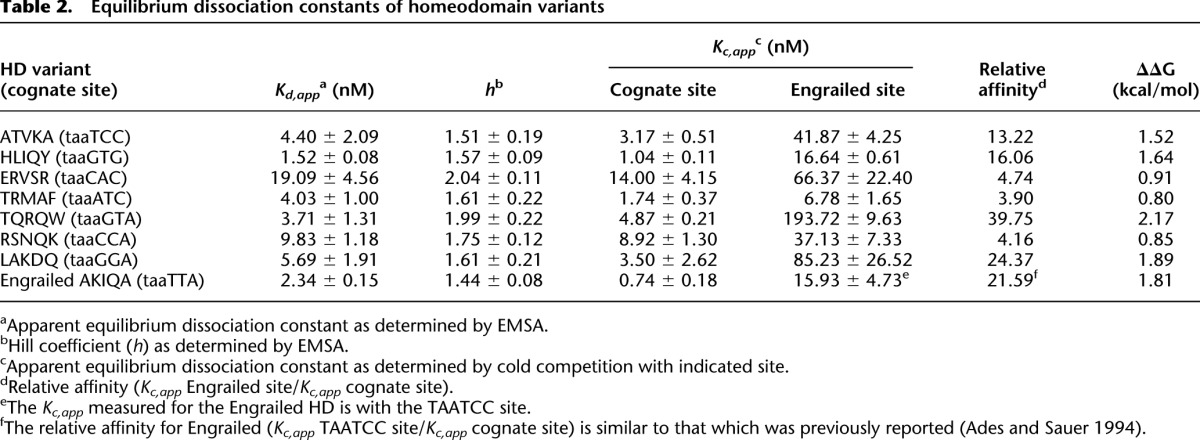
We determined the affinity and specificity of a subset of HD variants for different binding sites in vitro using electrophoretic mobility shift assays (EMSAs). For this analysis, a subset of seven HDs were chosen that span members with both well-defined and novel specificity determinants (Table 2). In all cases, the apparent equilibrium dissociation constant of each HD for its cognate site was similar to the affinity of Engrailed for its cognate site (Supplemental Fig. S6). Cold competition assays were employed to determine the degree of discrimination of each HD variant between its cognate site and the parent Engrailed binding site (Supplemental Fig. S7). The difference in the free energy of binding the cognate and parent site ranged from 0.8–2.2 kcal/mol, where binding the cognate site was always favored (Table 2). The degree of discrimination determined for Engrailed between its preferred site, TAATTA, and TAATCC (22-fold), which served as our internal control, was nearly identical to the difference previously reported by Sauer and colleagues (Ades and Sauer 1994). The TQRQW HD variant (selected HD variants are identified by the five amino acids selected at the randomized positions) has the greatest discrimination against the Engrailed site, displaying a 40-fold preference, while the TRMAF HD variant displays a modest fourfold preference for its target sequence. Thus, our selected HDs display a consistent preference for their identified cognate site outside the B1H system.
Table 2.
Equilibrium dissociation constants of homeodomain variants
Robust behavior of new specificity determinants
To determine if the newly observed specificity determinants are able to define similar DNA sequence preferences in the context of other HD backbones, we grafted the five key residues, residues 43, 46, 47, 50, and 54, from each of the seven HD variants within the sample set into three other Drosophila melanogaster HD backbones: DFD, SCR, and UBX. These HDs share 53%, 51%, and 46% identity with Engrailed, respectively. We then determined the DNA-binding specificity of all these variants using the B1H system (Fig. 4; Supplemental Fig. S8). In almost every instance, the grafted residues altered the DNA-binding specificity of each Hox factor in a predictable manner, in agreement with the previously defined DNA-binding specificity in the Engrailed backbone. In a few instances, such as HLIQY, the introduction of these residues into the Hox backbone slightly altered 5′ sequence preference. This alteration may indicate weak indirect effects of these altered determinants on the 5′ base preference, potentially through interactions with residues 51 and 55, which can influence 5′ specificity.
Figure 4.

Robust function of the new specificity determinants. Grafting key residues (43, 46, 47, 50, and 54) selected from the Engrailed library into the HD backbone of the Hox factor Deformed transforms its sequence preference to resemble the corresponding selected HD mutant.
We also examined the influence of different 5′ specificity determinants on the 3′ specificity of our selected HDs. Previous studies have shown that residues 3 and 55 influence the specificity at base 2, where the presence of K3 and R55 will preferentially recognize G over A (Passner et al. 1999; Piper et al. 1999; Noyes et al. 2008a). We introduced the mutations R3K and K55R into the Engrailed backbone for three HD variants (STRER, KVYER, and NRVMM) and determined their DNA-binding specificity (Supplemental Fig. S9). In all cases, we observe a shift in specificity from A to G at position 2 without substantial alteration in base preference at the other recognition positions. The robust behavior of our new specificity determinants suggests that they will serve as useful parameters for the prediction of DNA-binding specificity in extant HDs.
Computational models of the interactions mediating sequence-specific DNA recognition
We utilized the Rosetta molecular modeling package, which has recently undergone significant revision for protein–DNA complexes (Yanover and Bradley 2011), to predict the base-specific interactions between our sample set of seven HDs and their cognate sites. These structural calculations used a high-resolution Engrailed-DNA co-crystal complex as a starting model (Grant et al. 2000). In a number of instances, the calculated structural models yielded determinant–base interactions that are consistent with the correlated sequence preferences observed within our data set of selected HDs, allowing the potential roles of these determinants to be inferred (Fig. 5; Supplemental Fig. S10). For example, K47 in the LAKDQ–TAAGGA structural model positions the primary amine of this lysine between the O6 carbonyls of G4 and G5, mimicking the observed interaction of K50 with a pair of guanines on the complementary strand in the Q50K EN–DNA structure (Tucker-Kellogg et al. 1997).
Figure 5.

Modeling of HD variants. (A) Co-crystal structure of Engrailed bound to TAATTA (Fraenkel et al. 1998). (B) Model of HD variant STRER bound to its cognate site taaCGG. (C) Model of HD variant LAKDQ bound to its cognate site taaGGA. (D) Model of HD variant RSNQK bound to its cognate site taaCCA. (Dotted lines) Interactions between the protein and DNA (either hydrogen bonds or van der Waals interactions), where the numerical values indicate the distance in angstroms.
Improved predictive models of HD specificity
Previous efforts to predict the DNA-binding specificity of HDs based on their amino acid sequence have focused on nearest neighbor estimates of specificity (Noyes et al. 2008a; Alleyne et al. 2009). We have recently shown that when high-quality alignments of recognition motifs can be obtained, improved recognition models of HD specificity can be achieved using random forest–based methods (Christensen et al. 2012). This recognition model, which is trained on the existing data for extant HDs, is a poor predictor of DNA-binding specificity for our selected HDs (MSE = 0.053) (Supplemental Table S9). This deficit in predictive accuracy was expected given the increased diversity of recognition residues that are present in our selected HDs (Supplemental Fig. S11). Reassuringly, we found that a new recognition model trained only on the selected HDs performed reasonably well in the prediction of the extant HD set (MSE = 0.025; Supplemental Table S9), suggesting that much of the recognition repertoire that is present in the extant set is found in our selected HDs (Supplemental Fig. S12). In a 10-fold cross validation analysis, a joint recognition model between the selected and extant HDs provides excellent accuracy in the prediction of HD specificity within our mutant set (MSE = 0.014; Supplemental Table S9).
To facilitate the prediction of HD specificity, we have constructed a website (stormo.wustl.edu/PreMoTF) that incorporates our improved recognition model. Users can enter the amino acid sequence of a protein containing one or more HDs, and the algorithm will extract each HD sequence and generate a predicted recognition motif and representative position frequency matrix (PFM). When tested on mouse HDs, the predicted PFMs were very similar to those obtained by analysis of PBM data using BEEML-PBM (Zhao and Stormo 2011). By use of this model, we have also populated a page that displays predicted recognition motifs for the majority of the human HDs to facilitate the use of these data in constructing transcription regulatory networks within the human genome (Supplemental Data Set S1).
Discussion
In this study, we performed an unbiased assessment of the breadth of sequences that HDs can specify by selecting variants of Engrailed that would preferentially recognize each of the 64 possible TAANNN binding sites. By use of our selection system, we recovered HDs that preferentially recognized 44 of these sites (Fig. 2), a dramatic increase in the diversity of described recognition sequences. Many of these new sequence preferences are mediated by novel 3′ specificity determinants that are functional when incorporated into independent HD scaffolds (Fig. 4; Supplemental Figs. S8, S9).
Consistent with prior studies on HDs, MI analysis demonstrates critical overlapping roles for the residues at positions 47, 50, and 54 for 3′ base recognition. The overlap between these determinants may represent either direct or indirect effects, however at the level of individual subsites, one determinant typically dominates base preference at a specific subsite position. For example, while strong covariation is observed between residues 47 and 54, and base 4 (Table 1), K54 is highly preferred for recognition of CYN subsites, whereas the recovered residue at position 47 is more variable. The presence of a positively charged residue at positions 43 or 46 is anti-correlated over the entire data set (Supplemental Table S4), suggesting that these residues tune the overall affinity of the HD by adjusting electrostatic interactions with the phosphodiester backbone. These and other positions may also be responsible for more subtle sequence preferences that have been observed in protein binding microarray analysis of HD specificity (Berger et al. 2008) that potentially lead to discrimination of TFs between different binding sites of moderate affinity (Badis et al. 2009).
The diverse and potentially independent assortment of specificity determinants within our data set provides a foundation for constructing more accurate predictive models for 3′ DNA recognition by HDs. While significant prior effort has been expended on characterizing HD recognition, the functionality of specific determinants at critical recognition positions has remained poorly defined, and as a consequence, past predictive models of HD–DNA recognition have relied on nearest-neighbor type analyses (Noyes et al. 2008a; Alleyne et al. 2009). These models perform poorly when trying to predict the specificity of our selected HDs, which likely results from a lack of amino acid diversity at the key determinant positions within their training sets (Fig. 1). In the context of our improved predictive models, we can predict 3′ specificity of a representative set of extant HDs with reasonable accuracy (Supplemental Table S9), and a predictive model combining all of the available data provides superior performance in predicting HD specificity. Thus, selection-based interrogation of HD recognition can inform the construction of predictive models, much as it has for Cys2His2 zinc finger proteins (Benos et al. 2002; Kaplan et al. 2005; Liu and Stormo 2008; Persikov et al. 2009; Persikov and Singh 2011).
Our ability to select HDs with radically different specificity from characterized extant HDs, where novel sets of specificity determinants are employed, raises questions as to why extant HDs appear to be constrained in their diversity at the key recognition positions? Naively, we expect nature to exploit the full recognition potential of this domain to make a variety of orthogonal regulators for independent function in transcriptional regulatory networks. This characteristic is observed in the largest family of DNA-binding domains, Cys2His2 zinc fingers (Emerson and Thomas 2009), where comparison of zinc finger proteins across the mouse and human genomes indicates that this family is rapidly evolving within the finger arrays (Myers et al. 2010). The diversity in zinc finger protein (ZFP) recognition potential is even manifest within the human population, where differences in the fingers present in PRDM9 and their resulting specificity lead to differences in the location of meiotic recombination hotspots in individuals (Baudat et al. 2010). In this regard, ZFPs appear to be an outlier, as most other well-characterized families of DNA-binding domains–like HDs–display limited diversity in their core recognition motifs and the recognition residues that they employ (Deppmann et al. 2006; Wei et al. 2010; De Masi et al. 2011). It is possible that the recognition potential of these other families of DNA-binding domains are similarly constrained. For HDs, the source of the selective pressure limiting the employed diversity of recognition residues is unclear, but understanding its origin would provide insight into the fitness barriers that influence the evolution of novel transcriptional regulatory networks in organisms.
In many instances, HDs function as complexes with other DNA-binding domains to exert their gene regulatory function (Mann et al. 2009). This aspect of recognition is critical for the biological function of many of these factors, where complex formation can alter recognition preference of the component HDs. The most thoroughly characterized example of the influence of partner association on recognition is the Hox-Pbx heterodimer, where interactions between residues within and neighboring the N-terminal arm and minor groove features play critical roles in defining sequence preference for this complex (Joshi et al. 2007; Slattery et al. 2011). In general, the role of residues within the N-terminal arm in DNA recognition remains poorly defined, although there is evidence that sequence preference may be driven by complementarity to DNA sequence-dependent minor groove width (Rohs et al. 2009; Slattery et al. 2011). We have demonstrated that some of our selected HDs can tolerate changes that alter 5′ sequence recognition, but the degree of crosstalk between the recognition residues in the 5′ and 3′ segments of the binding site remains poorly defined. A selection-based analysis of the recognition potential of the N-terminal arm could help to clarify the roles of individual positions in minor groove recognition.
Our archive might present an opportunity to employ HDs as components of artificial transcription factors or endonucleases. The area of engineered DNA-binding domains has primarily been the purview of ZFPs (Urnov et al. 2010);however, efforts to engineer ZFPs to recognize a wide variety of target sites using public archives have been most successful for guanine-rich binding sites (Ramirez et al. 2008; Zhu et al. 2011a). HDs provide potential utility in the recognition of A-T-rich sequences and, in the context of zinc finger-HD chimeras (Pomerantz et al. 1995; Rivera et al. 1996), may have value in expanding the sequences that be efficiently targeted by zinc finger–based artificial nucleases.
Methods
Construction of the HD library
A pB1H2ω2-12En (pB1H2ω2-12En(SB)) (Noyes et al. 2008a) construct was created with the following modifications to the original en sequence: Restriction sites SacI and BamHI were installed for use with cassette mutagenesis of the recognition helix through introduction of a synonymous mutation at L38 and a T60G mutation, respectively (Supplemental Table S10). The randomized recognition helix was cloned into the SacI and BamHI sites of pB1H2ω2-12En(SB) by the direct ligation of the following phosphorylated and annealed three oligonucleotide: EN K55 library, EN Library 5p comp, and EN Library 3p comp (Supplemental Table S10). Following transformation into electrocompetent XL1Blue cells, the library was plated on 20 150-mm 2 × YT plates containing 100 μg/mL carbenicillin and incubated overnight at 37°C. The recovered library size was 1.3 × 108, where the theoretical library size, 3 × 107, was oversampled three- to fourfold.
Design of the target binding sites for the selection of HDs
The 64 target sites (GGCCGCnnnTTAGCTGGGCGGGACG) for use with the HD Library selections were cloned between the NotI and EcoRI site in pH3U3 (Noyes et al. 2008b). The bold nnnTTA element is the reverse complement of the 6-bp HD target site TAANNN, where the NNN represents each of the 64 possible 3-bp combinations. The bold TGGGCG element is the Zif12 binding site, which is positioned 10 bp upstream of the −35 box.
Bacterial-one hybrid (B1H) selections with the HD library
Each HD library/TAANNN selection in the B1H system was performed basically as previously described (Noyes et al. 2008b). For each selection, at least 1 × 108 dual transformants (of HD expression vector and binding site reporter vector into the selection strain) were plated on NM media supplemented with 1 μM IPTG and 200 μM uracil. The stringency of each selection was adjusted such that 1000–2000 colonies were recovered (Supplemental Fig. S2). About 24 colonies were initially sequenced to confirm the success of the HD selections. Subsequently, recovered HD library members were identified via Illumina sequencing. Surviving colonies from each selection were pooled and prepared for sequencing as previously described (Gupta et al. 2010). HD clones were amplified using a forward primer (CAAGCAGAAGACGGCATACGAGCTCTTCCGATCTATGCTTGCCCTGTCGAGTCC) and reverse primer (CTTAATGCGCCGCTACAGGGC), where the forward primer incorporated the Illumina P2-adapter sequence (bold). Each PCR product was then digested with either BamHI or XbaI for the ligation of barcoded P1 adapters (Supplemental Tables S1, S2) prior to Illumina library generation and sequencing.
MI and other statistical data analysis
The catalog of ∼44,000 selected HDs identified by Illumina sequencing for the 64 target sites was used to calculate MI between the randomized positions within the HD and base positions 4, 5, and 6 in the DNA target site according to the method previously described (Mahony et al. 2007). Significance was determined by calculating the MI for a set of randomly associated selected recognition helices to the 64 target sites performed 1000 times followed by a nonparametric test used to derive a null distribution where a P-value < 0.001 for each MI value was considered significant.
The two-sided Fisher exact test was applied to assess significant association between the positive charge status at position 43 and at position 46 for HDs recovered for each of the 64 binding sites and all binding sites combined. This statistical analysis was also applied to the correlation between the selected specificity determinants and a subset of recognition sequences. The odds ratio and its 95% of confidence interval were computed for each triplet and combined using the fisher_test function based on a conditional maximum likelihood estimation. These statistical analyses were performed using R, a system for statistical computation and graphics (Ihaka and Gentleman 1996). To adjust for multiple comparisons for the 64 binding sites, P-values were adjusted using the B-H method (Benjamini and Hochberg 1995), where sites with adjusted P-value < 0.05 were considered significant.
B1H selections of HD variants with the ZF10 library
All HD variants characterized from the HD library selections were sequences that were directly isolated from colonies on the selection plates, from either direct isolation of individual clones or the reconstruction of variants identified by Illumina sequencing through the ligation of phosphorylated and annealed oligonucleotides into pB1H2ω2-12En (Supplemental Table S11). Each ZF10 library/HD variant selection was performed as previously described (Noyes et al. 2008a) except that all selections were plated on NM media supplemented with 5 mM 3-AT, 1 μM IPTG, and 200 μM uracil. Recovered ZF10 library members were identified via Illumina sequencing as previously described (Gupta et al. 2010) except that the initial PCR product was digested with either BamHI or NcoI for the ligation of barcoded P1 adaptors (Supplemental Tables S1, S5). Overrepresented sequence motifs were identified using MEME (Bailey and Elkan 1994) from the top 1000 most frequently occurring unique sequences within the Illumina data set except for the grafted HDs, where the top 500 most frequently occurring unique sequences were used. Additional sequences were included in cases where they had the same number of reads as the 1000th (or 500th) sequence in the set. The input parameters used for MEME were zero or one motif per sequence (zoops), four bases as the width minimum, and 10 bases as the width maximum, while all other parameters retained the program default settings. Recognition motifs for each HD were then constructed as previously described (Zhu et al. 2011a) by weighting the number of reads for each sequence that comprise the most significant motif identified by MEME, where the number of sequences input for motif discovery and incorporated into each motif is reported in Supplemental Table 6
Expression and purification of proteins
Each HD variant was expressed in Rosetta2(DE3)pLysS cells as C-terminal fusions to a purification tag sequence consisting of a His-6 tag, maltose binding protein (MBP), and Tev protease cleavage site. Cells were lysed by sonication. Protein was purified from the lysates using Amylose Resin (New England Biolabs) and then was eluted from the amylose resin in binding buffer without BSA and IGEPAL CA-630 (25 mM NaCl, 10 mM Tris-HCl at pH 7.5, 0.1 mM EDTA, 1 mM DTT, and 5% glycerol) supplemented with 40 mM maltose. Protein concentrations were determined by absorbance at 280 nm. Single use aliquots of protein were stored at −80 prior to use.
Preparation of binding sites for EMSAs
Duplex binding sites were prepared by annealing the top oligonucleotide (GGGCAGNNNNNNGGACG) and bottom oligonucleotide (GGCGTCCNNNNNNCTGC) (Invitrogen) for a given binding site in annealing buffer (10 mM Tris-HCl, 50 mM NaCl, and 1 mM EDTA) to the final concentration of 40 μM dsDNA, where the N6 represents the 6-bp binding site used in a given EMSA. Initial single-stranded oligonucleotide concentrations were determined by absorbance at 260 nm. For detection, annealed oligonucleotides were radiolabeled with alpha-32P dCTP and Klenow (exo-) (New England Biolabs) followed by a MicroSpin G-25 column (GE Healthcare) purification.
Determination of apparent dissociation constant via EMSAs
Varying concentrations of a given purified HD variant were equilibrated with 40 pM of labeled oligonucleotide in binding buffer (25 mM NaCl, 10 mM Tris-HCl at pH 7.5, 0.1 mM EDTA, 1 mM DTT, 5% glycerol, 0.1 mg/mL BSA, and 0.1% IGEPAL CA-630) at room temperature for 4 h. Samples were loaded onto a 5% polyacrylamide gel without loading dye in 0.5× TBE buffer while running at 300 V at 4°C. Gels were run for 40 min following loading. Gels were dried and then exposed on phosphoimaging plates for 8–72 h. Plates were imaged using a Typhoon FLA 9000, and quantified using ImageGauge V4.22. The apparent equilibrium dissociation constants (Kd,app) were determined using the modified Hill equation:
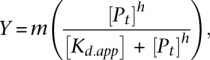 |
where Y is the fraction of bound DNA as determined by the ratio of the bound DNA band to the total (free + bound) bands, m is a normalization factor that represents Y max, [P]t is the total protein concentration, and h is the Hill coefficient.
Determination of apparent dissociation constant via competition binding assays
Competition assays were performed under the conditions described for the determination of apparent dissociation constant via EMSA except that varying concentrations of an unlabeled-annealed oligonucleotide were added to a subsaturating (70%–90%) amount of a given purified HD variant and 40 pM of labeled oligonucleotide prior to equilibration. The concentration of DNA that disrupts 50% of the bound labeled complex (IC50) was determined using a simplified sigmoidal dose-response curve (Ryder et al. 2008):
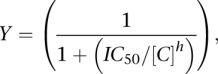 |
where Y is the fraction of bound DNA, C is the concentration of unlabeled competitor, and h is the Hill coefficient. The IC50 is then converted into the apparent equilibrium dissociation constant for the competitor (Kc,app) using the Lin and Riggs equation (Lin and Riggs 1972):
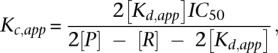 |
where P is the purified HD variant concentration, R is the concentration of the labeled oligonucleotide, and Kd,app is the apparent equilibrium dissociation constant of the HD for the labeled oligonucleotide as measured by EMSA.
Computational modeling of HD–DNA complexes
Modeling of mutant HD structures was performed with RosettaDNA, using the recently described flexible DNA protocol and scoring function (RosettaDNA executable and accompanying parameter sets kindly provided by Philip Bradley at the Fred Hutchinson Cancer Research Center, Seattle, Washington) (Yanover and Bradley 2011). Starting with the structure of the DNA-bound Engrailed Q50A HD (Grant et al. 2000), 20 models were generated by RosettaDNA for each DNA-bound mutant HD. Each model was minimized with flexible DNA backbone and bases, and side-chain packing was performed for residues adjacent to the DNA major groove (residues 31, 43–44, 46–51, 53–55, 57–58 in the crystal structure). Extended side-chain rotamer sets were used for buried residues having 15 neighbors within 10 Å (“-ex1 -ex2 -ex1aro::level 6 -extrachi_cutoff 15”), while extra DNA rotamers were used to sample base flexibility (“-exdna::level 2”). DNA backbone flexibility was specified for the 6-bp DNA target site plus 2 bp flanking each side of the site. For each mutant, the 20 models from RosettaDNA were rescored using DDNA, a knowledge-based energy potential developed to predict protein/DNA structures and binding affinities (Zhao et al. 2010), and the top DDNA score was used to select a structural model reflecting the anticipated interactions at the HD-DNA interface.
RF predictive modeling
Protein and PFM alignments and relative scaling of the PFMs used as inputs for the construction of a RF model were performed as previously described (Christensen et al. 2012). RF regression was performed as described using the previously identified determinant positions (3, 6, 19, 47, 50, 54, and 55) identified from the adjusted MI assessment of the 264 characterized extant HDs described in our previous study (Christensen et al. 2012). Models to test the utility of the extant HD specificity data from 246 mouse and fruit fly HDs (Berger et al. 2008; Noyes et al. 2008a,b; Zhu et al. 2011b) and the selected HDs in this study were trained as noted in Supplemental Table S9, where the evaluation incorporated 10-fold cross validation when the training set and prediction set overlapped. The reported mean squared error (MSE) values reflect the MSE per motif parameter in the predicted motif (Christensen et al. 2012).
Data access
Illumina data for the selected and characterized HDs have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE35806. A website (stormo.wustl.edu/PreMoTF.v2) provides user access to the predictive model of HD specificity and predictions for all of the annotated HDs in the human genome.
Acknowledgments
We thank Philip Bradley at the Fred Hutchinson Cancer Research Center for his generous contribution of RosettaDNA executable and parameter sets that allowed the calculation of our HD-DNA variant complexes. This research was supported by the US National Institutes of Health (NIH) (R01GM068110 [S.A.W.], R01GM084884 [Z.W.], and R01HG00249 [G.D.S.]).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.139014.112.
References
- Ades SE, Sauer RT 1994. Differential DNA-binding specificity of the engrailed homeodomain: The role of residue 50. Biochemistry 33: 9187–9194 [DOI] [PubMed] [Google Scholar]
- Ades SE, Sauer RT 1995. Specificity of minor-groove and major-groove interactions in a homeodomain-DNA complex. Biochemistry 34: 14601–14608 [DOI] [PubMed] [Google Scholar]
- Alleyne TM, Pena-Castillo L, Badis G, Talukder S, Berger MF, Gehrke AR, Philippakis AA, Bulyk ML, Morris QD, Hughes TR 2009. Predicting the binding preference of transcription factors to individual DNA k-mers. Bioinformatics 25: 1012–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. 2009. Diversity and complexity in DNA recognition by transcription factors. Science 324: 1720–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B 2010. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327: 836–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300 [Google Scholar]
- Benos PV, Lapedes AS, Stormo GD 2002. Probabilistic code for DNA recognition by proteins of the EGR family. J Mol Biol 323: 701–727 [DOI] [PubMed] [Google Scholar]
- Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, Pena-Castillo L, Alleyne TM, Mnaimneh S, Botvinnik OB, Chan ET, et al. 2008. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133: 1266–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burglin TR 2011. Homeodomain subtypes and functional diversity. Subcell Biochem 52: 95–122 [DOI] [PubMed] [Google Scholar]
- Christensen RG, Enuameh MS, Noyes MB, Brodsky MH, Wolfe SA, Stormo GD 2012. Recognition models to predict DNA-binding specificities of homeodomain proteins. Bioinformatics 28: i84–i89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly JP, Augustine JG, Francklyn C 1999. Mutational analysis of the engrailed homeodomain recognition helix by phage display. Nucleic Acids Res 27: 1182–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damante G, Pellizzari L, Esposito G, Fogolari F, Viglino P, Fabbro D, Tell G, Formisano S, Di Lauro R 1996. A molecular code dictates sequence-specific DNA recognition by homeodomains. EMBO J 15: 4992–5000 [PMC free article] [PubMed] [Google Scholar]
- De Masi F, Grove CA, Vedenko A, Alibes A, Gisselbrecht SS, Serrano L, Bulyk ML, Walhout AJ 2011. Using a structural and logics systems approach to infer bHLH-DNA binding specificity determinants. Nucleic Acids Res 39: 4553–4563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deppmann CD, Alvania RS, Taparowsky EJ 2006. Cross-species annotation of basic leucine zipper factor interactions: Insight into the evolution of closed interaction networks. Mol Biol Evol 23: 1480–1492 [DOI] [PubMed] [Google Scholar]
- Ekker SC, Jackson DG, von Kessler DP, Sun BI, Young KE, Beachy PA 1994. The degree of variation in DNA sequence recognition among four Drosophila homeotic proteins. EMBO J 13: 3551–3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson RO, Thomas JH 2009. Adaptive evolution in zinc finger transcription factors. PLoS Genet 5: e1000325 doi: 10.1371/journal.pgen.1000325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraenkel E, Rould MA, Chambers KA, Pabo CO 1998. Engrailed homeodomain-DNA complex at 2.2 A resolution: A detailed view of the interface and comparison with other engrailed structures. J Mol Biol 284: 351–361 [DOI] [PubMed] [Google Scholar]
- Gehring WJ, Affolter M, Burglin T 1994. Homeodomain proteins. Annu Rev Biochem 63: 487–526 [DOI] [PubMed] [Google Scholar]
- Grant RA, Rould MA, Klemm JD, Pabo CO 2000. Exploring the role of glutamine 50 in the homeodomain–DNA interface: Crystal structure of engrailed (Gln50→ala) complex at 2.0 A. Biochemistry 39: 8187–8192 [DOI] [PubMed] [Google Scholar]
- Gupta A, Meng X, Zhu LJ, Lawson ND, Wolfe SA 2010. Zinc finger protein-dependent and -independent contributions to the in vivo off-target activity of zinc finger nucleases. Nucleic Acids Res 39: 381–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanes SD, Brent R 1991. A genetic model for interaction of the homeodomain recognition helix with DNA. Science 251: 426–430 [DOI] [PubMed] [Google Scholar]
- Hovde S, Abate-Shen C, Geiger JH 2001. Crystal structure of the Msx-1 homeodomain/DNA complex. Biochemistry 40: 12013–12021 [DOI] [PubMed] [Google Scholar]
- Ihaka R, Gentleman R 1996. R: A language for data analysis and graphics. J Comput Graph Statist 5: 299–314 [Google Scholar]
- Joshi R, Passner JM, Rohs R, Jain R, Sosinsky A, Crickmore MA, Jacob V, Aggarwal AK, Honig B, Mann RS 2007. Functional specificity of a Hox protein mediated by the recognition of minor groove structure. Cell 131: 530–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan T, Friedman N, Margalit H 2005. Ab initio prediction of transcription factor targets using structural knowledge. PLoS Comput Biol 1: e1 doi: 10.1371/journal.pcbi.0010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissinger CR, Liu BS, Martin-Blanco E, Kornberg TB, Pabo CO 1990. Crystal structure of an engrailed homeodomain-DNA complex at 2.8 A resolution: A framework for understanding homeodomain-DNA interactions. Cell 63: 579–590 [DOI] [PubMed] [Google Scholar]
- Kornberg TB 1993. Understanding the homeodomain. J Biol Chem 268: 26813–26816 [PubMed] [Google Scholar]
- Lin SY, Riggs AD 1972. Lac repressor binding to non-operator DNA: Detailed studies and a comparison of equilibrium and rate competition methods. J Mol Biol 72: 671–690 [DOI] [PubMed] [Google Scholar]
- Liu J, Stormo GD 2008. Context-dependent DNA recognition code for C2H2 zinc-finger transcription factors. Bioinformatics 24: 1850–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony S, Auron PE, Benos PV 2007. Inferring protein-DNA dependencies using motif alignments and mutual information. Bioinformatics 23: i297–i304 [DOI] [PubMed] [Google Scholar]
- Mann RS, Lelli KM, Joshi R 2009. Hox specificity unique roles for cofactors and collaborators. Curr Top Dev Biol 88: 63–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bowden R, Tumian A, Bontrop RE, Freeman C, MacFie TS, McVean G, Donnelly P 2010. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327: 876–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyes MB, Christensen RG, Wakabayashi A, Stormo GD, Brodsky MH, Wolfe SA 2008a. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell 133: 1277–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyes MB, Meng X, Wakabayashi A, Sinha S, Brodsky MH, Wolfe SA 2008b. A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system. Nucleic Acids Res 36: 2547–2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passner JM, Ryoo HD, Shen L, Mann RS, Aggarwal AK 1999. Structure of a DNA-bound Ultrabithorax-Extradenticle homeodomain complex. Nature 397: 714–719 [DOI] [PubMed] [Google Scholar]
- Percival-Smith A, Muller M, Affolter M, Gehring WJ 1990. The interaction with DNA of wild-type and mutant fushi tarazu homeodomains. EMBO J 9: 3967–3974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persikov AV, Singh M 2011. An expanded binding model for Cys2His2 zinc finger protein–DNA interfaces. Phys Biol 8: 035010 doi: 10.1088/1478-3975/8/3/035010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persikov AV, Osada R, Singh M 2009. Predicting DNA recognition by Cys2His2 zinc finger proteins. Bioinformatics 25: 22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper DE, Batchelor AH, Chang CP, Cleary ML, Wolberger C 1999. Structure of a HoxB1–Pbx1 heterodimer bound to DNA: Role of the hexapeptide and a fourth homeodomain helix in complex formation. Cell 96: 587–597 [DOI] [PubMed] [Google Scholar]
- Pomerantz JL, Sharp PA 1994. Homeodomain determinants of major groove recognition. Biochemistry 33: 10851–10858 [DOI] [PubMed] [Google Scholar]
- Pomerantz JL, Sharp PA, Pabo CO 1995. Structure-based design of transcription factors. Science 267: 93–96 [DOI] [PubMed] [Google Scholar]
- Ramirez CL, Foley JE, Wright DA, Muller-Lerch F, Rahman SH, Cornu TI, Winfrey RJ, Sander JD, Fu F, Townsend JA, et al. 2008. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods 5: 374–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera VM, Clackson T, Natesan S, Pollock R, Amara JF, Keenan T, Magari SR, Phillips T, Courage NL, Cerasoli F Jr, et al. 1996. A humanized system for pharmacologic control of gene expression. Nat Med 2: 1028–1032 [DOI] [PubMed] [Google Scholar]
- Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B 2009. The role of DNA shape in protein–DNA recognition. Nature 461: 1248–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohs R, Jin X, West SM, Joshi R, Honig B, Mann RS 2010. Origins of specificity in protein–DNA recognition. Annu Rev Biochem 79: 233–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder SP, Recht MI, Williamson JR 2008. Quantitative analysis of protein-RNA interactions by gel mobility shift. Methods Mol Biol 488: 99–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery M, Riley T, Liu P, Abe N, Gomez-Alcala P, Dror I, Zhou T, Rohs R, Honig B, Bussemaker HJ, et al. 2011. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell 147: 1270–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steadman DJ, Giuffrida D, Gelmann EP 2000. DNA-binding sequence of the human prostate-specific homeodomain protein NKX3.1. Nucleic Acids Res 28: 2389–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treisman J, Gonczy P, Vashishtha M, Harris E, Desplan C 1989. A single amino acid can determine the DNA binding specificity of homeodomain proteins. Cell 59: 553–562 [DOI] [PubMed] [Google Scholar]
- Tucker-Kellogg L, Rould MA, Chambers KA, Ades SE, Sauer RT, Pabo CO 1997. Engrailed (Gln50→Lys) homeodomain–DNA complex at 1.9 A resolution: Structural basis for enhanced affinity and altered specificity. Structure 5: 1047–1054 [DOI] [PubMed] [Google Scholar]
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD 2010. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11: 636–646 [DOI] [PubMed] [Google Scholar]
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM 2009. A census of human transcription factors: Function, expression and evolution. Nat Rev Genet 10: 252–263 [DOI] [PubMed] [Google Scholar]
- Wei GH, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolma A, Varjosalo M, Gehrke AR, et al. 2010. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J 29: 2147–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolberger C, Vershon AK, Liu B, Johnson AD, Pabo CO 1991. Crystal structure of a MATα2 homeodomain-operator complex suggests a general model for homeodomain-DNA interactions. Cell 67: 517–528 [DOI] [PubMed] [Google Scholar]
- Yanover C, Bradley P 2011. Extensive protein and DNA backbone sampling improves structure-based specificity prediction for C2H2 zinc fingers. Nucleic Acids Res 39: 4564–4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Stormo GD 2011. Quantitative analysis demonstrates most transcription factors require only simple models of specificity. Nat Biotechnol 29: 480–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Yang Y, Zhou Y 2010. Structure-based prediction of DNA-binding proteins by structural alignment and a volume-fraction corrected DFIRE-based energy function. Bioinformatics 26: 1857–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Smith T, McNulty J, Rayla AL, Lakshmanan A, Siekmann AF, Buffardi M, Meng X, Shin J, Padmanabhan A, et al. 2011a. Evaluation and application of modularly assembled zinc-finger nucleases in zebrafish. Development 138: 4555–4564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ, Christensen RG, Kazemian M, Hull CJ, Enuameh MS, Basciotta MD, Brasefield JA, Zhu C, Asriyan Y, Lapointe DS, et al. 2011b. FlyFactorSurvey: A database of Drosophila transcription factor binding specificities determined using the bacterial one-hybrid system. Nucleic Acids Res 39: D111–D117 [DOI] [PMC free article] [PubMed] [Google Scholar]