

Abstract
Nutrient availability profoundly influences gene expression. Many animal genes encode multiple transcript isoforms, yet the effect of nutrient availability on transcript isoform expression has not been studied in genome-wide fashion. When Caenorhabditis elegans larvae hatch without food, they arrest development in the first larval stage (L1 arrest). Starved larvae can survive L1 arrest for weeks, but growth and post-embryonic development are rapidly initiated in response to feeding. We used RNA-seq to characterize the transcriptome during L1 arrest and over time after feeding. Twenty-seven percent of detectable protein-coding genes were differentially expressed during recovery from L1 arrest, with the majority of changes initiating within the first hour, demonstrating widespread, acute effects of nutrient availability on gene expression. We used two independent approaches to track expression of individual exons and mRNA isoforms, and we connected changes in expression to functional consequences by mining a variety of databases. These two approaches identified an overlapping set of genes with alternative isoform expression, and they converged on common functional patterns. Genes affecting mRNA splicing and translation are regulated by alternative isoform expression, revealing post-transcriptional consequences of nutrient availability on gene regulation. We also found that phosphorylation sites are often alternatively expressed, revealing a common mode by which alternative isoform expression modifies protein function and signal transduction. Our results detail rich changes in C. elegans gene expression as larvae initiate growth and post-embryonic development, and they provide an excellent resource for ongoing investigation of transcriptional regulation and developmental physiology.
Post-embryonic development of the roundworm Caenorhabditis elegans is governed by nutrient availability and other environmental conditions. High population density plus limited food causes developmental arrest as dauer larvae, an alternative to the third larval stage with significant morphological modification (Golden and Riddle 1983; Hu 2007). When larvae hatch in the absence of food, they arrest development in the first larval stage (L1 arrest or L1 diapause) without morphological modification (Baugh and Sternberg 2006). Microarray analysis of larvae hatching in the presence or absence of food revealed very different expression profiles in each condition (Baugh et al. 2009). Upon feeding, arrested L1s initiate growth and post-embryonic development, and their gene expression profile is similar to that of fed larvae after 3 h of recovery (Baugh et al. 2009). Genome-wide analysis of RNA polymerase II (Pol II) binding revealed that starved animals alter their pattern of transcription in response to feeding within 1 h of recovery (Baugh et al. 2009). This work revealed rapid recovery dynamics, but there has been no temporal analysis of mRNA levels during transition between arrest and full recovery. Furthermore, the microarrays used monitored gene expression but could not distinguish expression of individual transcript isoforms. L1 arrest and recovery provide a powerful model for nutritional control of development, and transcriptome analysis should elucidate molecular mechanisms governing quiescence and growth in response to nutrient availability.
Gene expression microarrays revolutionized biology by enabling measurement of mRNA expression levels genome-wide. More recent technological advances enabled measurement of mRNA expression levels by direct sequencing of the transcriptome with millions of short reads (RNA-seq). RNA-seq promises even better insight than microarrays with its ability to measure where the transcript of a gene starts, stops, and is spliced (Wang et al. 2009). In particular, when coupled with a statistical model, RNA-seq can estimate the levels of mRNA isoforms, a difficult task using microarrays. The sequence differences between isoforms can alter protein function by changing coding sequence (CDS); alter mRNA stability, localization, and translation by changing 3′ untranslated regions (UTRs); or reveal alternative promoter use (Zahler 2005). Since at least 25% (5210) of C. elegans’ genes produce multiple isoforms (WormBase 220), there is likely to be substantial regulation of the transcriptome that is invisible to traditional microarrays.
Previous studies have revealed important roles for alternative transcript isoforms. Surveys of alternative splicing in C. elegans identified hundreds of examples of splice forms that show alternative expression in development, including tissue-specific expression of transcript isoforms (Kuroyanagi et al. 2006; Hillier et al. 2009; Ramani et al. 2011). Intriguingly, genes involved in splicing are themselves often regulated by alternative splicing coupled to nonsense-mediated decay, suggesting post-transcriptional autoregulation (Sureau 2001; Ni et al. 2007; Barberan-Soler and Zahler 2008). Environmental control of alternative isoform expression has also been observed. For example, in Neurospora crassa, different isoforms of the frequency gene extend the temperature range of the circadian oscillator (Colot et al. 2005). Also, in plants, the large SR (serine/arginine rich) protein family of splicing factors is responsive to stress (Duque 2011). Finally, a survey of 21 alternative splicing events from 17 genes found relative expression levels of transcript isoforms to be well conserved between C. elegans and Caenorhabditis briggsae, suggesting that isoform expression levels are important for the fitness of the organism (Rukov et al. 2007). However, there has been no global analysis of nutritional control of alternative mRNA isoform expression in any system. The extent to which different isoforms are expressed in response to nutrient availability, the particular isoforms involved, and the functional consequences of these changes are unclear.
We used RNA-seq to characterize the poly-adenylated transcriptome during L1 arrest and recovery (after feeding with Escherichia coli). We used a combination of statistical approaches to identify differentially expressed genes and transcripts, and we connected these changes in expression with functional consequences by a variety of approaches. In particular, we used a pair of independent statistical tools (DEXSeq and Cufflinks) to identify genes with alternatively expressed transcript isoforms (Trapnell et al. 2010; Anders et al. 2012). This allowed us to corroborate the results of our analysis while analyzing temporal expression of exons, transcripts, and genes during L1 arrest and recovery. We tracked expression of alternative CDSs and 3′ UTRs, focusing on expression of predicted protein domains, phosphorylation sites, and miRNA binding sites. Our results shed light on the pervasive and extremely rapid changes in the C. elegans transcriptome as larvae recover from developmental arrest, connect these changes with specific functional consequences, and provide an excellent resource for future research.
Results
Detection and quantification of gene expression
We used RNA-seq to measure poly-adenylated RNA expression in C. elegans larvae during L1 arrest and recovery. We sampled biological replicates of L1 larvae starved for 12 h and subsequently fed for 1, 3, and 6 h (Fig. 1). We used Cufflinks 1.0.2 to determine gene expression levels from single-end, 50-nt RNA-seq data (Trapnell et al. 2010). Out of 19,518 genes annotated as “protein-coding” in WormBase 220, we detected 13,350 (68%) in at least one of the time points with a false discovery rate of 0.1% (Supplemental Tables 1, 2), indicating that our sequencing depth is sufficient to analyze transcriptome dynamics.
Figure 1.
Experimental design for RNA-seq analysis of L1 arrest and recovery. L1 larvae were starved for ∼12 h after hatching, and post-embryonic development was initiated by feeding them E. coli. Larvae were sampled for RNA-seq after 12 h of L1 arrest (0 h recovery) and after 1, 3, and 6 h of recovery.
Previous high-density oligonucleotide microarray analysis investigated gene expression during the onset of L1 arrest, during normal larval development and in a single 3-h time point after recovery from L1 arrest (Baugh et al. 2009). The fact that L1 arrest and 3-h recovery were analyzed in both cases presents the opportunity to validate the RNA-seq data and Cufflinks output. The microarray and RNA-seq data agree remarkably well in both time points, especially considering that independent biological samples were prepared for each in two different laboratories over 2 yr apart (Spearman’s r = 0.81 and 0.79) (Supplemental Fig. 1). This cross-platform comparison indicates that our RNA-seq methodology and analysis produces reliable gene expression measurements.
Differential expression and cluster analysis
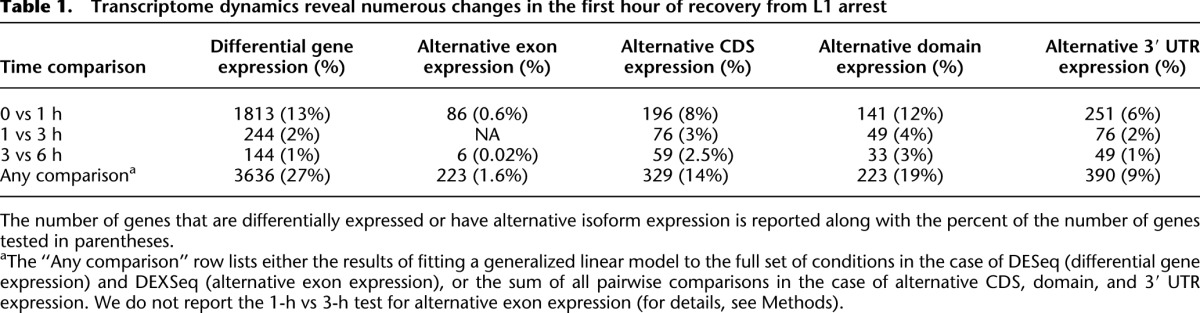
We used DESeq 1.6.1 to evaluate the statistical significance of differential gene expression (Anders and Huber 2010). We found that 3636 (27%) of detected protein-coding genes are differentially expressed during recovery from L1 arrest (χ2 test, FDR = 0.01%). Using pairwise tests between adjacent time points, we find that more than seven times the number of genes are differentially expressed in the first hour of recovery than in the next 2 h (Table 1). Furthermore, when the observations are projected onto the principal components of the data, fed and starved time points are clearly separated (Supplemental Fig. 2). Taken together, these data show that arrested and developing animals have distinct expression profiles and that this regulatory transition occurs largely within the first hour of feeding. This result is consistent with the rapid change in Pol II binding observed in response to feeding by Pol II ChIP-seq (Baugh et al. 2009), reflecting the paramount importance of nutrient availability on gene regulation.
Table 1.
Transcriptome dynamics reveal numerous changes in the first hour of recovery from L1 arrest
We used cluster analysis to reveal temporal patterns of gene expression during L1 arrest and recovery and to explore gene functions enriched among coregulated genes. We used a self-organizing map to generate 30 clusters of differentially expressed genes (Supplemental Fig. 3). Observed dynamics are relatively simple, with most clusters including only an increase or decrease in expression, and most of these changes occur in the first hour of recovery. Clusters that increase in the first hour are enriched for the Gene Ontology (GO) terms “positive regulation of growth rate” and “nematode larval development,” consistent with a fundamental role for these genes in initiation of growth and development (Table 2; Supplemental Fig. 4). Approximately 30% of differentially expressed genes belong to a cluster whose average trajectory shows a large inflection in the first hour of recovery (e.g., clusters 17, 11 in Fig. 2), suggesting that these genes function transiently during recovery from L1 arrest. Few clusters include changes in expression level occurring between 3 and 6 h of recovery, and most that do also change in the first hour. Cluster 29 is exceptional in that it includes large changes in expression only between 3 and 6 h of recovery (Fig. 2). Based on analysis of GO terms, this is the only cluster enriched for genes associated with the molting cycle (Table 2), which includes genes whose expression peaks near the molt (after ∼16 h of recovery), suggesting that this cluster includes genes involved in later larval development as opposed to initiation of growth and development. These results show that our data highlight broad-scale functional patterns associated with initiation and progression of L1 development.
Table 2.
Cluster analysis of differentially expressed genes reveals enrichment of functional GO terms among coregulated genes

Figure 2.

Cluster analysis reveals predominant expression patterns during recovery from starvation. Genes were clustered using a self-organizing map on mean normalized data corrected for heteroscedasticity using a variance stabilizing function implemented in DESeq (see Methods), and FPKM is plotted. Only select clusters are presented; see Supplemental Figure 3 for the complete set of clusters identified.
Operons and trans-splicing
Nematodes and some other invertebrate genomes contain operons that are transcribed to produce poly-cistronic pre-mRNA. Nematodes also use trans-splicing to add a short (22 nt) leader sequence to the 5′ end of most (∼70%) mRNA transcripts (Blumenthal 2005). There are two major types of spliced leader in C. elegans: SL1 and SL2. SL1 is most common, and it is thought to be specific to mono-cistronic transcripts and the first transcript in operons. In contrast, SL2 is added during processing of poly-cistronic pre-mRNAs from operons such that it is spliced to transcripts that come from the inside of operons (Blumenthal 2005).
Consistent with previous analysis (Zaslaver et al. 2011), annotated operon genes are significantly up-regulated relative to non-operon genes in the first hour of recovery (ANOVA p < 2 × 10−16) (Fig. 3A). By including tobacco acid pyrophosphatase in our sequencing protocol, we were able to sequence the 5′ end of transcripts. This allowed us to determine if they are trans-spliced to SL1 or SL2, inferring operon genes from our data rather than annotation (see Methods). SL2 bearing transcripts are significantly up-regulated relative to SL1 or non-trans-spliced transcripts in the first hour of recovery (ANOVA, p < 2 × 10−16) (Fig. 3B). These results support the importance of operons in recovery from developmental arrest (Zaslaver et al. 2011).
Figure 3.

Genes predicted to be in operons and transcripts trans-spliced to SL2 are significantly up-regulated in the first hour of recovery. (A) The expression of genes in operons and those not in operons is plotted. (B) The expression of genes with transcripts spliced to either SL1, SL2, both, or neither at their 5′ ends is plotted. Average log2 mean normalized expression is plotted for each group. A transcript is considered to be trans-spliced to a particular trans-spliced leader if it received more than 10 reads bearing its sequence. Error bars are 95% confidence intervals of the mean.
Consistent with previous analysis (Allen et al. 2011), we found that some SL2-bearing transcripts are also robustly spliced to SL1. This likely reflects activity of an internal promoter producing mono-cistronic messages despite the gene being part of an operon (Allen et al. 2011). Curiously, we found such transcripts to be more highly expressed than those with only SL1, SL2, or neither (Fig. 4A). Furthermore, the expression level of these transcripts is bimodal and correlates with the ratio of SL1/SL2 (Fig. 4B). These observations suggest that the exceptionally high levels of expression are the result of two strong promoters acting simultaneously. Eighteen of the top 25 (hypergeometric test for enrichment, p < 2 × 10−16) most highly expressed of these transcripts encode ribosomal proteins, consistent with dual promoters driving expression of high-abundance proteins.
Figure 4.

The expression level of genes with transcripts spliced to both SL1 and SL2 reads is bimodal and correlates with the ratio of SL1/SL2 reads. Data for the first hour of recovery are plotted, but this pattern is also present in all other time points (data not shown). Correlation between high expression and high SL1 trans-splicing in addition to SL2 splicing suggests that the activity of an internal promoter in addition to the operon promoter contributes to expression of these most highly expressed genes. (A) The empirical density function of the expression level of transcripts trans-spliced to SL1, SL2, neither, or both is plotted. Trans-spliced transcripts are more highly expressed than transcripts without trans-spliced leaders. (B) The correlation of gene expression with the ratio of SL1/SL2 is plotted for the set of genes with transcripts trans-spliced to both SL1 and SL2. A transcript is considered to be trans-spliced to a particular trans-spliced leader if it received more than 10 reads bearing its sequence. Error bars are 95% confidence intervals of the mean. Of the genes, 2488 genes are in the “no spliced leader” group, 1045 are in the SL1 group, 184 are in the SL2 group, and 80 are in the “both” group.
Identifying alternative exon and isoform expression
Approximately 25% of C. elegans' protein-coding genes are predicted to encode multiple transcript isoforms (WormBase 220). Our data present an opportunity to analyze dynamics of gene expression at the isoform level during a major physiological transition. We used a pair of independent statistical approaches to address reliability of the results, and we mined a variety of databases to assess their functional significance.
Combinations of exons not annotated as transcript isoforms are coexpressed, indicating that annotation of the C. elegans transcriptome is incomplete (Gerstein et al. 2010). We therefore used the Bioconductor package DEXSeq to look for alternative isoform expression without relying on transcript isoform models. DEXSeq fits a generalized linear model to the number of reads mapping to an exon and looks for a significant interaction term between the identity of the exon and experimental condition. We tested 13,374 detected protein-coding genes for alternative exon expression between adjacent time points and across the entire time series. Two hundred twenty-three show alternative exon expression (FDR of 5%) (Table 2). Of these, 72 have only one transcript isoform model in WormBase 220. Of these, 34 were also reported by the modENCODE Consortium to have more than one isoform (Gerstein et al. 2010). These results further show that annotation of the C. elegans transcriptome is incomplete, and they demonstrate the value of an approach not based on transcript isoform models (i.e., DEXSeq).
Interpretation of alternative exon expression based on DEXSeq can be challenging for genes with many exons and many transcript isoforms. In contrast, Cufflinks estimates the most likely concentration of transcript isoforms present in a sample that would generate the observed RNA-seq read patterns given a set of transcript models (Trapnell et al. 2010). We used Cufflinks with transcript annotations from WormBase 220 to estimate the abundance of each annotated transcript isoform. Transcript isoforms can differ by CDS or UTRs. Alternative expression of CDSs can change the function of the protein, and alternative UTRs (especially 3′ UTRs) can alter the stability, localization, and translation of transcripts (Zahler 2005). We used Cufflinks three different ways to identify genes with alternative isoform expression after grouping their transcript isoforms based on (1) CDSs, (2) predicted protein domains, and (3) 3′ UTRs. We used Cuffdiff from the Cufflinks suite to implement an entropy-based statistical test to assign significance to alternative isoform expression (Trapnell et al. 2010). In addition, we looked at the fraction of a gene's expression represented by each isoform or group of isoforms (e.g., those with common CDS), which we refer to as “fractional representation.” Fractional representation is arguably more intuitive and biologically relevant than entropy, but it is not associated with a statistical test. Therefore, we combined these approaches to identify genes that have at least a twofold change in fractional representation between adjacent time points that are also statistically significant according to Cuffdiff. Genes identified this way include more than half of the genes identified by DEXSeq that were also tested by Cuffdiff (Fig. 5). Although we used a much lower FDR than DEXSeq, Cuffdiff considers many more genes to have alternative isoform expression than DEXSeq. However, the overlap between these two very different approaches is highly significant (Fisher's exact test, p < 2 × 10−16), and the functional enrichments they identify are similar (see below).
Figure 5.

Overlap between three metrics for identifying alternative isoform expression. Genes identified by DEXSeq with alternative exon expression are generally identified by Cuffdiff with significant alternative isoform expression. However, nearly half of the genes identified by Cuffdiff have less than a twofold change in isoform expression. The set of genes identified by Cuffdiff with alternative isoform expression is the union of genes with a significant change in CDS, protein-coding domains, or 3′ UTR. Likewise, the set of genes with twofold change in fractional isoform representation is the union of genes with twofold change in their representation in any of those three categories. The set of genes identified by DEXSeq with alternative exon expression (FDR 5%) is the result of a single test over the entire time series. The set of genes used to make the diagram is the intersection of genes tested by both Cuffdiff and DEXSeq for alternative isoform or exon expression, respectively.
Extent and dynamics of alternative isoform expression
Alternative transcript isoform expression has similar dynamics as differential gene expression (Table 1). There were more genes with alternative exon expression during the first hour of recovery than any other tested time interval, demonstrating the very rapid changes to the L1 transcriptome that take place as the worm recovers from arrest. However, many genes that are differentially expressed go from “on” to “off” and vice versa, but only three genes switched the predominant isoform they express from <5% to >95% representation. This suggests that “all-or-none” regulation of isoforms across the whole organism is rare, although we cannot rule out such regulation in specific tissues. To elucidate the biological significance of alternative isoform expression, we examined the affected genes for significant GO term enrichments. Supplemental Table 3 shows enrichments in the set of genes with alternative exon and isoform expression across the entire time series. Genes annotated with the GO terms “nematode larval development” and “growth” are enriched in each set of genes with alternative exon, CDS, and 3′ UTR expression. Interestingly, genes associated with “splicing” and “translation” are also enriched in both the set of genes with differential 3′ UTR use and those with alternative exon expression (Supplemental Table 3). These data suggest that nutritional control of alternative isoform expression affects fundamental biological processes central to growth and post-embryonic development.
Alternative protein domain and 3′ UTR expression
Alternative CDS expression does not necessarily imply expression of proteins with different functional properties. To address functionality, we used the Pfam and Phospho-pep databases of predicted protein domains and phosphorylation sites to group isoforms by shared functional domains. We then ran Cuffdiff to examine protein domain expression during recovery from L1 arrest. One thousand one hundred ninety-nine genes encode multiple transcript isoforms that differ in predicted protein domains. There were no significant GO terms enriched among this set of genes, but we identified a common pattern of domain expression where a change in phosphorylation site is accompanied by a constant functional domain. This pattern appears in 30% of the genes with alternative protein domain expression (for examples, see Fig. 6), and it is statistically significant (Fisher's exact test, p < 2 × 10−16). “Phosphorylation site” is also the only protein domain significantly enriched in the set of genes with alternative CDS expression across the time series (Fishers' exact test, q < 2 × 10−16), and it is enriched in the set of genes with alternative exon expression (Fisher's exact test, q = 7.2 × 10−10). Finally, genes with protein domains annotated with the term “RNA recognition motif” are enriched in the set of genes with alternative exon expression (Fisher's exact test, q = 2.9 × 10−6). These findings suggest that changing phosphorylation sites is a common way to alter protein function and that genes with RNA binding domains are disproportionately affected by alternative exon expression.
Figure 6.

Genes with alternative predicted domain expression often alter expression of a phosphorylation site while other domains remain constant. Six representative genes are plotted. Note that since not all transcripts contain annotated protein-coding domains, the fractional representation of the protein domain configurations does not necessarily sum to 1.
Alternative transcript isoforms may also affect post-transcriptional regulation independent of effects on protein function. We therefore examined potentially functional consequences of alternative isoform expression by examining fractional representation of 3′ UTR sequences. We were able to test 4292 genes for alternative 3′ UTR expression using Cuffdiff. Three hundred ninety (9%) of these genes have statistically significant, twofold or greater changes in fractional representation of 3′ UTRs (Table 1). We tested this set of genes for enrichment of GO terms (see above) and predicted miRNA binding sites. The most enriched predicted miRNA binding site was for miR-34, with a threefold enrichment of its targets in the set of genes with alternative 3′ UTR expression (Fisher's exact test, q = 3 × 10−5) (Supplemental Fig. 5; Supplemental Table 5). miR-34 binding sites were also highly enriched in the set of genes with alternative exon expression (q = 2 × 10−4) (Supplemental Table 5). Binding sites for 16 other miRNAs were enriched in either the set of genes with alternative exon expression or the set with alternative 3′ UTR expression using a FDR cutoff of 1% (Supplemental Table 5). The function of these miRNAs in this context is unclear, but these results suggest a complex interplay between transcriptional and post-transcriptional regulation.
Regulation of mRNA metabolism by alternative exon expression
The serine/arginine (SR)–rich family of proteins regulates several aspects of mRNA metabolism, including alternative splicing and nuclear export (Long and Cáceres 2009). Four of the eight genes encoding the C. elegans homologs of SR proteins are regulated by alternative exon expression during recovery from starvation. Three (rsp-1, 3, and 6) show highly significant alternative exon expression (q = 1.2 × 10−5, 5.2 × 10−8, and 1.8 × 10−5, respectively), and a fourth (rsp-5) shows less significant alternative exon expression (q = 0.0011). Using Cufflinks (see below), we estimate that ∼26% of the gene products of rsp-3 have an intermediate length 3′ UTR during L1 arrest (Fig. 7D–F). However, within 1 h of feeding this drops to <1%. In contrast, during starvation, 66% and 69% of the gene products for rsp-1 (Fig. 7A–C) and rsp-6 (Fig. 7G–I), respectively, encode proteins with an RNA recognition motif and a C-terminal domain rich in arginine and serine required for splicing and phosphorylation (Longman 2000; Long and Cáceres 2009). Within 1 h of recovery, 85% of rsp-6 gene products contain both domains; this fraction remains constant for the rest of the experiment. Similarly, rsp-1 transcripts containing both domains also increase in the first hour of recovery to 80%, eventually rising to comprise 89% of the gene's expression. Previous work demonstrated functional redundancy for most SR protein-encoding genes with the exception of rsp-3 (Longman 2000); likewise, our results show that rsp-1 and rsp-6 are regulated in similar fashion and that rsp-3 is relatively exceptional.
Figure 7.

Genes encoding SR (serine/arginine rich) proteins are regulated by alternative exon expression during recovery from starvation. Each column shows the mean coverage of reads over the gene model for each condition (A,D,G), the fitted expression for each exon from the generalized linear model used by DEXSeq (B,E,H), and Cufflinks' estimate of the expression level of each isoform in the gene model (C,F,I). To highlight relative changes in exon expression, gene browser coverage is scaled to the maximum coverage for the gene. Exons identified by DEXSeq as being alternatively expressed (FDR 5%) are highlighted in magenta in the gene model. Transcript sequence annotated as “RNA-recognition motif” by Pfam is highlighted in orange in the transcript models. Transcript sequence rich in serine and arginine (the SR proteins' RS domains) is highlighted in blue. (Dotted line) Untranslated regions. Error bars on Cufflinks' estimates of isoform levels are 95% confidence intervals of the mean. All genes are plotted with their 5′ ends to the left, regardless of strand.
SR protein expression is regulated by inclusion of premature stop codons, making them targets of nonsense-mediated decay (Long and Cáceres 2009). The rsp-1 and rsp-6 exons we identify as alternatively expressed (Fig. 7) have been shown to include premature stop codons and trigger nonsense-mediated decay (Morrison et al. 1997; Barberan-Soler et al. 2009; Ramani et al. 2009), demonstrating that inclusion of premature stop codons is under nutritional control. In summary, our results show that factors controlling mRNA metabolism are themselves regulated by alternative isoform expression in response to nutrient availability.
Regulation of translation by alternative isoform expression
Protein synthesis is a fundamental aspect of gene regulation and growth control. Multiple lines of evidence suggest that alternative isoform expression affects translation (Fig. 8; Supplemental Tables 3, 4), and translational regulators and machinery are both involved. DEXSeq identified 18 genes annotated with the GO term “translation” with alternative exon expression (Supplemental Table 4). Ten of these encode ribosomal proteins, and the others regulate translation. Cuffdiff identified 29 “translation” genes with alternative 3′ UTR expression, six of which were also identified by DEXSeq. Twenty of the genes that Cuffdiff identified encode ribosomal proteins, and the others regulate translation. The overlap between “translation” genes identified by DEXSeq and Cuffdiff is statistically significant (Fisher's exact test, p < 2 × 10−16). Ribosomal protein gene expression tends to be regulated by alternative 3′ and 5′ UTR expression, although there are cases in which the CDS is alternatively expressed (Supplemental Table 4). Several ribosomal protein genes regulated by alternative exon expression appear to encode unannotated transcript isoforms, underscoring the novelty of our experimental design and data as well as the value of an exon-based approach (Fig. 8). These results suggest that translation is regulated by alternative isoform expression during recovery from L1 arrest, consistent with the paramount physiological significance of translational control.
Figure 8.

Novel ribosomal isoforms are expressed during recovery from L1 arrest. The genes rps-19 (A), rpl-31 (B), and rpl-38 (C) were identified by DEXSeq as displaying alternative exon expression. However, examination of read coverage in the genome browser reveals changes inconsistent with annotated gene models (WS220). The patterns observed also do not correspond well to modENCODE annotations. All genes are plotted with their 5′ ends to the left, regardless of strand. (Dotted line) Untranslated regions.
Discussion
Appropriate developmental responses to nutrient availability are critical to organismal fitness, and transcriptional regulation is an essential mediator of such responses. Microarray analysis revealed dramatic differences in gene expression between L1 arrest and L1 development (Baugh et al. 2009), but the dynamics of L1 arrest recovery were not captured, and transcript isoforms were not distinguished. Alternative isoforms are expected to provide functional diversity to the transcriptome, and studies using RNA-seq and splice junction–sensitive microarrays have revealed extensive alternative isoform expression during C. elegans development (Barberan-Soler and Zahler 2008; Gerstein et al. 2010; Ramani et al. 2011). We used RNA-seq to measure the extent and dynamics of alternative isoform expression in the first hours of recovery from L1 arrest. Our analysis reveals the dynamics of isoform-specific expression during a major physiological transition, providing the first genome-wide analysis of environmental control of alternative isoform expression.
We used two independent methods (DEXSeq and Cufflinks) to examine alternative isoform expression during recovery from L1 arrest. The genes identified by these two very different approaches agree reasonably well (Fig. 5). Furthermore, in cases in which alternative exon expression is easily interpretable as a correlate of the alternative expression of a particular isoform, the results of DEXSeq and Cufflinks are consistent with each other. For example, the rsp-3 isoform with an intermediate length 3′ UTR is clearly less abundant than other isoforms during recovery from starvation (Fig. 7). This is visible in the genome browser, Cuffdiff considers rsp-3 to have alternative 3′ UTR expression, and DEXSeq considers the exons making up the 3′ UTR to be alternatively expressed (Fig. 7). Notably, despite using a much more restrictive FDR for Cuffdiff than DEXSeq (10−9% and 5%, respectively), Cuffdiff considers many more genes to have alternative isoform expression than DEXSeq (Fig. 7). Cuffdiff may identify more genes because it uses reads spanning splice junctions in addition to exon coverage in its calculations, because it performs poorly using our library preparation protocol (single-end, 50-nt reads), or because it does not take into account biological variability in its statistical test (Anders et al. 2012). However, despite some disagreement between the two methods, they converge on common genes and functional classes in several databases (Fig. 7; Supplemental Table 3). Based on our analysis, we believe that DEXSeq is a more conservative test of alternative isoform expression than Cuffdiff. However, our results highlight the complementarity of these two fundamentally different statistical approaches and show that they identify common genes and biological processes.
Our results show that substantial changes to the C. elegans transcriptome occur rapidly in response to feeding during recovery from L1 arrest (Table 1). Transcriptional resources are limiting during transition between arrest and development, and it has been suggested that this limitation contributed to the evolution of metazoan operons as a way to extend transcriptional resources and accelerate recovery from arrest (Zaslaver et al. 2011). Consistent with this hypothesis, operon genes and genes trans-spliced to SL2 are up-regulated relative to non-operon genes during recovery from L1 arrest. We also found that expression of genes trans-spliced to both SL1 and SL2 is bimodal, suggesting the activity of a strong internal promoter in addition to the operon promoter, consistent with the observations of Allen et al. (2011).
Temporal patterns of gene expression during recovery from L1 arrest are relatively simple: A gene is either up-regulated or down-regulated, and most differential expression is in the first hour of recovery (Fig. 2; Supplemental Fig. 3). This pattern extends to isoform expression as well: Many more genes show alternative isoform and exon expression in the first hour than in later time intervals. Furthermore, in cases we have examined closely (e.g., SR protein genes), the difference between starvation and 1 h of feeding is readily apparent, whereas subsequent changes are relatively subtle (Fig. 7). Although these changes are very rapid, few genes completely switch the isoform they express during recovery, consistent with analysis of various larval stages and conditions (Gerstein et al. 2010). Given dramatic changes in the transcriptome at the level of the gene, this is somewhat surprising. This may reflect our inability to detect tissue-specific isoform expression with whole animal measurements, which is expected to be substantial (Zahler 2005). Nevertheless, the genes with alternative isoform expression are enriched for GO terms like “larval development,” “mRNA metabolism,” and “translation” (Supplemental Table 3), supporting the conclusion that these changes are functionally significant and impact processes fundamental to growth and development.
Regulation of genes involved with mRNA splicing by alternative isoform expression suggests that splicing factors regulate their own gene products. The C. elegans genome encodes eight serine/arginine (SR)–rich proteins, which function in splice site selection as well as the transcription and export of mRNAs (Long and Cáceres 2009). Many of these genes are regulated by alternative splicing linked to nonsense-mediated decay (Ni et al. 2007; Barberan-Soler et al. 2009). It has been suggested that this is a form of autoregulation in that high levels of a splicing factor lead to increased inclusion of highly conserved exons with premature stop codons, triggering nonsense-mediated decay (Sureau 2001). Consistent with autoregulation, we find that half of C. elegans SR proteins show alternative exon expression in the first hour of recovery and that 3′ UTR and protein domain expression is altered (Fig. 7). Both rsp-1 and rsp-6 encode transcripts targeted by nonsense-mediated decay that change fractional representation in the first hour of recovery, showing that this mode of regulation occurs rapidly in response to nutrient availability. Although we highlight SR protein-encoding genes, we also detected changes in isoform expression of other important regulators of splicing such as asd-2, hel-1, and uaf-1 (Supplemental Fig. 6) as well as W04D2.6, which has been shown to interact with rsp-6 to control splicing (Longman et al. 2001; Fortes et al. 2007). In addition, the protein domain “RNA recognition motif” is enriched among genes with alternative exon expression. These results further support the conclusion that splicing factors are themselves regulated by alternative splicing, revealing acute effects of nutrient availability on mRNA metabolism.
Translation is tightly regulated during growth and development, and our results suggest that it is controlled by alternative isoform expression. In yeast, the energy homeostasis regulators TOR and PKA affect transcriptional regulation of ribosomal proteins, linking their expression to nutrient availability (Martin et al. 2004). Post-transcriptional regulation of ribosomal proteins is also important, as exemplified by the mammalian and yeast homologs of C. elegans rpl-3 and rpl-32, respectively, which catalyze their own splicing in a post-transcriptional feedback circuit (Vilardell and Warner 1997; Russo et al. 2011). Regulation by nonsense-mediated decay is also common (Cuccurese et al. 2005). We find frequent alternative isoform and exon expression among ribosomal proteins (Fig. 8; Supplemental Table 4). Notably, we identify several alternatively expressed isoforms corresponding to unannotated transcripts, including one expressed transiently at 1 h of recovery (Fig. 8A). Although the function of these isoforms is unclear, our results suggest that alternative isoform expression alters ribosomal protein function, contributing to translational regulation in response to nutrient availability.
Our results provide the first genome-wide survey of environmental control of transcript isoform expression, characterizing rich changes in the C. elegans transcriptome during recovery from starvation and initiation of growth and post-embryonic development. Our results suggest that nutrient availability impinges on transcriptional and post-transcriptional gene regulation. We previously showed post-recruitment, nutrient-dependent regulation of RNA polymerase II elongation (Baugh et al. 2009). Taken together, these results show that nutrient availability impacts RNA polymerase II recruitment and elongation as well as mRNA splicing, export, and translation. Our study demonstrates the ability of RNA-seq to elucidate transcriptome complexity and dynamics, and it provides an excellent resource for ongoing investigation of transcriptional regulation of growth and development.
Methods
Worm culture
Wild-type C. elegans strain N2 was used for RNA-seq. The N2 stock used was from the Sternberg collection at the California Institute of Technology; this strain was obtained from the Caenorhabditis Genetics Center in 1987, expanded, and frozen. Nematodes were maintained on standard NGM plates with E. coli OP50 as food (Lewis 1995), but liquid culture was used to prepare RNA-seq samples. A starved 5-cm plate was used to inoculate 25 mL of liquid culture (S-complete plus 40 mg/mL E. coli HB101) (Lewis 1995). The culture was incubated for 65 h at 20°C and 180 rpm and then bleached to produce a clean preparation of embryos. Embryos were suspended in S-complete at 5 eggs/μL and incubated for 24 h at 20°C and 180 rpm so that they hatch and enter L1 arrest. After 24 h, cultures were fed with 40 mg/mL HB101 to initiate synchronous development, and they were incubated for 57 h at 20°C and 180 rpm. In these conditions, the first eggs are fertilized at ∼53 h, and when bleached at 57 h the yield is typically about 10 eggs per worm. After 57 h, the cultures were bleached, and the eggs were suspended in S-complete at 10 eggs/μL. Animals were incubated for 24 h at 20°C and 180 rpm so that they hatch and enter L1 arrest. The 0-h time point was collected 24 h after bleaching, corresponding to ∼12 h of L1 arrest. The remainder of the culture was fed with 25 mg/mL E. coli HB101 and incubated at 20°C and 180 rpm with collections at 1, 3, and 6 h after feeding. Larvae were collected by centrifugation and washed three times in S-basal before being flash-frozen. Three biological replicates were collected at each time point. However, one replicate from 1 and 3 h after feeding was discarded due to low library quality.
RNA extraction
RNA was prepared using TRIzol (Invitrogen) according to the manufacturer's protocol with minor modifications. One milliliter of TRIzol was used per sample, and homogenization was supplemented with 100 μL of acid-washed sand. Poly-adenylated mRNA was isolated from total RNA using Dynal oligo(dT) magnetic beads (Invitrogen) according to the manufacturer's protocol. Two hundred nanograms of poly-adenylated RNA was used in a Tobacco Acid Pyrophosphatase reaction (Epicentre) according to the manufacturer's protocol in order to remove 5′ caps. The product was purified with phenol:chloroform extraction and ethanol precipitation using GlycoBlue as a coprecipitant (Ambion). This purified product was then used in the RNase III fragmentation reaction at the beginning of the Solid Total RNA-Seq Kit Whole Transcriptome protocol (Applied Biosystems). The manufacturer's instructions were followed for the remainder of the library preparation process. Fragmentation efficiency was analyzed with the BioAnalyzer (Agilent), and one-half of each sample (corresponding to ∼100 ng of RNA) was used for adapter ligation. cDNA was gel-purified to capture inserts of RNA fragment size 100–200 nt. Twelve PCR cycles were used to amplify the libraries. Libraries were processed and sequenced on the Solid 4 system according to the manufacturer's protocols.
Read mapping to the genome and transcriptome
We first mapped SOLiD-generated reads to the WormBase 210 version of the genome (WS210) in color space using Bowtie (v. 0.12.7) (Langmead et al. 2009). Reads that did not map in this step were mapped to the WormBase 220 predicted transcriptome, whose coordinates had been mapped back to the WS210 coordinate system (see Supplemental Table 1 for read mapping summary). We used Bowtie to map to the transcriptome as opposed to TopHat (Trapnell et al. 2009) because we found that many reads mapped to transcriptome-derived splice junctions with Bowtie were not mapped with TopHat (data not shown).
Approximately 70% of genes in C. elegans are spliced in trans to a 22-nt sequence donated by a snRNA called a “trans-spliced leader” (Blumenthal 2005). The vast majority of trans-spliced leaders come from two different sequences: either spliced leader 1 or 2 (SL1 or SL2) (Blumenthal 2005). Reads that did not map to either the genome or the transcriptome were stripped of the first (5′) 22 nt of sequence and remapped to determine if these reads came from the 5′ end of trans-spliced mRNAs. We determined whether the stripped 22-nt sequence corresponded to SL1 or SL2 for the reads that mapped after stripping. Reads that mapped after stripping and began with GGTTTAATTACCCAAGTTTGAG were counted as being spliced to SL1, whereas those that started with GGTTTTAACCCAGTTACTCAAG were counted as being spliced to SL2. Although other trans-spliced leaders have been identified (SL3, SL4, etc.), they are very rare in comparison. We detected these sequences much less frequently than SL1 and SL2 and therefore did not include them in our analysis. We created alignment files of reads mapping to the genome and transcriptome along with an index linking the IDs of reads containing spliced leaders to the type of spliced leader contained in the read (i.e., SL1 or SL2).
We assigned trans-spliced reads to gene models by creating clusters of trans-spliced reads (the upstream edge of which putatively represent trans-splice sites). We required each cluster to have coverage of five reads, and for there to be no more than a 10-bp gap between reads in the cluster. We then compared these clusters to annotated transcript start sites in WS220. We assigned trans-splicing clusters to genes that had one and only one trans-splicing cluster within 100 bp of its annotated start site. One thousand six hundred eighty-one genes were mapped to trans-spliced reads using this method. A gene was considered to receive SL1 or SL2 reads if it received more than 10 reads in this way.
Detection and differential expression
To look at the reproducibility of the replicates for each time point and to generate the principal components analysis plot, we used Cufflinks 1.0.2 (Trapnell et al. 2010) and a GTF of the WS220 transcriptome as detailed above. Each count file from Cufflinks was then corrected for heteroscedasticity using DESeq. PCA was performed using these pseudocounts after mean correction. For all further analysis, we used Cuffdiff, a program included in the Cufflinks suite, to determine the FPKM (fragments per kilobase per million) of genes and transcripts at each time point. We estimated the mean of the fragments to be 150 bp with a standard deviation of 40 bp, based on Bioanalyzer traces, and we used this information to run Cuffdiff. We did not use sequence-specific bias correction because we found this to decrease the correlation between replicates (data not shown).
To determine whether a gene was detected, we first discarded all genes where Cufflinks could not determine an FPKM value, or where that value was equal to infinity. Cufflinks assigns confidence intervals to FPKM estimates. These estimates for FPKM should be normally distributed (Trapnell et al. 2010), so we calculated the estimated standard deviation using the formula:
where FPKMhi is the 95% upper bound of the FPKM confidence interval and FPKM is the estimated FPKM value. A P-value was calculated using a one-sided z-test against the null hypothesis that the FPKM was zero. The resulting P-values were then corrected for multiple testing using the Benjamini-Hochberg method. Genes with a Q-value ≤0.1% in any time point were considered detected.
Differential expression was assessed using the R package DESeq (version 1.6.1) (Anders and Huber 2010) using the raw counts of sequences Cufflinks assigned to gene models. A negative binomial generalized linear model including time was compared with a null model (no change in average expression) using a χ2 test. Genes with a false discovery rate (FDR) <0.01% (determined using the Benjamini-Hochberg method) were considered differentially expressed. We chose this cutoff to allow for only one false positive. Pairwise tests between times 0 and 1, 1 and 3, and 3 and 6 h were also performed with the same FDR cutoff.
Cluster analysis, principal components analysis, and operon analysis
Gene set analysis for expression clusters and genes with alternative isoform expression was done using the Bioconductor package Category (Gentleman et al. 2004). Only “biological process” terms within the GO hierarchy were analyzed. Gene clusters were tested for enrichment using the set of differentially expressed genes as the gene universe. Clustering was done with the R package “kohonen,” which implements a self-organizing map on variance-stabilized pseudocounts generated from DESeq in order to avoid bias in clustering from the heteroscedasticity of RNA-seq data. Principal components analysis was also performed using these data. However, all plots of genes are FPKM, not variance-stabilized pseudocounts. The choice of cluster number was found empirically to be the lowest number of clusters that would make consistent, distinct clusters. Operon information was downloaded using WormMart from WormBase 220 in April 2011.
Alternative isoform expression
Significant alternative isoform expression was determined using the program Cuffdiff from the Cufflinks package. Cuffdiff can test for alternative expression of isoforms grouped by either transcriptional start site (TSS) or CDS. Since trans-splicing obscures the true TSS for most genes (Allen et al. 2011), we did not analyze isoforms grouped by TSS. Rather, we tested for alternative isoform expression after grouping them by annotated coding sequence, shared predicted protein domains, and 3′ UTR. In each case, the “coding sequence” slot (p_id) in the annotated GTF file was changed so the transcript belonged to the correct UTR or CDS group. In each case, we used a FDR cutoff of 10−12 to define genes with significant alternative isoform expression. This was the lowest FDR reported by the software. Isoform fractional representation was calculated as the isoform's (or group of isoforms', such as those grouped by CDS, predicted domains, or common 3′ UTRs) expression level at a certain time point divided by the expression level of the gene at that time point.
We also ran Cufflinks with and without including the transcript annotations from the “Aggregate Integrated Transcript Set” from the modENCODE consortium (Gerstein et al. 2010). The modENCODE annotations increase the number of transcript models, but many of these models are very similar to each other, differing by as little as a single nucleotide. To assess the effect of including these annotations on our analysis, we looked at the pairwise correlations between three starved replicates with and without including the modENCODE annotation. Although the average correlation of isoforms between replicates was high in either case (r = 0.87 with modENCODE annotations, r = 0.94 without), isoforms with low expression levels showed reduced correlation between replicates when the additional annotations were included (r = 0.25 with, r = 0.68 without). This result indicates that Cufflinks' estimation of isoform-specific expression levels is less reliable when the additional modENCODE transcript models are included. As a result, we chose to use only WormBase 220 annotations for our analysis.
Alternative exon expression
To test for alternative exon expression, we used the Bioconductor package DEXSeq version 1.0.2. We used the same GTF file used to run Cuffdiff to define the exons tested in the analysis; however, to help with multiple testing, we only tested for alternative exon expression of protein-coding genes that were also detected in at least one time point. All counting of exons was done using scripts provided with the DEXSeq package. We used a FDR cutoff of 5% to define significant alternative exon expression. This cutoff was chosen based on the paper describing the development of DEXSeq (Anders et al. 2012), which used an FDR of 10%. More than 400 genes showed alternative exon expression in the pairwise test of 1 and 3 h of recovery. Since this number is more than the number identified as significant across the whole time series, and because this was the only pairwise test to have only two replicates in each condition, we suspect that DEXSeq lost control of the false-positive rate in this case and did not analyze this comparison further. To test for GO term enrichments in the set of genes identified by DEXSeq as being regulated by alternative exon expression, we restricted the universe of genes to those with GO “biological process” annotations.
Protein domain and miRNA binding site annotation
Protein domain CDSs were defined using data downloaded from the Phospho-pep and Pfam databases using WormMart 220 in June 2011. A protein domain CDS was defined as a unique set of protein domains. Information about 3′ UTRs of transcripts was extracted from WS220 using WormMart. To avoid considering very similar UTRs as distinct, UTRs differing by <22 nt were lumped together. Predicted miRNA binding domains are predictions from Miranda (Betel et al. 2008) with an mirsvr_score (a measure of the likelihood that a miRNA targets a certain sequence) less than −0.4. Additionally, we only considered miRNA binding domains in the 3′ UTR of genes. Enrichment for particular protein domains in the set of genes was performed using a Fisher's exact test. We restricted the universe of genes to test for enrichment to those where Cuffdiff tested for alternative expression and where there was protein domain annotation (871 genes). We only used protein annotations where the number of genes containing the protein domain was greater than five. For each time point comparison, we tested for significant association between the two variables' sets. We tested 57/911 protein domains this way with Cuffdiff, but we tested 868/911 with DEXSeq. We report all enrichments using a FDR cutoff of 5%. To test for enrichment of miRNA binding domains, we likewise restricted the set to genes where Cuffdiff was able to test for alternative 3′ UTR expression (4292 genes).
Data access
Raw reads, mapped reads, annotation files, and exon, isoform, and gene expression estimates are available at the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE33023.
Acknowledgments
We thank Lisa Bukovnik and Nick Hoang of the Duke IGSP Genome Sequencing and Analysis Core Resource. This work was supported by the Ellison Medical Foundation and the National Science Foundation (IOS-1120206).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.133587.111.
References
- Allen MA, Hillier LW, Waterston RH, Blumenthal T 2011. A global analysis of C. elegans trans-splicing. Genome Res 21: 255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106 doi: 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Reyes A, Huber W 2012. Detecting differential usage of exons from RNA-seq data. Genome Res (this issue). doi: 10.1101/gr.133744.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberan-Soler S, Zahler AM 2008. Alternative splicing regulation during C. elegans development: Splicing factors as regulated targets. PLoS Genet 4: e1000001 doi: 10.1371/journal.pgen.1000001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberan-Soler S, Lambert NJ, Zahler AM 2009. Global analysis of alternative splicing uncovers developmental regulation of nonsense-mediated decay in C. elegans. RNA 15: 1652–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh LR, Sternberg PW 2006. DAF-16/FOXO regulates transcription of cki-1/Cip/Kip and repression of lin-4 during C. elegans L1 arrest. Curr Biol 16: 780–785 [DOI] [PubMed] [Google Scholar]
- Baugh LR, DeModena J, Sternberg PW 2009. RNA Pol II accumulates at promoters of growth genes during developmental arrest. Science 324: 92–94 [DOI] [PubMed] [Google Scholar]
- Betel D, Wilson M, Gabow A, Marks DS, Sander C 2008. The microRNA.org resource: Targets and expression. Nucleic Acids Res 36: D149–D153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal T. 2005. http://www.wormbook.org Trans-splicing and operons. In Wormbook (ed. The C. elegans Research Community). [Google Scholar]
- Colot HV, Loros JJ, Dunlap JC 2005. Temperature-modulated alternative splicing and promoter use in the circadian clock gene frequency. Mol Biol Cell 16: 5563–5571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuccurese M, Russo G, Russo A, Pietropaolo C 2005. Alternative splicing and nonsense-mediated mRNA decay regulate mammalian ribosomal gene expression. Nucleic Acids Res 33: 5965–5977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duque P 2011. A role for SR proteins in plant stress responses. Plant Signal Behav 6: 49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortes P, Longman D, McCracken S, Ip JY, Poot R, Mattaj IW, Cáceres JF, Blencowe BJ 2007. Identification and characterization of RED120: A conserved PWI domain protein with links to splicing and 3′-end formation. FEBS Lett 581: 3087–3097 [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. 2004. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol 5: R80 doi: 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. 2010. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE Project. Science 330: 1775–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden JW, Riddle DL 1983. The Caenorhabditis elegans dauer larva: Developmental effects of pheromone, food, and temperature. Dev Biol 102: 368–378 [DOI] [PubMed] [Google Scholar]
- Hillier LW, Reinke V, Green P, Hirst M, Marra MA, Waterston RH 2009. Massively parallel sequencing of the polyadenylated transcriptome of C. elegans. Genome Res 19: 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu PJ. 2007. http://www.wormbook.org Dauer. In Wormbook (ed. The C. elegans Research Community). [Google Scholar]
- Kuroyanagi H, Kobayashi T, Mitani S, Hagiwara M 2006. Transgenic alternative-splicing reporters reveal tissue-specific expression profiles and regulation mechanisms in vivo. Nat Methods 3: 909–915 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg S 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 doi: 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JA, Fleming JT 1995. Basic culture methods. In Caenorhabditis elegans: Modern biological analysis of an organism (ed. HF Epstein, DC Shakes), pp. 4–27. Academic Press, San Diego [Google Scholar]
- Long JC, Cáceres JF 2009. The SR protein family of splicing factors: Master regulators of gene expression. Biochem J 417: 15–27 [DOI] [PubMed] [Google Scholar]
- Longman D 2000. Functional characterization of SR and SR-related genes in Caenorhabditis elegans. EMBO J 19: 1625–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longman D, McGarvey T, McCracken S, Johnstone IL, Blencowe BJ, Cáceres JF 2001. Multiple interactions between SRm160 and SR family proteins in enhancer-dependent splicing and development of C. elegans. Curr Biol 11: 1923–1933 [DOI] [PubMed] [Google Scholar]
- Martin DE, Soulard A, Hall MN 2004. TOR regulates ribosomal protein gene expression via PKA and the Forkhead transcription factor FHL1. Cell 119: 969–979 [DOI] [PubMed] [Google Scholar]
- Morrison M, Harris KS, Roth MB 1997. smg mutants affect the expression of alternatively spliced SR protein mRNAs in Caenorhabditis elegans. Proc Natl Acad Sci 94: 9782–9785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O'Brien G, Shiue L, Clark TA, Blume JE, Ares M 2007. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev 21: 708–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani A, Nelson A, Kapranov P, Bell I, Gingeras T, Fraser A 2009. High resolution transcriptome maps for wild-type and nonsense-mediated decay-defective Caenorhabditis elegans. Genome Biol 10: R101 doi: 10.1186/gb-2009-10-9-r101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani AK, Calarco JA, Pan Q, Mavandadi S, Wang Y, Nelson AC, Lee LJ, Morris Q, Blencowe B, Zhen M, et al. 2011. Genome-wide analysis of alternative splicing in Caenorhabditis elegans. Genome Res 21: 342–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rukov JL, Irimia M, Mørk S, Lund VK, Vinther J, Arctander P 2007. High qualitative and quantitative conservation of alternative splicing in Caenorhabditis elegans and Caenorhabditis briggsae. Mol Biol Evol 24: 909–917 [DOI] [PubMed] [Google Scholar]
- Russo A, Catillo M, Esposito D, Briata P, Pietropaolo C, Russo G 2011. Autoregulatory circuit of human rpL3 expression requires hnRNP H1, NPM and KHSRP. Nucleic Acids Res 39: 7576–7585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureau A 2001. SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J 20: 1785–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL 2009. TopHat: Discovering splice junctions with RNA-seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardell J, Warner JR 1997. Ribosomal protein L32 of Saccharomyces cerevisiae influences both the splicing of its own transcript and the processing of rRNA. Mol Cell Biol 17: 1959–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M 2009. RNA-seq: A revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler AM. 2005. Alternative splicing in C. elegans. In Wormbook (ed. The C. elegans Research Community). http://www.wormbook.org. [Google Scholar]
- Zaslaver A, Baugh LR, Sternberg PW 2011. Metazoan operons accelerate recovery from growth-arrested states. Cell 145: 981–992 [DOI] [PMC free article] [PubMed] [Google Scholar]