

Abstract
The genetics of aging in the yeast Saccharomyces cerevisiae has involved the manipulation of individual genes in laboratory strains. We have instituted a quantitative genetic analysis of the yeast replicative lifespan by sampling the natural genetic variation in a wild yeast isolate. Haploid segregants from a cross between a common laboratory strain (S288c) and a clinically derived strain (YJM145) were subjected to quantitative trait locus (QTL) analysis, using 3048 molecular markers across the genome. Five significant, replicative lifespan QTL were identified. Among them, QTL 1 on chromosome IV has the largest effect and contains SIR2, whose product differs by five amino acids in the parental strains. Reciprocal gene swap experiments showed that this gene is responsible for the majority of the effect of this QTL on lifespan. The QTL with the second-largest effect on longevity was QTL 5 on chromosome XII, and the bulk of the underlying genomic sequence contains multiple copies (100–150) of the rDNA. Substitution of the rDNA clusters of the parental strains indicated that they play a predominant role in the effect of this QTL on longevity. This effect does not appear to simply be a function of extrachromosomal ribosomal DNA circle production. The results support an interaction between SIR2 and the rDNA locus, which does not completely explain the effect of these loci on longevity. This study provides a glimpse of the complex genetic architecture of replicative lifespan in yeast and of the potential role of genetic variation hitherto unsampled in the laboratory.
Aging is determined by genes, environment, and chance. The budding yeast Saccharomyces cerevisiae (baker's yeast) has been used as a model organism in the genetics of aging and longevity since the early 1990s (Jazwinski 2004), and lifespan has been analyzed in two different ways in this organism. Replicative lifespan (RLS) refers to the number of times an individual cell divides, while chronological lifespan (CLS) denotes the time spent in stationary culture before a cell loses viability. The number of genes implicated in the RLS and in the CLS of yeast grows continuously (Steinkraus et al. 2008), and a search of the Saccharomyces Genome Database reveals some 242 genes to date (see http://www.yeastgenome.org). This progress has been made through the genetic manipulation of laboratory yeast strains, but not all of the genetic effects on longevity that have been described are identically operable across all laboratory strain backgrounds examined. An informative case in point is the extension of RLS in yeast strains devoid of mitochondrial DNA (Kirchman et al. 1999), in which the retrograde response is commonly induced (Liu and Butow 2006). The retrograde response is an intracellular signaling pathway from the mitochondrion to the nucleus that is triggered by mitochondrial dysfunction and results in the activation of numerous nuclear genes that compensate for this dysfunction (Liu and Butow 2006; Miceli et al. 2011). A comparison of four strains showed varying effects on RLS, spanning RLS extension through curtailment. However, extension was found in each strain when the retrograde response was activated, which in some strains is glucose repressed (Kirchman et al. 1999). This effect has been confirmed by others (e.g., Heeren et al. 2009). It constitutes an example of the impact of gene interactions in determining longevity (Flint and Mackay 2009).
Laboratory strains are derived from wild isolates adapted to growth in the laboratory. For yeast, this generally means rapid growth and smooth colony morphology that facilitates picking and streaking (cloning). The evolutionary rate of laboratory strains surpasses that of some wild isolates (Gu et al. 2005; Ronald et al. 2006). Furthermore, the process of picking single colonies, all the cells of which are genetically identical, results in population bottlenecks, which favor the accumulation of deleterious mutations (Hartl and Clark 1997). This raises the possibility that the longevity genes isolated in laboratory strains may simply correct the effects of these deleterious mutations.
Yeasts appear to be clonal in the wild, implying little opportunity for sexual reproduction and the attendant genetic segregation and recombination (Tibayrenc et al. 1991). This is not surprising because S. cerevisiae is homothallic in nature. A single haploid spore on germination quickly gives rise to cells that switch mating type and reconstitute the predominant diploid in a homozygous state (Herskowitz 1988). The distinction between wild and laboratory strains is that the former are continuously subjected to selective forces imposed by the ecological niche in which they reside. These forces are likely to be different than those normally found in the laboratory. Even in the laboratory, the imposition of modest selective pressure results in waves of mutations waxing and waning across a yeast population (Adams 2004).
The above considerations imply that genes implicated in aging and longevity in laboratory strains may not play the same role in wild yeast strains and that genetic analysis of wild strains may reveal a different set of genetic determinants of longevity. This is not a trivial alternative that simply expands the repertoire of longevity genes. The question of what genes can affect lifespan is quite different from the question of what genes actually do normally affect lifespan. This is highlighted by the recent demonstration that 65% of the long-lived mutants in a laboratory strain showed reduced fitness relative to the wild type (Delaney et al. 2011). Similar results have been obtained in the Caenorhabditis elegans model system (Jenkins et al. 2004). The other major genetic models used in aging research, Drosophila melanogaster and Mus musculus, are also inbred and adapted to the laboratory, raising identical concerns, while the focus on natural genetic variation in human aging is an obvious contrast.
Natural yeast variation has been queried to identify quantitative trait loci (QTLs) and genes that play a role in heat resistance, multidrug resistance, ethanol tolerance, acetic acid production during alcoholic fermentation, gene expression, spontaneous mitochondrial genome instability, proteome variation, and telomere length (Winzeler et al. 1998; Steinmetz et al. 2002; Brem et al. 2005; Gatbonton et al. 2006; Foss et al. 2007; Hu et al. 2007; Marullo et al. 2007; Dimitrov et al. 2009). Most of these studies have utilized gene expression microarrays that can query several thousands of single-nucleotide polymorphisms across the yeast genome (Winzeler et al. 1998). Here, we have used this strategy to map QTLs associated with RLS in S. cerevisiae. We sampled the natural genetic variation in a clinically derived S. cerevisiae strain, YJM789, by analyzing haploid segregants generated from a cross with the standard laboratory strain S288c (Steinmetz et al. 2002). The genomic sequences of both YJM789 (Wei et al. 2007) and S288c (Goffeau et al. 1996) have been determined. YJM789 and S288c differ at 60,000 bp throughout their genomes.
Results
Genetic variation in longevity genes
To investigate the role of natural genetic variation associated with longevity, we crossed the laboratory strain S96 (MATa ho lys5), a derivate of S288c, and the strain YJM789 (MATα ho∷hisG lys2 cyh), a haploid segregant of the homozygous diploid strain YJM145, which was derived from the clinical isolate YJM128 (Steinmetz et al. 2002). The S96 × YJM789 diploid was sporulated, and 54 tetrads were dissected into their four haploid spores. On germination, the resulting 216 haploid segregants were characterized. We established 39 of these haploid segregants, each from a separate tetrad, as recombinant lines for further analysis. Each was mating-type a (MATa) and was easily micromanipulated. Their mean RLSs (mRLSs) are shown in Table 1 and Supplemental Figure S1. The mRLS of S96 and of the parental diploid are included for comparison. We could not determine the mRLS of YJM789, as it is not possible to micromanipulate this strain in lifespan determinations, because it displays filamentous growth. DNA was isolated from these recombinant lines, fragmented using DNase I, labeled with biotin, and hybridized to Affymetrix S98 yeast gene chips. Hybridization intensity data were uploaded to the program Allelescan to determine the genotype of each strain (Steinmetz et al. 2002). A critical F-test value of 35 was used to identify 3048 molecular markers in five separate hybridizations for DNA prepared from the two parental strains (Supplemental Fig. S2) for use in genotyping of the recombinant lines. The genotyping data and the mRLS of the 39 genotyped lines were processed using Windows QTL cartographer (Basten et al. 1994), with a permutation value of 1000 and a significance level of 0.01 chosen. A total of five significant QTL related to RLS were thus identified. Their logarithm-of-the-odds (LOD) scores were greater than the genome-wide significance threshold of 2.5 (Table 1; Fig. 1).
Table 1.
Mean RLS of 41lines/strains and the parental origin of the longevity QTL
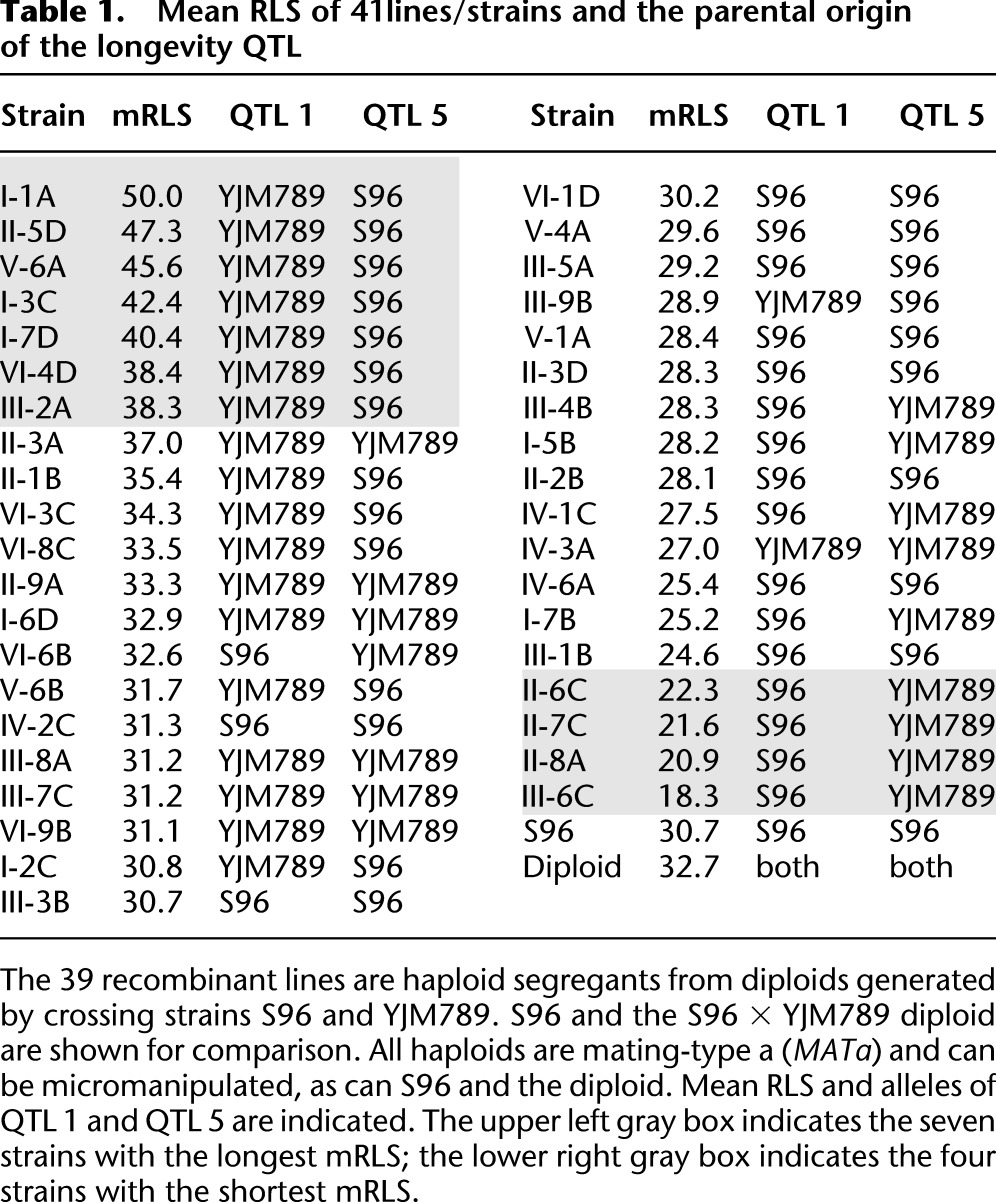
Figure 1.

Composite interval mapping of RLS QTL. (A) The upper panel depicts all QTL in all 16 yeast chromosomes, each indicated by a roman numeral. (B) QTL 1, 2, and 3 on chromosome IV have LOD scores of 9.27, 5.02, and 2.60, respectively. (C) QTL 4 and 5 on chromosome XII have LOD scores of 2.70 and 3.68, respectively. In all panels, the top graphs show the LOD score (y-axis) versus genetic distance in centimorgans (x-axis). A LOD score above the genome-wide significance threshold of 2.5 is evidence for a QTL. The lower graphs show the “additive effect.” Positive or negative values indicate that the parental YJM789 or S96 allele, respectively, is associated with longer RLS. The open and closed arrowheads in A indicate the positions of AMN1 and MATa on chromosomes II and III, respectively. The QTL are numbered in B and C.
After we identified significant QTL using composite interval mapping (CIM), we employed multiple interval mapping (MIM) to further estimate the effects and interactions of significant QTLs and their contribution to the genetic variance. MIM uses multiple marker intervals simultaneously to fit multiple putative QTLs directly in the genetic model. The MIM results showed that QTL 1 accounts for 50% of the genetic effect on the variation of RLS, and QTL 5 accounts for another 20% of the genetic effect. This is only an estimate, however. Overall, the genetic effects explained 67% of the variation in the RLS. The MIM analysis detected no statistically significant interactions between QTLs, but interactions could be missed given the relatively small number of recombinant lines. We focused attention on the two QTLs with the greatest contribution to longevity.
The QTL with the largest effect on RLS is located on chromosome IV (QTL 1, LOD score = 9.27) (Fig. 1A,B). It contains 12 genes within a region of 18 kbp (Fig. 2A). The 13 recombinant lines with the longest RLS contain QTL 1 from parental strain YJM789, whereas the seven strains with the shortest RLS contain QTL 1 from parental strain S96 (Table 1). This agrees with the positive signal in the lower graph of Figure 1B. The LOD score of the QTL located on chromosome XII (QTL 5) is 3.68 (Fig. 1A,C). It comprises >1 Mbp of DNA, including 23 protein-coding genes, one tRNA, some autonomously replicating sequence (ARS) consensus sequences (ACSs), and the rDNA cluster (Fig. 2B).
Figure 2.

Genomic regions of QTL 1 and QTL 5. (A) QTL 1 is located on chromosome IV (bp 370,000–387,742). It contains 12 genes within a region of 18 kbp; one of them is SIR2. (B) QTL 5 is located on chromosome XII (bp 446,389–487,500). It contains 23 protein-coding genes, one tRNA gene, some ARS consensus sequences, and the rDNA cluster (RDN1). Two copies of the rDNA are shown here, but the rDNA cluster contains between 100 and 200 repeats. The x-axes are in kilobase pairs. (Large dark dots) Centromeres; (vertical dashed lines) QTL boundaries. The diagrams were adapted from the Saccharomyces Genome Database (www.yeastgenome.org) (Cherry et al. 1997).
SIR2 is part of QTL 1
One of the genes contained in QTL 1 is SIR2, a well-known regulator of RLS in laboratory strains of S. cerevisiae (Kaeberlein et al. 1999; Kim et al. 1999). In S96 and YJM789, Sir2p has a length of 562 amino acids (AA) but differs in five AA: P65T, V92E, D158V, K320N, and M424V. None of the five altered AA possesses an assigned function (Garcia and Pillus 2002; for review see Sauve et al. 2006; Choy et al. 2011; Froyd and Rusche 2011). Nevertheless, SIR2 was chosen as a candidate for gene swap experiments. The SIR2 open reading frame (ORF) of strain S96 was substituted by the YJM789 allele between +75 and +1323. This part of the gene contains all five polymorphisms. We then compared the RLS of S96 and S96 possessing the SIR2 gene of YJM789 (S96 sir2Δ∷SIR2[YJM789]). The mean RLS of S96 was 31.7 generations, and for S96 sir2Δ∷SIR2(YJM789) it was 46.9 generations, which is 48% longer (Fig. 3A).
Figure 3.

Cumulative survival curves. (A) The RLSs of 49 cells of strain S96 and 60 cells of strain S96 sir2Δ∷SIR2(YJM789) were determined. The mRLS of S96 was 31.7 generations, and for S96 sir2Δ∷SIR2(YJM789) it was 46.9 generations. The difference in RLS was significant (P < 5 × 10−6). (B) The RLS of 81 cells each was determined. For line I-1A, the mRLS was 49.5 generations; for strain I-1A sir2Δ∷SIR2(S96), it was 41.6. The difference in RLS was significant (P = 0.0079). For S96, the mRLS was 30.7 generations, and for S96 sir2Δ∷SIR2(YJM789) it was 43.7 generations. The difference in RLS was significant (P < 5 × 10−6). I-1A is the recombinant line with the longest RLS. (C) The RLS of 45 cells each was determined. For strain YFS7 (YJM789 background with RDN1 from YJM789) the mRLS was 33.8 generations, and for strain YFS9 (YJM789 background with RDN1 from S96) it was 40.0 generations. The difference in RLS was significant (P = 0.030). (D) The RLS of 40 cells each was determined. For strain S96, the mRLS was 29.8 generations, and for strain YFS1 (S96 background with RDN1 from YJM789) it was 24.6 generations. The difference in RLS was significant (P = 0.032). The x-axis shows the RLS in generations and the y-axis shows the percentage of surviving cells.
We next conducted the reverse experiment. Strain YJM789 displays filamentous growth; it cannot be micromanipulated nor can its RLS be determined. Therefore, the recombinant line I-1A was used for another SIR2 ORF-swap experiment. I-1A has the longest mRLS (50.0 generations) of all analyzed recombinant lines; it has the YJM789 allele of QTL 1 (Table 1). We generated strain I-1A sir2Δ∷SIR2(S96) by substituting the SIR2(YJM789) by the S96 allele. Strains S96 and S96 sir2Δ∷SIR2(YJM789) served as additional controls in the subsequent RLS determination. Strain I-1A had a mRLS of 49.5 generations, and its corresponding mutant I-1A sir2Δ∷SIR2(S96) had a mRLS of 41.6, a decrease of 16% (Fig. 3B). S96 and S96 sir2Δ∷SIR2(YJM789) had a mRLS of 30.7 and 43.7 generations, respectively, an increase of 42% in the latter strain in this experiment (Fig. 3B). These results were reproducible (data not shown) and in agreement with the previous experiment (Fig. 3A). From both RLS determinations, we conclude that one or more of the AA polymorphisms in SIR2 are responsible for the large differences in mRLS.
The effect of the SIR2 allele on RLS was confirmed by reciprocal hemizygosity analysis (RHA) (Steinmetz et al. 2002). This was performed by generating diploid strains (S96 × YJM789) in which either the S96 or the YJM789 SIR2 gene was deleted and replaced by the KanMX drug-resistance marker, which does not affect RLS. To examine, the potential effect of copy number, an additional diploid that had two copies of the YJM789 SIR2, one of which replaced the S96 allele, was created. The hemizygous SIR2 allele from S96 (strain YFS90) curtailed RLS compared with the heterozygote (strain YFS97), while the hemizygous SIR2 allele from YJM789 (strain YFS70) extended it (Supplemental Fig. S3). An additional copy of the YJM789 allele of SIR2 in the diploid (strain YFS77) had no further effect on this RLS extension. Thus, SIR2 is the major RLS locus in QTL 1, and its alleles from the two parental strains YJM789 and S96 show incomplete dominance (semi-dominance). Indeed, the heterozygous diploid had a mRLS and maximum RLS of 30.6 and 57 generations, respectively, which was very close to the arithmetic average of those values for the hemizygous strains YFS70 and YFS90. These results suggest that the SIR2 from S96 not only cannot fill the role of the YJM789 allele in determining RLS but that it has a deleterious effect, perhaps by competition at the protein level.
We know that aging processes and the production of extrachromosomal rDNA circles (ERCs) in yeast are connected (Sinclair and Guarente 1997). High levels of ERCs are responsible for accelerated aging. To investigate whether the AA polymorphisms in Sir2p influence ERC production, we conducted Southern blot analyses with a probe directed against the rDNA in the strains S96, S96 sir2Δ∷SIR2(YJM789), I-1A, and I-1A sir2Δ∷SIR2(S96). The levels of ERCs were quantified using the ACT1 gene as reference (Fig. 4). S96 has a shorter mRLS than S96 sir2Δ∷SIR2(YJM789) and showed higher levels of ERCs. Similarly, I-1A sir2Δ∷SIR2(S96) has a shorter mRLS than I-1A and its ERC levels were higher. This experiment suggests that RLS is indirectly proportional to the levels of ERCs. It is known that overexpression of SIR2 increases RLS (Kaeberlein et al. 1999). We did not alter the endogenous SIR2 promoter region in any of the strains we generated. Nevertheless we performed qRT-PCR to determine the SIR2 expression level in S96 and YJM789. No significant differences were found (Fig. 5). This suggests that the differences in mRLS are caused by the altered AA of Sir2p.
Figure 4.

Southern analysis of ERC levels. S96, S96 sir2Δ∷SIR2(YJM789), I-1A, and I-1A sir2Δ∷SIR2(S96) were compared. YFS7 (YJM789 background with RDN1 from YJM789) and YFS9 (YJM789 background with RDN1 from S96), as well as S96 and YFS1 (S96 background with RDN1 from YJM789) were compared separately. I-1A is the recombinant line with the longest RLS. (Upper membranes) Genomic DNA of the eight strains was prepared, cut with the restriction endonuclease SpeI and separated by electrophoresis. The DNA was hybridized with a probe specific for 35S rDNA. The brightest signal on each membrane corresponds to the genomic rDNA locus (arrowheads); additional signals correspond to circular multimers or monomers of ERC (arrows). (Lower membranes) The blots were hybridized with a probe to the single-copy gene ACT1 to allow quantification. For the quantification of ERC, the signal intensities were normalized to the hybridization signals of ACT1 (values at bottom).
Figure 5.

SIR2 expression. The expression level of SIR2 mRNA is shown for strains S96, S96 sir2Δ∷SIR2(YJM789), YJM789, I-1A, and I-1A sir2Δ∷SIR2(S96). I-1A is the recombinant line with the longest RLS. Standard error bars are shown for three biological replicates each; the differences are not statistically significant.
QTL 5 consists mainly of the rDNA locus
In addition to QTL 1, other QTLs impact RLS, the most prominent of which is QTL 5. About 97%–99% of QTL 5 consists of tandem repeats of rDNA depending on the copy number of the rDNA array (Fig. 2B). This and the effect of ERCs on RLS in laboratory strains (Sinclair and Guarente 1997) drew our attention to this locus. Of the 39 recombinant lines, the seven with the longest RLS contain QTL 5 from S96, while the four shortest-lived lines contain QTL 5 from YJM789 (Table 1). This suggests that the S96 variant of QTL 5 may prolong the RLS compared with the YJM789 variant. Therefore, we decided to create strains in which the rDNA cluster of strain YJM789 is substituted by the S96 rDNA cluster. Because the rDNA cluster segregates as a unit during meiosis (Petes et al. 1978), this was accomplished by introgressing the S96 rDNA cluster into YJM789 through seven successive backcrosses and the selection of the S96 rDNA cluster after each backcross. Finally, strains which contained statistically 99.25% of their genome from YJM789 were selected for both versions of the rDNA cluster. These strains were MATa to allow a comparison with all previously analyzed strains. An RLS analysis was performed with the congenic strains YFS7 and YFS9. The former contains 99.25% DNA from YJM789 including the rDNA, whereas the latter differs by carrying the rDNA cluster from S96 (Table 2). The mRLS of YFS7 was 33.8 generations, and the mRLS of strain YFS9 was 40.0 (Fig. 3C), which is 18.3% (6.2 generations) longer. This analysis reveals that the possession of the S96 rDNA cluster extends longevity compared with the rDNA of YJM789. RLS determinations with other clones of these strains have been conducted and replicate these findings (data not shown).
Table 2.
Background, mRLS and QTL of wild-type, recombinant line, and mutant strains
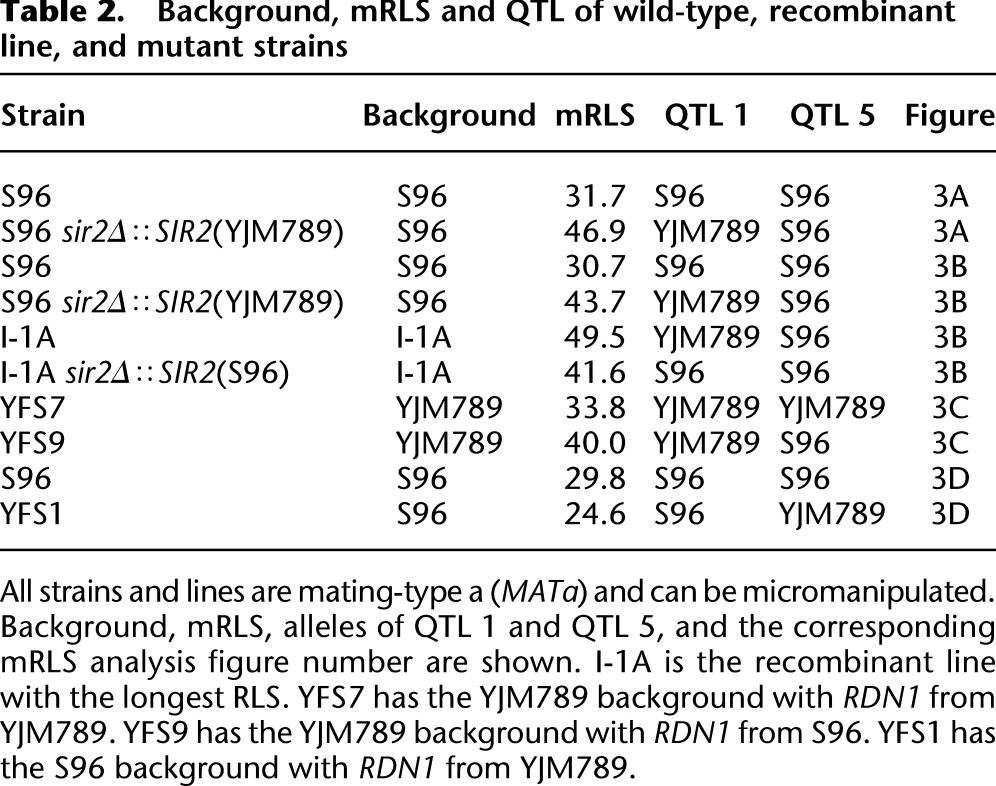
To further support our findings, we replaced the S96 rDNA in strain S96 by the rDNA cluster from strain YJM789. The same introgression strategy was used, but the seven backcrosses were into strain S96 with selection of the YJM789 rDNA cluster after each backcross. The resulting strain YFS1 contains statistically 99.25% of its genome from strain S96. It is MATa, and it has the rDNA cluster from YJM789 (Table 2). The mRLS of S96 was 29.8 generations, and the mRLS of strain YFS1 was 24.6 (Fig. 3D), which is a decrease of 17.4% (5.2 generations). Thus, the YJM789 rDNA cluster reduces RLS of strain S96.
The rDNA repeats of S96 and YJM789 are different
To gain insight into the effects of the rDNA cluster on mRLS and ERC production, we compared the sequences of the rDNA of strains S96 and YJM789. The rDNA sequence of strain S96 can be accessed in the Saccharomyces Genome Database (http://www.yeastgenome.org/). We sequenced the rDNA of YJM789 in our laboratory (GenBank accession number JQ277730). Comparison of the rDNA of both strains reveals a variety of polymorphisms that may account for functional differences. Interestingly, the sequences of the rRNA-encoding genes RDN25, RDN58, RDN18, and RDN5 are identical in both strains, as are the ORF of the polymerase II transcribed genes TAR1, YLR154W-E, YLR154W-F, and the ETS2-1 sequence. On the other hand, polymorphisms were found in ITS2, ITS1, ETS1-1, NTS2-1, and NTS1-2 sequences (Supplemental Figs. S4, S5). Two polymorphisms are located in the 107-bp ARS1200. Compared to the 11-bp ACS, the ninth base pair of the corresponding sequence of YJM789 is a C instead of a T (Supplemental Fig. S6), which would most likely alter its function. We would expect decreased ARS function to result in lower ERC levels (Ganley et al. 2009). Comparison of strains YFS7 and YFS9 suggests a decrease in ARS function for the ARS from strain YJM789, but the opposite conclusion arises from the comparison of strains S96 and YFS1 (Fig. 4). SIR2 has been shown to suppress ERC levels in a laboratory strain (Kaeberlein et al. 1999), and the SIR2 from YJM789 is more effective in this regard (Fig. 4). However, the parental origin of SIR2 is only one of the differences between the strains in the comparisons of YFS7 with YFS9 and S96 with YFS1 in Figure 4. Thus, it appears that genetic variation outside the SIR2 and rDNA loci also contributes to ERC levels. In any case, RLS does not appear to simply be a function of ERC levels (Figs. 3, 4).
The 9.1-kb rDNA repeat in S. cerevisiae gives rise to several primary transcripts (Supplemental Fig. S5). We determined the transcript levels of 5S rRNA and 18S rRNA, which are transcribed by polymerase III and I, respectively, because we found polymorphisms in spacers and nontranscribed sequences that might affect their transcription (Supplemental Figs. S4, S5). The polymerase II transcript TAR1 was examined for comparison. Strains harboring rDNA clusters from S96 showed an increase in the 5S rRNA transcripts but not in 18S rRNA or TAR1 transcripts (Fig. 6). This suggests that polymorphisms in the NTS2-1 region, which is the promoter region of RDN5, are responsible for its altered transcription levels. It is not immediately obvious, however, how this would affect RLS or ERC levels.
Figure 6.

Transcription of 18S rRNA, 5S rRNA, and TAR1. The transcript levels of 18S rRNA (A), TAR1 (B), and 5S rRNA (C) of strains S96, YFS1, YFS7, and YFS9 were determined using qRT-PCR. No significant differences were found for 18S rRNA and TAR1 transcripts in A and B, respectively. Significant differences were found between strains S96 and YFS1 (S96 background with RDN1 from YJM789) (P < 0.05), and between strains YFS7 (YJM789 background with RDN1 from YJM789) and YFS9 (YJM789 background with RDN1 from S96) (P < 0.05), for 5S rRNA transcripts in C. Standard error bars are shown for n = 3 biological replicates.
We also examined the rDNA copy number quantitatively in the parental strains S96 and YJM789, as well as in several of the other strains and lines generated in this study (Supplemental Fig. S7). Total rDNA and genomic rDNA paralleled each other, with the difference attributed to the ERC copy number, which was also determined in this analysis. Total rDNA copy number did not correlate with mRLS (Supplemental Fig. S7). Strain YFS9 had 88% as many ERCs as genomic rDNA copies. Yet, its mRLS was 81% that of the longest-lived line (I-1A) and 63% longer than that of the shortest-lived strain that was analyzed for rDNA copy number (YFS1). Thus, there was no direct relationship between ERC levels and mRLS.
Discussion
We have found five QTLs associated with RLS, representing genetic differences between a laboratory yeast strain and a clinically derived strain. These QTLs account for two-thirds of the variation in RLS in the cross between these strains. The two QTLs with the largest effects on longevity contain genes implicated in RLS in previous studies with laboratory strains. They are QTL 1, which has the largest impact on longevity accounting for 50% of the genetic effect, followed by QTL 5, which accounts for 20%. The naturally occurring variant of the SIR2 gene was responsible for the majority of the effect on RLS of the first of these QTL, as determined in the gene swap experiments and confirmed by RHA (Fig. 3A,B; Supplemental Fig. S3). Interestingly, it was the laboratory variant of the RDN1 (rDNA) locus that accounted for the majority of the effect of the second QTL, as demonstrated by the reciprocal introgression experiments (Fig. 3C,D). The results suggest that the wild SIR2 variant compensates for the negative effect of the wild RDN1 variant on RLS. One negative effect of rDNA is the generation of ERCs, which curtail RLS (Sinclair and Guarente 1997). The wild SIR2 variant appears to compensate for this, both for the wild and for the laboratory variants of rDNA, with the expected effects on RLS (Figs. 4 [S96, S96 sir2Δ∷SIR2(YJM789), I-1A, I-1A sir2Δ∷SIR2(S96)]; 3A,B). This represents a gene-by-gene interaction in determining yeast longevity and an example of heterosis (hybrid vigor). Although this conclusion regarding the interaction of the rDNA locus and SIR2 is broadly correct, there appear to be some modifying effects of the genetic background, as the ERC levels in strains YSF7 and YSF9 seem to indicate (Fig. 4). In any case, RLS does not appear to simply be a function of ERC levels, and SIR2 seems to have longevity effects in addition to those impinging on the rDNA locus, based on the results presented here. This conclusion is supported by both the complex molecular biology of the interaction of SIR2 with rDNA (Ganley et al. 2009; Ha and Huh 2011) and evidence pointing to a role in RLS for SIR2 outside rDNA (Kaeberlein et al. 1999; Laskar et al. 2011; Wang et al. 2011).
SIR2 has been found abundantly expressed in long-lived compared with short-lived yeast strains (Guo et al. 2011). In addition, young cells contain greater than 10 times more Sir2p than older cells (Lindstrom et al. 2011). As a consequence, histone H4 lysine-16 of older cells remains heavily acetylated. This hyperacetylation is thought to be one factor contributing to senescence (Dang et al. 2009). It was previously shown that the introduction of a second copy of SIR2 in yeast strain W303R resulted in ∼30% extension of mRLS (Kaeberlein et al. 1999). Our qRT-PCR experiments show that the laboratory strain S96 and the clinically derived strain YJM789 displayed equal levels of SIR2 expression (Fig. 5). On the other hand, a change in five AA in the SIR2 ORF led to a 48% increased mRLS (Fig. 3A), a larger effect than seen due to overexpression of the gene (Kaeberlein et al. 1999). Figure 4 (S96, S96 sir2Δ∷SIR2[YJM789], I-1A, I-1A sir2Δ∷SIR2[S96]) shows reduced ERC levels in strains carrying the SIR2 ORF from strain YJM789 compared with strains with the S96 allele, irrespective of the source of the rDNA. We conclude that one or more of the five changed AA strengthens the silencing of the rDNA cluster, which in turn leads to stabilized rDNA and thus to increased mRLS. However, it is possible that the reduced ERC amplification and the increased RLS are independent effects of the altered SIR2 in these strains.
Four of the five polymorphic AA of strains S96 and YJM789 (Pro65/Thr65, Val92/Glu92, Asp158/Val158, and Met424/Val424) are not conserved in other species, since their SIR2 homologs lack the corresponding protein domains (Sauve et al. 2006). We compared S96 and YJM789 with four other S. cerevisiae strains (FostersB, FostersO, JAY291, RM11-1a). Apart from YJM789, no other strain contains Glu92 or Val158. One of the other strains contains either Thr65 or Val424. Strains like S. cerevisiae RM11-1a or the species Saccharomyces bayanus—but only a few higher eukaryotes—contain Asn320. Unlike the four other altered AA, only Lys320/Asn320 is located in a conserved domain of the Sir2 protein. The identity of the variant(s) critical for RLS and ERC production thus remains an open question.
The effects on the RLS and ERC of the SIR2 and RDN1 variants and their mutual interactions appear to be modified by the genetic background. The replacement of the SIR2 ORF in S96 by the one from YJM789 increases the mRLS by 15 generations (Fig. 3A) in the presence in both strains of the rDNA cluster from S96. However, if the SIR2 ORF of recombinant line I-1A is replaced by the S96 variant, the difference in mRLS is only eight generations (Fig. 3B), even though both strains carry the S96 variant of the rDNA. Obviously, the RLS extension mediated by the SIR2 variant from YJM789 depends partly on the rDNA cluster from strain S96, but the effects that lead to an altered lifespan are probably more complex in this case. This complexity is likely dependent upon gene interactions within QTL 1 and QTL 5 and/or interactions with loci outside the five QTLs we have identified, as MIM analysis showed no detectable interactions between the five QTLs. However, MIM provides only an estimate of interaction effects. Furthermore, this analysis was performed on a relatively small number of recombinant lines, and therefore, interactions could have been missed. In turn, interactions within QTLs have been demonstrated for other quantitative traits in yeast (Steinmetz et al. 2002; Sinha et al. 2006).
The presence of the S96 rDNA cluster leads to increased mRLS (Fig. 3C,D). But what causes the RLS extension? We found many sequence changes between the rDNA clusters of S96 and YJM789. One candidate for the polymorphism that affects RLS is within ARS1200 where, in YJM789, a conserved T in the ACS is replaced by a C that may reduce the recognition of ARS1200. The activity of the replication origin of the rDNA affects the levels of ERC (Ganley et al. 2009), suggesting that the rDNA cluster of YJM789 would give rise to fewer ERCs than its S96 counterpart. This is only the case in the YJM789 background, but not in the S96 background (Fig. 4; Table 2). However, in both backgrounds, the possession of the S96 rDNA cluster increases mRLS significantly (Fig. 3). Therefore, we exclude the effect of the sequence changes in ARS1200 on ERC levels as the cause of the observed RLS extension. We postulated that the polymorphism in the ARS1200 sequence influences the transcription rate of the rDNA. The promoter region of the 5S rRNA is located in the NTS2-1 region, which also contains the ARS1200 polymorphism. Significantly increased transcription was only found for 5S rRNA in strains harboring the rDNA cluster from strain S96 (Fig. 6).
There is another spacer of interest. The most prominent polymorphisms in the NTS1-2 region are four deletions in the sequence of YJM789 (Supplemental Fig. S4). In the sequence of S96, the spacing between these deletions is ∼190 bp, the distance between two nucleosomes. The NTS1-2 region contains the 400-bp EXP region adjacent to the replication fork barrier (RFB) sequence (Supplemental Fig. S4). EXP is needed for FOB1-dependent repeat expansion of the rDNA (Kobayashi et al. 1992, 2001). The EXP region contains seven polymorphisms, but we saw no gross differences in rDNA unit copy number (Fig. 4). Furthermore, quantitative analysis showed that there was no direct relationship between rDNA copy number and mRLS (Supplemental Fig. S7). Two rDNA polymorphisms are close to the Fob1p binding site, which itself is conserved (Supplemental Fig. S4). Since DNA wraps around Fob1p in a nucleosome-like manner (Kobayashi 2003), adjacent polymorphisms may be sufficient to affect Fob1p function. Fob1p has multiple functions, among them replication fork arrest and rDNA silencing (Bairwa et al. 2010). Therefore, altered Fob1p binding can have different effects. ERC levels seem to be affected, but in a genetic background–dependent manner (see above). On the other hand, the increased transcription rate of the 5S rRNA may lead to increased displacement of Fob1p at the RFB sites, which in turn may reduce the amount of Fob1p-induced double-strand breaks (Weitao et al. 2003). This is in agreement with the extended RLS in strains harboring rDNA from S96. Alternatively, a process involving condensin could be involved in the genetic effects described here (Machín et al. 2004; Johzuka and Horiuchi 2009). FOB1 is ∼200 cM centromere-proximal to QTL 3 on chromosome IV, suggesting that these potential mechanisms must operate independently of any variation in FOB1 itself.
The various recombinant lines carrying the SIR2 ORF of YJM789 and rDNA of S96 display differences in mRLS up to 21 generations (I-1A and III-9B in Table 1). This suggests that QTL 2, 3, and 4 contain genes influencing RLS significantly, none of which have been identified before in the laboratory. It is likely that other RLS QTL segregate in the S96 × YJM789 cross but are masked by the major effect QTL and epistasis (Sinha et al. 2008), although our MIM analysis seems to marginalize the latter effect. The complex genetic architecture of QTL has been amply demonstrated (Steinmetz et al. 2002; Sinha et al. 2006). This architecture includes genetic interactions in cis and in trans, as well as complicated background effects. It also encompasses haplotypic effects that involve tightly linked genes that alone are neither necessary nor sufficient to elicit the phenotype. These considerations suggest that our study only scratches the surface of the quantitative genetics of RLS in yeast.
The quantitative genetic analysis described here does not assess the potential contributions to RLS of the mating-type MAT locus or of loci responsible for filamentous growth. The parental strains S96 and YJM789 are MATa and MATα, respectively, and the MAT locus segregates in the cross between them. We focused our attention on MATa segregants exclusively to decrease the sample size needed to detect QTLs for RLS elsewhere in the genome, leaving any contributions of the mating-type locus for future studies. It is worth noting, however, that no QTL was detected in the vicinity of the MAT locus on the right arm of chromosome III (Fig. 1A). It is possible to draw some conclusions regarding an association of filamentous growth and RLS. Because S96 does not display filamentous growth, we searched for the presence in all 39 recombinant lines of the same region(s) of the S96 genome identified by contiguous stretches of one or more of the 3048 markers used for genotyping. Only one such region was found, encompassing 11 markers. It was on the right arm of chromosome II between base pairs 562,406 and 582,418 (Fig. 1A), 4208 bp downstream from AMN1 and encompassing 13 genes. However, there was only one recombinant line that could potentially possess a crossover within AMN1, and there were no QTL for RLS on chromosome II. AMN1 was identified in a large-scale analysis of filamentous growth (Jin et al. 2008). A dedicated search will be necessary to identify QTL that affect filamentous growth. In sum, we have not ruled out the possibility that the MAT locus or a filamentous growth QTL might affect RLS, and as discussed above, QTL1 and QTL5 identified here may contain RLS genes in addition to SIR2 and RDN1, respectively.
S. cerevisiae is an opportunistic pathogen (Muller and McCusker 2009). The natural variation in RLS that we have sampled comes from one clone of a clinical isolate of S. cerevisiae. A population genetic analysis has shown that three distinct groups characterize this species. These include vineyard and brewery strains, clinical and fruit isolates, and soil isolates (Diezmann and Dietrich 2009). Interestingly, the parental strain S96 is a laboratory derivative of a yeast isolated from a rotten fig (Mortimer and Johnston 1986) and together with YJM789 belongs to the second group. This leaves the genetic variation in the other two groups unsampled in our analysis. This variation is likely to reveal additional genes and pathways that determine longevity.
Methods
Gene chip sample preparation
Genomic DNA was isolated using Qiagen Genomic-tip 100/G columns. All procedures for DNA isolation were followed as published in the manufacturer's instructions, except incubation times were extended from 30 to 45 min. Purified samples had an A260/A280 of 1.7–1.9. Eleven micrograms of genomic DNA was digested with 0.165 units of DNase I (Invitrogen) for 3–10 min at 37°C. DNase I was then heat inactivated for 15 min at 99°C. A 0.5 μg aliquot of fragmented DNA was loaded onto a 2% agarose gel to determine fragment lengths and sample homogeneity by electrophoresis. Sybr green II was added to samples to visualize the DNA, and a 25-bp oligonucleotide marker (Invitrogen) was electrophoresed alongside to locate and quantify 25- to 50-bp fragments. Digested DNA was labeled using 50 units of terminal transferase in 1× TdT reaction buffer (Roche), 2.5 mM CoCl2, and 1 nmol biotin-N6-ddATP (Enzo) for 1.5 h at 37°C. To stop the labeling reaction, 25 mM EDTA was added.
Array hybridization, washing, and scanning
Reagents for hybridization, wash, and staining were purchased from Affymetrix (GeneChip HT Hybridization, Wash and Stain kit) unless otherwise stated. The Affymetrix gene chip expression analysis manual was followed for sample preparation. Each sample was included in a hybridization cocktail containing 30 pM control oligonucleotide B2, 1× eukaryotic hybridization controls (bioB, bioC, bioD, cre), 0.1 mg/mL herring sperm DNA (Promega), 0.5 mg/mL acetylated BSA (Invitrogen), and 1× hybridization buffer. Hybridization samples were heated for 5 min to 99°C, incubated for 5 min at 45°C, and centrifuged for 5 min at 20,000g, to remove any precipitate. Each Affymetrix Yeast S98 GeneChip (AYGC) was equilibrated to room temperature prior to hydration with 200 μL 1× MES hybridization buffer and then incubated for 10 min at 45°C with rotation. The hybridization buffer was removed from the AYGC, and the hybridization cocktail was applied to the gene chip and incubated for 16 h at 45°C, while mixing on a rotisserie at 60 rpm. After hybridization, samples were inserted into the Affymetrix fluidics station. The stain solution, containing 1× stain buffer, 2 mg/mL acetylated BSA, and 10 μg/mL streptavidin-phycoerythrin (SAPE) was inserted into the sample holder of the fluidics station. The protocol for washing and staining (EukGE-WS2v4) of the AYGC was followed according to the Affymetrix Gene chip expression analysis manual. Parameters using the Gene Chip Operating Software (GCOS) version 1.4 were as follows: fluid extension value of 450, target signal of 250, and normalization value of 1.
Data analysis
Raw output CEL files were converted from compressed binary CEL files to CEL text files using the Affymetrix CEL file conversion tool (available at https://www.affymetrix.com/support/file_download.affx?onloadforward=/support/developer/downloads/Tools/CelFileConversion.ZIP). CEL files were imported into Allelescan (available for download at http://med.stanford.edu/sgtc/research/download/), and data quality was determined for parental strains and recombinant lines by using Allelescan's quality check for controls, markers, hybridizations, and replicates. Samples were excluded from the data set if the comparative analysis indicated an outlier, as described in the Allelescan manual. A critical F-test value of 35 and marker score filter value of 0.50 was used to identify approximately 3048 molecular markers found between the two parentals, constituting an estimated 3.9% of the total genetic variation between the two strains (Winzeler et al. 1998). The genotype of each haploid segregant was confirmed using the molecular marker data to determine the inherited regions from each parental strain (Supplemental Fig. S2).
QTL mapping
Molecular marker designations and crossover locations were determined using the Allelescan software. Molecular marker positions and segregant genotypes were then used to compile input files for Windows QTL Cartographer version 2.5 (available at http://statgen.ncsu.edu/qtlcart/WQTLCart.htm), and along with RLS data, they were analyzed using CIM to find DNA regions associated with RLS (Basten et al. 1994, 2004). The walking step for the mapping was set at 0.5 cM. The LOD value 2.5 was determined as the genome-wide significance threshold for statistical significance of the QTL identified. The effect of QTL on RLS and QTL interactions were further estimated using MIM provided by Windows QTL Cartographer (Zeng 1994).
RLS analysis
Lifespan analysis was performed as described (Kim et al. 1999). Briefly, virgin cells (new buds) from an exponentially growing overnight culture were used for each RLS determination. These cells were deposited in a row in isolated spots on an YPD (2% peptone, 1% yeast extract, 2% glucose) plate. The number of buds removed by microdissection prior to cell death is the cell's RLS in generations. The Mann-Whitney test was used to assess statistical significance of differences between the lifespan of different strains in an experiment. All RLS analyses were repeated at least two times. Statistical analysis was done using Statmost (DataMost). Reported P-values are two-sided.
Construction of strains S96 sir2Δ∷SIR2(YJM789) and I-1A sir2Δ∷SIR2(S96)
The ura3-1 of strain W303, obtained by PCR amplification using primers URA3-F (5′-ATTGAGGCTACTGCGCCAAT-3′) and URA3-R (5′-GGAGTTCAATGCGTCCATCT-3′), was transformed into strain S96 and line I-1A. Transformants were selected on synthetic complete (SC) medium (Guthrie and Fink 1991) containing 1 g/L 5-fluoroorotic acid (5-FOA). The SIR2 loci of strains S96 and YJM789, amplified using PCR primers 5′- TATTACGTCGACCCTGCAACTCCTCAATGT-3′ and 5′-ATGATTGTCGACTGCTGTTCCACCTGCCCTTC-3′, were ligated into plasmid pUC19 previously cut with SalI, resulting in plasmids pUC5S3 and pUC5M3, respectively. The SIR2 ORF of plasmids pUC5S3 and pUC5M3 were replaced (between the BsaAI and BstXI sites) by a URA3 cassette (obtained by PCR amplification using plasmid pCM297, kindly provided by Mark Rinnerthaler (University of Salzburg, Austria), and primers 5′-AAGGTTCACGTAGCTGTGGTTTCAGGGTCCATAA-3′ and 5′-CATTATCCACATTTTTGGGACCGAGATTCCCGGGTAATAA-3′), resulting in plasmids pUC5U3 and pUCMUMi, respectively. Plasmids pUC5U3 and pUCMUMi were cut with SalI and transformed into strains S96 ura3-1 and I-1A ura3-1, respectively. The selection of S96 ura3-1 sir2Δ∷URA3 and I-1A ura3-1 sir2Δ∷URA3 transformants was conducted on SC -uracil medium. The SIR2 ORF (S96) of plasmid pUC5S3 was replaced by the SIR2 ORF (YJM789) of plasmid pUC5M3 between BsaAI and BstXI sites, resulting in plasmid pUCSMS. The same was done in the opposite direction, resulting in plasmid pUCMSMi containing the S96 SIR2 ORF. Plasmids pUCSMS and pUCMSMi were cut with SalI and transformed into strains S96 ura3-1 sir2Δ∷URA3 and I-1A ura3-1 sir2Δ∷URA3, respectively. Selection of S96 ura3-1 sir2Δ∷SIR2(YJM789) and I-1A ura3-1 sir2Δ∷SIR2(S96) transformants was carried out on 5-FOA medium. Both strains were transformed by the URA3 locus of strain S96, amplified using PCR primers URA3-F and URA3-R, to restore the URA3 locus. Selection of S96 sir2Δ∷SIR2(YJM789) and I-1A sir2Δ∷SIR2(S96) transformants was done on SC-uracil medium. The entire SIR2 loci of strains S96 sir2Δ∷SIR2(YJM789) and I-1A sir2Δ∷SIR2(S96) were verified by sequencing. (Underlined primer sequences indicate sites for SalI, BstXI, and BsaAI, respectively.)
Construction of strains YFS1, YFS7, and YFS9
Strain YJM789 was crossed with strain S96. The diploid was sporulated, and a haploid segregant was backcrossed to S96, followed by isolation of a haploid segregant. This backcross procedure was repeated five more times to introgress the YJM789 RDN1 cluster into the S96 background in seven steps, yielding strain YFS1. The introgression was carried out in the opposite direction to introduce the S96 RDN1 cluster into the YJM789 background, yielding strains YFS7 and YFS9. In each step, strains with yeast-like growth type and the desired RDN1 (rDNA cluster) allele were selected. This was done by PCR amplification and restriction analysis of RNH203, which is located 12,241 bp upstream of the rDNA cluster. Primers 5′-ATGACCAAAGATGCCGTGAAT-3′ and 5′-TTACTGATTTATGACATCGATGAG-3′ were used. The resulting 333-bp product can be cut into 184 bp and 149 bp by Cac8I only when derived from YJM789. The rDNA cluster of the final strains was partially sequenced for verification.
ERC detection
Cells were grown overnight in YPD. DNA was isolated using the spheroplast method, as described (Medvedik and Sinclair 2007). Three micrograms of DNA was cut with SpeI, which does not cut in the rDNA, but releases a 4.1-kb fragment of ACT1 used for quantification. DNA was electrophoresed in a 0.7% agarose gel at 0.9 V/cm for 18 h and subsequently transferred to a nylon membrane, which was hybridized with the ACT1 and 35S rDNA [α-32P]dCTP labeled probes. Preparation of the probes using the RediPrime kit (GE Healthcare) was described previously (Borghouts et al. 2004). Blots were scanned using the Typhoon (GE Healthcare), and quantification was performed using ImageQuant version 5.1 software (Molecular Dynamics). ERC copy number was quantified by normalizing to the specific activity of the ACT1 and 35S rDNA probes. The probes were prepared by nick translation, in this case, and they were ∼500 bp. Total rDNA copy number was similarly determined. However, DNA was digested with KpnI, which cuts once within each rDNA repeat and once within ACT1 (Kim et al. 2006). The genomic rDNA copy number was the difference between total rDNA and ERC copy number.
qRT-PCR analysis
For the preparation of cDNA, the RNase-free DNase set (Qiagen) and the TaqMan kit (Applied Biosystems) were used according to the manufacturer's manual. For amplification of cDNA, primers 5′-CCTGAGAAACGGCTACCACATC-3′ and 5′-ATTGTCACTACCTCCCTGAATTAGGA-3′ (18S rRNA), 5′-TTACCACCCACTTAGAGCTGCAT-3′ and 5′-CGAGGAGTGCGGTTCTTTGT-3′ (TAR1), 5′-GGTTGCGGCCATATCTACCA-3′ and 5′-ACTACTCGGTCAGGCTCTTACCA-3′ (5S rRNA), and 5′-TTCCATCCAAGCCGTTTTGT-3′ and 5′-CAGCGTAAATTGGAACGACGT-3′ (ACT1) were used in the same PCR. qRT-PCR were carried out in an Applied Biosystems 7300 real-time PCR machine. PCR conditions were chosen according to the manufacturer's manual. The Newman-Keuls post hoc test was used to determine the significance of specified differences in transcript levels. P-values are two-sided.
Sequencing
The equivalent of one repeat of the RDN1 locus of strain YJM789 was sequenced. Parts of the rDNA of ∼1 kbp were PCR subcloned at least three times for sequencing. Subsequently, they were sequenced in both directions. Proofreading DyNazyme and Phusion polymerases were utilized (NEB). Sequences with stretches of the same base were sequenced at least an additional three times. All strains generated in this study have been verified by sequencing altered loci plus 1 kbp upstream and downstream.
Data access
Array genotyping data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE37590. The GenBank (http://www.ncbi.nlm.nih.gov/genbank/) accession number for the sequence of YJM789 rDNA is JQ277730.
Acknowledgments
We thank Abha Tiwari, Liliana Cosenza, and Justin Manges for expert technical assistance. We thank Ronald W. Davis (Stanford University) for providing strains S96 and YJM789. This work was supported by grant AG-SS-1461-05 from the Ellison Medical Foundation and grant AG006168 from the National Institutes of Health.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.136549.111.
References
- Adams J 2004. Microbial evolution in laboratory environments. Res Microbiol 155: 311–318 [DOI] [PubMed] [Google Scholar]
- Bairwa NK, Zzaman S, Mohanty BK, Bastia D 2010. Replication fork arrest and rDNA silencing are two independent and separable functions of the replication terminator protein Fob1 of Saccharomyces cerevisiae. J Biol Chem 285: 12612–12619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basten CJ, Weir BS, Zeng Z-B 1994. Zmap-a QTL cartographer. In Proceedings of the fifth world congress on genetics applied to livestock production: Computing strategies and software (ed. C Smith et al.), Vol. 22, pp. 65–66. Department of Animal & Poultry Science, University of Guelph, Ontario, Canada [Google Scholar]
- Basten CJ, Weir BS, Zeng Z-B 2004. QTL cartographer, version 1. 17. Department of Statistics, North Carolina State University, Raleigh, NC [Google Scholar]
- Borghouts C, Benguria A, Wawryn J, Jazwinski SM 2004. Rtg2 protein links metabolism and genome stability in yeast longevity. Genetics 166: 765–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem RB, Storey JD, Whittle J, Kruglyak L 2005. Genetic interactions between polymorphisms that affect gene expression in yeast. Nature 436: 701–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JM, Ball C, Weng S, Juvik G, Schmidt R, Adler C, Dunn B, Dwight S, Riles R, Mortimer RK, et al. 1997. Genetic and physical maps of Saccharomyces cerevisiae. Nature 387: 67–73 [PMC free article] [PubMed] [Google Scholar]
- Choy JS, Acuña R, Au WC, Basrai MA 2011. A role for histone H4K16 hypoacetylation in Saccharomyces cerevisiae kinetochore function. Genetics 189: 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, Berger SL 2009. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459: 802–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney JR, Murakami CJ, Olsen B, Kennedy BK, Kaeberlein M 2011. Quantitative evidence for early life fitness defects from 32 longevity-associated alleles in yeast. Cell Cycle 10: 156–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diezmann S, Dietrich FS 2009. Saccharomyces cerevisiae: Population divergence and resistance to oxidative stress in clinical, domesticated and wild isolates. PLoS ONE 4: e5317 doi: 10.1371/journal.pone.0005317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov LN, Brem RB, Kruglyak L, Gottschling DE 2009. Polymorphisms in multiple genes contribute to the spontaneous mitochondrial genome instability of Saccharomyces cerevisiae S288C strains. Genetics 183: 365–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J, Mackay TF 2009. Genetic architecture of quantitative traits in mice, flies, and humans. Genome Res 19: 723–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss EJ, Radulovic D, Shaffer SA, Ruderfer DM, Bedalov A, Goodlett DR, Kruglyak L 2007. Genetic basis of proteome variation in yeast. Nat Genet 39: 1369–1375 [DOI] [PubMed] [Google Scholar]
- Froyd CA, Rusche LN 2011. The duplicated deacetylases sir2 and hst1 subfunctionalized by acquiring complementary inactivating mutations. Mol Cell Biol 31: 3351–3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley AR, Ide S, Saka K, Kobayashi T 2009. The effect of replication initiation on gene amplification in the rDNA and its relationship to aging. Mol Cell 35: 683–693 [DOI] [PubMed] [Google Scholar]
- Garcia SN, Pillus L 2002. A unique class of conditional sir2 mutants displays distinct silencing defects in Saccharomyces cerevisiae. Genetics 162: 721–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatbonton T, Imbesi M, Nelson M, Akey JM, Ruderfer DM, Kruglyak L, Simon JA, Bedalov A 2006. Telomere length as a quantitative trait: Genome-wide survey and genetic mapping of telomere length-control genes in yeast. PLoS Genet 2: e35 doi: 10.1371/journal.pgen.0020035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, et al. 1996. Life with 6000 genes. Science 274: 563–567 [DOI] [PubMed] [Google Scholar]
- Gu Z, David L, Petrov D, Jones T, Davis RW, Steinmetz LM 2005. Elevated evolutionary rates in the laboratory strain of Saccharomyces cerevisiae. Proc Natl Acad Sci 102: 1092–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Adomas AB, Jackson ED, Qin H, Townsend JP 2011. SIR2 and other genes are abundantly expressed in long-lived natural segregants for replicative aging of the budding yeast Saccharomyces cerevisiae. FEMS Yeast Res 11: 345–355 [DOI] [PubMed] [Google Scholar]
- Guthrie C, Fink GR 1991. Guide to yeast genetics and molecular biology. Academic Press, San Diego [Google Scholar]
- Ha CW, Huh WK 2011. Rapamycin increases rDNA stability by enhancing association of Sir2 with rDNA in Saccharomyces cerevisiae. Nucleic Acids Res 39: 1336–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl DL, Clark AG 1997. Principles of population genetics, 3rd ed. Sinauer, Sutherland, MA [Google Scholar]
- Heeren G, Rinnerthaler M, Laun P, von Seyerl P, Kössler S, Klinger H, Hager M, Bogengruber E, Jarolim S, Simon-Nobbe B, et al. 2009. The mitochondrial ribosomal protein of the large subunit, Afo1p, determines cellular longevity through mitochondrial back-signaling via TOR1. Aging 1: 622–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskowitz I 1988. Life cycle of the budding yeast Saccharomyces cerevisiae. Microbiol Rev 52: 536–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XH, Wang MH, Tan T, Li JR, Yang H, Leach L, Zhang RM, Luo ZW 2007. Genetic dissection of ethanol tolerance in the budding yeast Saccharomyces cerevisiae. Genetics 175: 1479–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazwinski SM 2004. Yeast replicative life span: The mitochondrial connection. FEMS Yeast Res 5: 119–125 [DOI] [PubMed] [Google Scholar]
- Jenkins NL, McColl G, Lithgow GJ 2004. Fitness cost of extended lifespan in Caenorhabditis elegans. Proc Biol Sci 271: 2523–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Dobry CJ, McCown PJ, Kumar A 2008. Large-scale analysis of yeast filamentous growth by systematic gene disruption and overexpression. Mol Biol Cell 19: 284–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johzuka K, Horiuchi T 2009. The cis element and factors required for condensin recruitment to chromosomes. Mol Cell 34: 26–35 [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L 1999. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13: 2570–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Benguria A, Lai CY, Jazwinski SM 1999. Modulation of life-span by histone deacetylase genes in Saccharomyces cerevisiae. Mol Biol Cell 10: 3125–3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y-H, Ishikawa D, Ha HP, Sugiyama M, Kaneko Y, Harashima S 2006. Chromosome XII context is important for rDNA function in yeast. Nucleic Acids Res 34: 2914–2924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchman PA, Kim S, Lai CY, Jazwinski SM 1999. Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics 152: 179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T 2003. The replication fork barrier site forms a unique structure with Fob1p and inhibits the replication fork. Mol Cell Biol 23: 9178–9188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Hidaka M, Nishizawa M, Horiuchi T 1992. Identification of a site required for DNA replication fork blocking activity in the rRNA gene cluster in Saccharomyces cerevisiae. Mol Gen Genet 233: 355–362 [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Nomura M, Horiuchi T 2001. Identification of DNA cis elements essential for expansion of ribosomal DNA repeats in Saccharomyces cerevisiae. Mol Cell Biol 21: 136–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskar S, Bhattacharyya MK, Shankar R, Bhattacharyya S 2011. HSP90 controls SIR2 mediated gene silencing. PLoS ONE 6: e23406 doi: 10.1371/journal.pone.0023406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom DL, Leverich CK, Henderson KA, Gottschling DE 2011. Replicative age induces mitotic recombination in the ribosomal RNA gene cluster of Saccharomyces cerevisiae. PLoS Genet 7: e1002015 doi: 10.1371/journal.pgen.1002015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Butow RA 2006. Mitochondrial retrograde signaling. Annu Rev Genet 40: 159–185 [DOI] [PubMed] [Google Scholar]
- Machín F, Paschos K, Jarmuz A, Torres-Rosell J, Pade C, Aragón L 2004. Condensin regulates rDNA silencing by modulating nucleolar Sir2p. Curr Biol 14: 125–130 [PubMed] [Google Scholar]
- Marullo P, Aigle M, Bely M, Masneuf-Pomarède I, Durrens P, Dubourdieu D, Yvert G 2007. Single QTL mapping and nucleotide-level resolution of a physiologic trait in wine Saccharomyces cerevisiae strains. FEMS Yeast Res 7: 941–952 [DOI] [PubMed] [Google Scholar]
- Medvedik O, Sinclair DA 2007. Caloric restriction and life span determination of yeast cells. Methods Mol Biol 371: 97–109 [DOI] [PubMed] [Google Scholar]
- Miceli MV, Jiang JC, Tiwari A, Rodriguez-Quiñones JF, Jazwinski SM 2011. Loss of mitochondrial membrane potential triggers the retrograde response extending yeast replicative lifespan. Front Genet 2: 102 doi: 10.3389/fgene.2011.00102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer RK, Johnston JR 1986. Genealogy of principal strains of the yeast genetic stock center. Genetics 113: 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller LA, McCusker JH 2009. Microsatellite analysis of genetic diversity among clinical and nonclinical Saccharomyces cerevisiae isolates suggests heterozygote advantage in clinical environments. Mol Ecol 18: 2779–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petes TD, Hereford LM, Botstein D 1978. Simple Mendelian inheritance of the repeating yeast ribosomal DNA genes. Cold Spring Harb Symp Quant Biol 42: 1201–1207 [DOI] [PubMed] [Google Scholar]
- Ronald J, Tang H, Brem RB 2006. Genomewide evolutionary rates in laboratory and wild yeast. Genetics 174: 541–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve AA, Wolberger C, Schramm VL, Boeke JD 2006. The biochemistry of sirtuins. Annu Rev Biochem 75: 435–465 [DOI] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L 1997. Extrachromosomal rDNA circles: A cause of aging in yeast. Cell 91: 1033–1042 [DOI] [PubMed] [Google Scholar]
- Sinha H, Nicholson BP, Steinmetz LM, McCusker JH 2006. Complex genetic interactions in a quantitative trait locus. PLoS Genet 2: e13 doi: 10.1371/journal.pgen.0020013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha H, David L, Pascon RC, Clauder-Münster S, Krishnakumar S, Nguyen M, Shi G, Dean J, Davis RW, Oefner PJ, et al. 2008. Sequential elimination of major-effect contributors identifies additional quantitative trait loci conditioning high-temperature growth in yeast. Genetics 180: 1661–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinkraus KA, Kaeberlein M, Kennedy BK 2008. Replicative aging in yeast: The means to the end. Annu Rev Cell Dev Biol 24: 29–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz LM, Sinha H, Richards DR, Spiegelman JI, Oefner PJ, McCusker JH, Davis RW 2002. Dissecting the architecture of a quantitative trait locus in yeast. Nature 416: 326–330 [DOI] [PubMed] [Google Scholar]
- Tibayrenc M, Kjellberg F, Arnaud J, Oury B, Brenière SF, Dardé ML, Ayala FJ 1991. Are eukaryotic microorganisms clonal or sexual? A population genetics vantage. Proc Natl Acad Sci 88: 5129–5133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Hua CY, Hsu HE, Hsu CL, Tseng HY, Wright DE, Hsu PH, Jen CH, Lin CY, Wu MY, et al. 2011. The C-terminus of histone H2B is involved in chromatin compaction specifically at telomeres, independently of its monoubiquitylation at lysine 123. PLoS ONE 6: e22209 doi: 10.1371/journal.pone.0022209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, McCusker JH, Hyman RW, Jones T, Ning Y, Cao Z, Gu Z, Bruno D, Miranda M, Nguyen M, et al. 2007. Genome sequencing and comparative analysis of Saccharomyces cerevisiae strain YJM789. Proc Natl Acad Sci 104: 12825–12830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitao T, Budd M, Campbell JL 2003. Evidence that yeast SGS1, DNA2, SRS2, and FOB1 interact to maintain rDNA stability. Mutat Res 532: 157–172 [DOI] [PubMed] [Google Scholar]
- Winzeler EA, Richards DR, Conway AR, Goldstein AL, Kalman S, McCullough MJ, McCusker JH, Stevens DA, Wodicka L, Lockhart DJ, et al. 1998. Direct allelic variation scanning of the yeast genome. Science 281: 1194–1197 [DOI] [PubMed] [Google Scholar]
- Zeng Z-B 1994. Precision mapping of quantitative trait loci. Genetics 136: 1457–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]