

Abstract
We lack a deep understanding of genetic and metabolic attributes specializing in microbial consortia for initial and subsequent waves of colonization of our body habitats. Here we show that phylogenetically interspersed bacteria in Clostridium cluster XIVa, an abundant group of bacteria in the adult human gut also known as the Clostridium coccoides or Eubacterium rectale group, contains species that have evolved distribution patterns consistent with either early successional or stable gut communities. The species that specialize to the infant gut are more likely to associate with systemic infections and can reach high abundances in individuals with Inflammatory Bowel Disease (IBD), indicating that a subset of the microbiota that have adapted to pioneer/opportunistic lifestyles may do well in both early development and with disease. We identified genes likely selected during adaptation to pioneer/opportunistic lifestyles as those for which early succession association and not phylogenetic relationships explain genomic abundance. These genes reveal potential mechanisms by which opportunistic gut bacteria tolerate osmotic and oxidative stress and potentially important aspects of their metabolism. These genes may not only be biomarkers of properties associated with adaptation to early succession and disturbance, but also leads for developing therapies aimed at promoting reestablishment of stable gut communities following physiologic or pathologic disturbances.
Ecologists studying colonization of macro-ecosystems have built a conceptual framework that can be applied to microbial community assembly in our various human body habitats. Establishment of plants in diverse habitats involves systematic species turnover (“primary succession”) with subsequent development into a relatively stable configuration. Disturbing this stable configuration triggers secondary succession, where pioneer or opportunistic species can thrive (McCook 1994). Some biological attributes of pioneer species are habitat specific, such as low shade tolerance in plants, while others are universal. Biological properties that plant and microbial opportunists may share include rapid growth on limiting or labile (evanescent) resources (McCook 1994; Sigler and Zeyer 2004; Fierer et al. 2010; Vieira-Silva and Rocha 2010), high-dispersal capabilities facilitated by cosmopolitan distribution (Fierer et al. 2010), and high tolerance to environmental stresses (Callaway and Walker 1997; Sigler and Zeyer 2004; Fierer et al. 2010). We know little about the specific attributes that typify early microbial colonizers of our bodies. Here we examine this question in the context of the human gut.
The adult human distal gut contains most of the microbes in our bodies, and is dominated by bacteria in two phyla: Firmicutes and Bacteroidetes (Eckburg et al. 2005). The gut microbiota harvests energy from otherwise indigestible dietary components (Sonnenburg et al. 2005), shapes immune system development (Tsuda et al. 2010; Olszak et al. 2012), and protects against enteropathogen invasion (Bartlett 2002). Mechanisms explaining how this community assembles are poorly understood, although gut microbial communities increase in diversity and stability over time (Palmer et al. 2007; Dominguez-Bello et al. 2011), and early colonizers generally grow rapidly (Vieira-Silva and Rocha 2010) and have high tolerance to stress (Koenig et al. 2011), including oxygen (Stark and Lee 1982). Disturbances such as acute diarrhea or antibiotic treatment often alter community composition and initially decrease diversity (Young and Schmidt 2004; Mai et al. 2006)—a characteristic of secondary succession (Schoonmaker and Mckee 1988). Ecological questions about succession in the gut thus arise: Does a subset of our normal microbiota generally associate with early succession? If so, does this subset increase in abundance during gut disturbance?
Culture-independent methods, including 16S rRNA gene sequencing, have described bacterial community composition across human populations as a function of age, physiologic status, and disease (e.g., Hayashi et al. 2003; Ley et al. 2006; Frank et al. 2007; Turnbaugh et al. 2009a,b; Biagi et al. 2010; Joly et al. 2010; Koenig et al. 2011), and these data are complemented by hundreds of annotated gut bacterial genomes (Nelson et al. 2010). These data sets offer an opportunity to identify genomic determinants of successional order and to ascertain whether these determinants also identify opportunistic pathogens.
Species in Clostridium cluster XIVa (phylum Firmicutes) provide an attractive model for studying adaptation to stages of community succession and the evolution of opportunistic pathogens. Species in this cluster are among the most prevalent bacteria in the distal gut of healthy adult humans (Maukonen et al. 2006), but can also cause systemic infections (Elsayed and Zhang 2004; Finegold et al. 2005; Decousser et al. 2007; Huh et al. 2010). Furthermore, flagellins related to those in species in cluster XIVa have been implicated in the pathogenesis of irritable bowel syndrome (Schoepfer et al. 2008) and IBD (Duck et al. 2007). Here we show that species in Clostridium cluster XIVa adapt to different stages of community succession, and that suspected pathobiont species in this group (i.e., gut symbionts that can cause disease in permissive genetic or environmental circumstances) are often adapted to early succession. Our approach was to first use a co-occurrence network to reveal how species in Clostridium cluster XIVa codistribute across healthy and diseased individuals, and then test for niche differentiation of these microbes during colonization of an infant human gut as well as the guts of gnotobiotic mice that have received a human fecal microbiota transplant. Next, we used over/underrepresentation of genes in Clostridial genomes to relate distribution patterns to biological traits. Finally, we determined whether these genes are similarly represented in unrelated enteric opportunistic pathogens to test whether the adaptations are lineage specific or related to opportunism in general. This analytic approach may have general applicability.
Results
Defining a co-occurrence network in human gut microbiota
To determine which Clostridium cluster XIVa species share ecological properties and hence distribution patterns, we generated a co-occurrence network from relative abundances of 155 bacterial species with sequenced genomes (Supplemental Table S1) reported in analysis of the fecal microbiomes of 124 unrelated European adults by the MetaHIT consortium of investigators (Qin et al. 2010). This study included 42 obese individuals (BMI>30 kg/m2) and 25 with IBD. Relative abundances of these bacterial species in each individual were estimated by Qin and coworkers using 576.7 Gbp of shotgun sequence generated from DNAs prepared from single fecal samples, as the number of sequence reads uniquely aligned to each genome at >90% identity threshold (normalized across individuals to account for differences in sequencing depth). These 155 genomes were selected from a larger set of 932 publicly available genomes because they were nonredundant and prevalent in the gut samples (see Methods).
A co-occurrence network is built by computing a score for each species pair from their abundance profiles, then thresholding these scores to remove chance links. Several similarity scores exist, including correlation (Qin et al. 2010), hypergeometric distribution for species presence/absence data (Chaffron et al. 2010; Freilich et al. 2010), and mutual information (Date and Marcotte 2003). Different scores have different strengths and weaknesses. For example, Bray-Curtis is recommended for species abundances because unlike measures not designed to handle the particular problems of ecological data (e.g., Euclidean distances), it moderates the impact of highly abundant species and of “double absences” where neither member of a species pair was observed (Legendre and Legendre 1998). Spearman and Kendall correlations, robust to outliers and nonparametric, are recommended for non-normalized data. Because the 155 species differed in normality, prevalence, and other attributes across the fecal samples, we combined three correlation scores (Pearson, Spearman, Kendall), two scores of distance and dissimilarity (Euclidean and Bray Curtis), and one similarity score (mutual information, implemented in Meyer et al. 2008) to capture diverse ecological relationships. We also implemented a bootstrap-based multiple testing correction procedure (BS_FD: BootStrap with control of False Discoveries) (Lallich et al. 2006) that is stricter than a permutation test (see Methods). The multiple testing correction threshold allowed only 1% of co-occurrence network links to be present by chance. As expected, agreement among scores increased with stricter thresholds.
We detected only significantly positive and no significantly negative co-occurrence between taxa. Positive co-occurrence patterns were partly explained by phylogenetic relationships (Fig. 1; Supplemental Fig. S1): The fraction of species-pairs in the same co-occurrence module decreased as phylogenetic distance increased, up to ∼88% pairwise nucleotide sequence identity of the 16S rRNA gene (%ID) (Supplemental Fig. S2; see the Methods section for %ID calculation details). Taxon pairs at >98.5%ID (n = 45) were always in the same co-occurrence module (Supplemental Fig. S2). From 97% to 98.5% ID (n = 69), only 77% of pairs co-occurred, indicating that even within the %ID typically used to denote the same species, ecological differences can cause differing distribution patterns. Positive co-occurrence between closely related taxa suggests that habitat filtering, where species sharing traits persist in the same habitat, drives distribution patterns more than competitive exclusion, which would cause closely related taxa to negatively co-occur more or to positively co-occur less than more distantly related taxa (see the Supplemental Discussion).
Figure 1.

Co-occurrence network of human gut bacteria, based on a relative abundance matrix previously reported in Qin et al. (2010). The nodes represent species whose genomes have been sequenced. The size of the nodes indicates the average relative abundance across the 124 individuals in the MetaHIT cohort, and the color of the node reflects taxonomic information. Species with significantly positive co-occurrence for any of six measures used (Pearson, Spearman, Kendall, Bray-Curtis, Euclidean, and mutual information) are joined with an edge. Co-occurrence modules are defined as a set/collection of species that are connected among each other (directly or via several steps), but not to any other species in the network, and are labeled with Mx. Species' full names are shown in Supplemental Table S1, and phylogenetic relationships among them as determined with 16S rRNA are shown in Supplemental Fig. S1. For further discussion of this network, see the Supplemental Material.
Species from the same taxonomic group, e.g., Clostridiales (including Clostridium cluster XIVa), Bacteroidales, or Actinobacteriales, have %ID values, suggesting that they would co-occur (Supplemental Fig. S2). However, phylogeny predicted concentration in co-occurrence modules poorly, instead suggesting niche diversification and convergence (Supplemental Fig. S1). For example, the 19 Clostridium cluster XIVa species have >92%ID and occur in four different modules (M1–M4 in Fig. 1): This is fewer modules than expected (P < 0.001 by Monte Carlo simulations; in 1000 random assignments of species to modules, the 19 Clostridium cluster XIVa species were always found in more than four). However, within these four modules, phylogenetic relationships predict membership poorly (Fig. 2).
Figure 2.

Phylogenetic relationships between species in Clostridium cluster XIVa. Bootstrap support (based on 1000 replicates) is indicated on the branches of the 16S rRNA NJ tree when >40% (except for Ruminococcus sp. SR1/5, as this was added by parsimony insertion after the initial tree creation, since only a short region of 16S rRNA sequence was available; see Methods). The species names are colored according to their module in the co-occurrence network: M1 (blue), M2 (turquoise), M3 (red), and M4 (green). Species that were evaluated in the network analysis but showed no significant co-occurrence are in black text. Species that were not evaluated for co-occurrence are colored purple. These were added to further support that the species in this group that can cause disease form a clade with expanded genome size. The branches are colored by genome size rank, with the red branches representing the largest genomes and the blue branches the smallest. The genome size in Mb is listed after the species name. Species that have been recovered from clinical samples (e.g., bacteremia) are marked with a red circle, and those reported to be in increased abundance with IBD are marked with a blue circle. Species that contain the genomic machinery for a flagellum are marked with a blue curved line. Note that the network diagram only shows species with significant co-occurrence with at least one other species; thus 36.7% (11/30) of evaluated species in Clostridium cluster XIVa, are not shown. Details about the evaluated species are provided in Table 1.
Table 1.
Summary of the Clostridium cluster XIVa species that were evaluated for co-occurrence
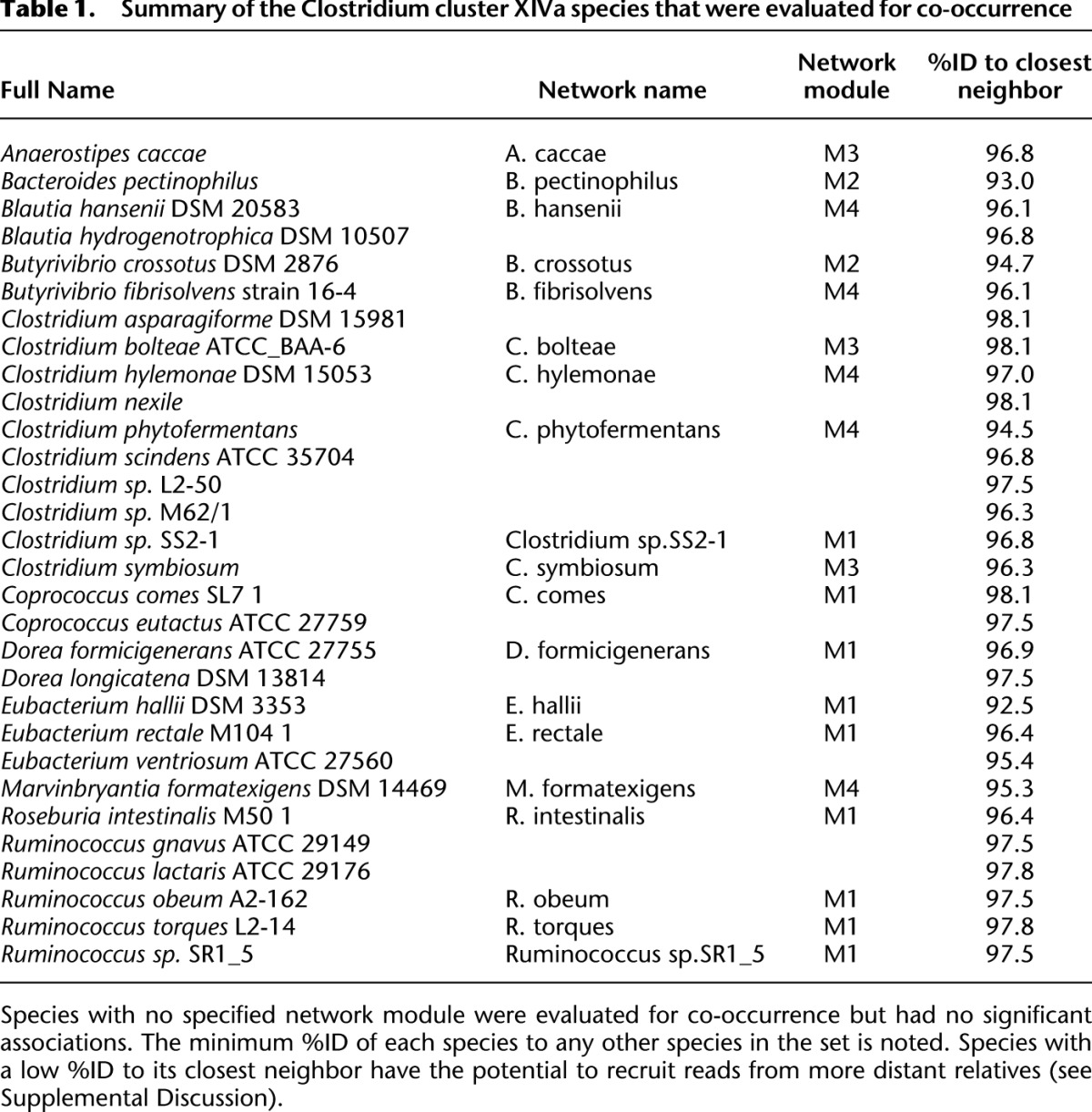
If Clostridium cluster XIVa species in the same co-occurrence module were also more closely related phylogenetically, vertically inherited biological traits would likely drive the distribution. Because species in the same co-occurrence module (M1–M4) do not group phylogenetically (Fig. 2), convergence in biological attributes may drive distribution. This is consistent with reports of high horizontal gene transfer (HGT) among gut microbes (Zaneveld et al. 2010; Smillie et al. 2011).
Clostridium cluster XIVa species associated with systemic infections co-occur in M3, including Clostridium bolteae (Finegold et al. 2005) and C. symbiosum (Elsayed and Zhang 2004; Decousser et al. 2007; Huh et al. 2010). These species group within a 16S rRNA phylogeny that contains C. clostridioforme and C. hathewayi—species also associated with opportunistic infections including bacteremia, intra-abdominal abscess, and wound infection (Fig. 2; Finegold et al. 2005). This observation suggests that this clade evolved a predisposition to virulence. Genome size in this clade is approximately double that in other gut-associated cluster XIVa members (Fig. 2).
The biological properties of the five Clostridium cluster XIVa species in M4 suggest that syntrophy, not shared ecological attributes, drive some of these interactions. For example, Marvinbryantia formatexigens, which grows faster in vitro with formate using the acetate-producing Wood-Ljungdahl pathway (Wolin et al. 2003), co-occurs positively with Butyrivibrio fibrisolvens 16/4 (Diez-Gonzalez et al. 1999): The latter species is related to a strain that grows >50% faster and increases formate production when acetate is present, suggesting syntrophic formate-acetate exchange (see the Supplemental Discussion).
Distribution of Clostridium cluster XIVa species in health and disease
M1 species were more abundant than M3 and M4 species, representing nine of the 17 most abundant species in the 155 human gut-associated genomes considered in our analysis of the MetaHIT fecal microbiome data set. To verify that patterns in this 124-subject cohort are relevant to other populations, and to better understand host properties influencing distribution, including age or health status, we examined Clostridium cluster XIVa distribution patterns in five additional 16S rRNA gene surveys of the human gut (Supplemental Table S2). In these analyses, species-per-sample relative abundances were estimated using sequencing reads with >98%ID to the species' 16S rRNA gene (see Methods). This 98%ID threshold approximates the degree of species specificity in Clostridium cluster XIVa likely attained in the relative abundance table from the MetaHIT study from which we generated the co-occurrence network (Qin et al. 2010; see the Supplemental Discussion).
The numerical dominance of M1 over M3 members observed in the MetaHIT data set was even stronger in two independent 16S rRNA surveys of healthy human adult gut microbiota conducted by two other groups (Fig. 3A; Supplemental Table S2; Eckburg et al. 2005; Turnbaugh et al. 2009a). Thus, only a subset of species in the numerically dominant Clostridium cluster XIVa group appear to be abundant in the healthy adult gut; these are phylogenetically interspersed with absent or low-abundance species.
Figure 3.

Relative abundance of species in M1 and M3 in various gut samples. Sample categories are detailed in Supplemental Table S2. (A) Average relative abundance across samples from individuals with and without gastrointestinal disease. (Blue circles) M1 species; (red squares) M3 species. The SEM for each treatment is plotted. For healthy, estimated using data from (1) stool samples from obese and lean twins and their mothers (Turnbaugh et al. 2009a) (Turnbaugh Feces), (2) samples from three healthy adults from six mucosal sites along the length of the colon (Eckburg et al. 2005) (Eckburg Mucosal), (3) fecal samples from the same three individuals as in Eckburg Mucosal (Eckburg Feces). For diseased, averaged relative abundance across (1) colon (Frank Colon), (2) small intestine (Frank SI), and (3) MLNs (Frank MLN) from individuals with gastrointestinal disease including Crohn's disease, ulcerative colitis, and colon cancer from Frank et al. (2007) (B) Results from humanized gnotobiotic mice (Turnbaugh et al. 2009b). Fecal samples from the healthy human donor (donor, [1]) and from the recipient gnotobiotic mice ([2] stomach, [3] small intestine [SI], [4] cecum, [5] colon, and [6] feces). The points representing R. torques, which was an outlier in this analysis, are marked with a green circle. Error bars represent the median and interquartile range. (C) Age trends in M1 and M3 species in a single infant using data from Koenig et al. ( 2011). The x-axis in each plot is the age in days and the y-axis is the relative abundance in a single sample. The species in M1 have series colored in blue, M3 red, and those with no detected co-occurring microbes are in black. The relative abundance of each OTU in the mother is plotted at day 950 in light green. The period before the introduction of solid food is shaded in blue and between then and the switch from breast milk to formula is shaded in yellow. Ruminococcus sp. SR 1/5 was not evaluated because sequence information for the V2 region of its 16S rRNA is incomplete. Coprococcus comes is not shown because it was absent across the infant timeseries and in the mother.
In contrast, M3 species were rare in healthy but relatively abundant in diseased guts. In a 16S rRNA data set generated from colons, small intestines, and mesenteric lymph nodes (MLNs) of 27 patients with Crohn's disease, 45 with ulcerative colitis, and 41 with other gastrointestinal disorders, primarily colorectal cancer (Frank et al. 2007), C. bolteae abundance was about average for M1 species in this cohort (Fig. 3A). C. bolteae was detected in mucosa harvested from colons or ceca of four of 27 (14.8%) Crohn's disease patients, seven of 45 (15.6%) ulcerative colitis patients, and four of 41 (9.8%) non-IBD samples, even though these samples were sequenced at only ∼80 bacterial 16S rRNA reads per sample. All three M3 species (A. caccae, C. symbiosum, and C. bolteae) had comparable abundance to M1 species in the small intestines of the same subjects. M1 species were never detected in MLNs, but all three M3 species were detected in at least one of the five MLN samples (Fig. 3A). Detection of C. symbiosum and C. bolteae in MLNs agrees with previous reports that these species can invade the gut mucosa and cause systemic infection (Elsayed and Zhang 2004; Finegold et al. 2005; Decousser et al. 2007). Matched small intestine samples indicated that these bacteria are sometimes abundant (up to 16.5%) at intestinal disease sites, where translocation to MLNs presumably occurred (Supplemental Table S3).
Clostridium cluster XIVa distribution patterns in gut colonization
Disturbance-associated (secondary) succession is associated with a compromised stable/complex gut consortium, perhaps indicating a parallel with primary succession, e.g., during development of the infant gut microbiota. We compared M1 and M3 prevalence during colonization events to test the hypothesis that M3 species adapt to early succession, while M1 species thrive only in late successional, stable gut communities. To test this hypothesis, we used two data sets generated by pyrosequencing V2 amplicons from bacterial 16S rRNA genes. The first data set was produced from mucosal samples collected along the length of the guts of adult gnotobiotic C57Bl/6J mice 3 mo after introduction of adult human fecal microbiota (Turnbaugh et al. 2009b); the second was from fecal samples collected during the first 2.3 yr of life from a single human infant (Koenig et al. 2011; Supplemental Table S2). This infant was breast fed exclusively until day 134, when solid food was introduced. On day 161, the child was first given infant formula. The mother's stool was also analyzed.
Both data sets supported our hypothesis about adaptation to different successional stages. When germ-free mice were colonized by gavage of freshly voided human fecal sample, 88% of genus-level bacterial taxa in the donor microbiota survived transfer (Turnbaugh et al. 2009b). M3 species were significantly more likely than M1 to colonize the mice when introduced as part of a complex community, and to appear in fecal samples collected from the “humanized” gnotobiotic animals (P = 0.0032, G-test for independence). Seven of nine M1 species were detected in the human donor feces, with variable relative abundances (0.086 ± 0.079% of the total population for R. intestinalis to 2.6% ± 0.76 for E. hallii). Only one M1 species, Ruminococcus torques, colonized the guts of gnotobiotic mice at the threshold level of detection (Fig. 3B): Its proportional representation was significantly greater in gnotobiotic mice (17.7% of V2-16S rRNA sequences generated from small intestine; ∼9% from cecum, colon, and feces) compared with the fecal microbiota of the human donor (P = 0.00042, t-test) (Fig. 3B). In contrast, all three M3 species colonized these same mice. C. bolteae and C. symbiosum significantly increased in relative abundance in the feces of humanized mice relative to human donor feces (P < 0.0034; t-test); both species showed no strong biogeographical trends and comprised 0.5%–1.4% of 16S rRNA sequences in communities distributed along the celphalocaudal axis of the gut. A. caccae was detected in low abundance in mouse feces (<0.06%), but at a greater average abundance than in the human donor (0.007%), although this difference was not statistically significant (Fig. 3B).
Consistent with specialization to later successional stages, in the human infant time-series several M1 species were detected in the mother but colonized the infant late (E. rectale, E. hallii, and D. formicigenerans) or never (R. obeum) (Fig. 3C). In contrast, M3 species were rare or undetected in the mother, but colonized the infant. A. caccae colonized just before the introduction of solid foods, and was then quickly lost. Detection of C. bolteae and C. symbiosum coincided with introduction of formula at ∼5-½ mo, and both species persisted through the first 2 yr of postnatal life (Fig. 3C). M1 species observed in this infant peaked in relative abundance at a later age (666.5 ± 199.3 d) than M3 (187 ± 57.9 d; P = 0.011; t-test).
Ruminococcus gnavus falls outside M3, but showed a strong age trend coinciding with introduction of solid food, peaking at 35.6% of the population, then rapidly declining (Fig. 3C). R. gnavus was among the few Clostridium cluster XIVa species found in MLN of IBD patients (Supplemental Table S3) that colonized humanized gnotobiotic mice, supporting its identification as opportunistic. Interestingly, R. gnavus and R. torques are more abundant in IBD patients' mucosa (Png et al. 2010), and R. gnavus increases specifically in patients with ileal Crohn's disease (Willing et al. 2010). R. torques is in M1, but nevertheless became dominant when introduced into gnotobiotic mice. Success in primary succession and increased abundance with IBD suggests that the same species may thrive in healthy infants and disturbed guts. R. gnavus and R. torques both metabolize human mucins (Png et al. 2010), suggesting that adaptive foraging of host mucosal glycans contributes to their success.
Evaluating opportunistic communities in a larger context
Adaptation to different stages of community succession likely requires evolutionary tradeoffs. We previously suggested that selection between fast-growing “weedy” pioneers and complex interrelated bacterial consortia explains global bacterial diversity patterns (Ley et al. 2008). This observation was based on principal coordinates analysis of 464 published samples from diverse free-living (e.g., freshwater, saltwater, soils) and host-associated (e.g., mammals and insects) bacterial assemblages. Although the first principal coordinate axis (PC1) highlighted differences between the vertebrate gut and other communities, the second axis (PC2) separated environments where opportunistic/pioneer organisms might thrive (e.g., samples cultured before DNA sequencing, or from marine ice) versus conditions in which more complex communities could develop (e.g., culture-independent soil surveys) (Supplemental Fig. S3). Previous studies suggested that high culturability on nonselective media indicates opportunism and early succession in soil bacteria (Garland et al. 2001; Sigler and Zeyer 2004).
Human gut samples also spread across this second axis: infant samples from two studies and one sample from an individual with antibiotic-induced diarrhea grouped with opportunistic consortia (e.g., samples from cultured isolates) along PC2, whereas most human gut samples resembled other complex consortia (e.g., mature soils and rumen samples) (Supplemental Fig. S3). Elderly individuals (ages 75–94) (Hayashi et al. 2003) also had an opportunistic “signature,” matching reports that centenarians have more gut pathobionts than younger adults (ages 20–40) (Biagi et al. 2010). To determine whether succession and antibiotic-induced community changes within an individual shared these patterns, we added samples from the infant time-series (Koenig et al. 2011): the mother's sample plus the infant at postnatal day 3 (meconium), day 100 (before establishing adult-like microbiota as determined in Koenig et al. 2011), day 206 (after establishing adult-like microbiota), and day 432 (during treatment with the broad-spectrum antibiotic Cefnidir) (see Methods). Early samples (days 3 and 100), and samples from antibiotic-induced disturbance (day 432), had opportunistic signatures; the maternal sample's signature was least opportunistic; and the child's day 206 sample was intermediate (Supplemental Fig. S3).
Comparative genomic analysis of M1 versus M3
A general challenge in genomics/metagenomics is determining which genes are overrepresented in a group because they encode a biologically relevant trait, and which simply occur in the same genome as the genes that encode traits that drive distribution. Using phylogenetic relationships to determine genomic characteristics better explained by environmental distribution than phylogeny provides a powerful correction for genomic background (Lozupone et al. 2008). Because A. caccae and C. bolteae/C. symbiosum are phylogenetically interspersed with M1 species, we can identify genomic adaptations differentiating M1 from M3. Gene families selected for in M3 genomes must be conserved in C. bolteae and C. symbiosum, and independently selected in A. caccae. We assigned each genome's genes to orthology groups defined in the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto 2000) by comparing them to a database of all KEGG genes with KO assignments using BLAST (we assigned each gene to its best hit if below a 10−10 E-value threshold). To determine which gene families differed in the presence/absence or abundance between M1 and M3, we used t-tests, correcting for multiple comparisons by false discovery rate (FDR) (Benjamini and Hochberg 1995). We only evaluated genes present in more than two of 12 genomes (1672 gene families). The results revealed 41 gene families that were significantly more abundant in M3 species' genomes than M1 (Supplemental Table S4). None were significantly less abundant in M3, suggesting that ecological changes in M3 members were due primarily to new gene acquisition, or retention of genes lost in M1.
Because genomes were available for multiple strains of several species, we evaluated the conservation of gene counts across all available strains (Supplemental Fig. S5). This control was needed because reads from all strains could contribute to relative abundance estimates used for the Clostridium cluster XIVa co-occurrence network (see the Supplemental Discussion). For each of 41 significant genes, we did the t-test again using the average of counts across available strains. Because some species in M3 and M1 are in bootstrap-supported clades excluding species in the other module so gene gain/loss may not have been independent, we also performed t-tests using a single average count for these species (Supplemental Table S4). These controls had little effect on the overall patterns (Supplemental Table S4).
Many overrepresented genes in M3 are important for virulence in diverse gut pathogens (Supplemental Table S4). Selection for these genes in M3 likely promotes adaptation to a disturbed/inflamed gut, and need not indicate virulence in these particular species. Consistent with detection of M3 species in MLNs, overrepresented genes include genes important for survival in macrophages, including genes involved in resisting pore-forming antimicrobial peptides (pilB; K02652), resisting attack with peroxide (peroxyredoxin; ahpC; K03386), and acquisition of Mg2+ (Mg2+ transporter-C mgtC family; K07507; macrophages represent low Mg2+ environments, and mgtC is required by Salmonella for macrophage survival [Supplemental Table S4; Blanc-Potard and Groisman 1997]). Consistent with reports that osmotic stress responses are important for some enteric pathogens (Sleator and Hill 2002), M3 genomes overrepresent an inducible P-type ATPase K+ importer with high K+ affinity (Epstein 1992), whose primary function in bacteria may be protection from environmental stress (Chan et al. 2010; Supplemental Table S4).
M3 genomes overrepresent other genes known to confer oxidative stress resistance, including class I ribonucleotide reductase (RNR) genes and proteins that repair oxidative DNA damage (Supplemental Table S4). Some aerotolerance may be essential for establishing blood and tissue infections (Jean et al. 2004) and A. caccae has far more aerotolerance than two species in M1 (Flint et al. 2007). Increased aerotolerance may be key in the infant gut: Culture-based studies show the anaerobe/aerobe ratio increases in formula-fed infant feces during the first year of life (Stark and Lee 1982).
The observation that genes selected for in M3 versus M1 may promote virulence led us to more systematically examine their representation in the genomes of enteric pathogens. We determined counts of homologs to the 41 discriminatory genes in a phylogenetically diverse collection of 15 enteric pathogens, including unrelated Clostridia (C. difficile, C. perfringens, and C. tetani), Enterobacteria (pathogenic Escherichia coli and Salmonella enterica), Streptococcus sanguinis, Campylobacter jejuni, Helicobacter hepaticus, Lawsonia intracellularis, and Bacteroides fragilis. Although a few of these genes are overrepresented only in M3 (Supplemental Fig. S4), the majority are in greater abundance in some of the enteric pathogens compared with the M1 species. Genes in M3 and in almost all opportunistic pathogens surveyed included those encoding class I RNR (K00525-6) and peroxiredoxin (K05386), indicating that oxidative stress tolerance may be generally important for opportunism in diverse pathogens and that some molecular mechanisms conferring this biological property are general. Genes overrepresented in a subset of pathogens include S. enterica mgtC (K07507 in the KEGG database), involved in Mg2+ import and required for survival in macrophages (Blanc-Potard and Groisman 1997) (present in Clostridial and Enterobacterial pathogens) and the P-type ATPase K+ importer (K01546-8; present in Clostridial and Enterobacterial pathogens and B. fragilis) (Supplemental Fig. S4). Because flagella are also important for virulence and an important antigen, we explored their distribution across Clostridium cluster XIVa genomes. Their distribution is noted in Figure 2 and discussed in the Supplemental Material.
Metabolic adaptations in M3 could impact succession and virulence
We used Cytoscape (Cline et al. 2007) to visualize metabolic reactions overrepresented in M3 versus M1 (Fig. 4). KEGG families in each genome were converted to a reaction list by parsing the KEGG KO file (downloaded from KEGG version 49.0). The KEGG reaction_mapformula.lst file was used to build a network where nodes are compounds and reactions edges, using all reactions present in any M1 or M3 genome, and colored by overrepresentation in M3.
Figure 4.

Metabolic network. Generated based on the combined metabolic network of M1 and M3 genomes (see Methods). The nodes (circles) are compounds and the edges are reactions. Edges colored blue have more copies or are more likely present in M3 genomes compared with M1. Red edges are reactions known to occur, but no described enzymes that perform them are in any of these genomes. The node color indicates the fraction of M3 genomes in which that compound is found with white being 100% and black being 0%. The thick green edges indicate that branches of the network emanating from that node were eliminated from the figure.
The overrepresented genes suggest that M3 genomes were selected for rapid turnover and biosynthesis of RNA, perhaps due to selection for faster growth rates—a phenotype previously suggested to be important in early successional species (McCook 1994), opportunistic pathogens (Vieira-Silva and Rocha 2010), and bacteria in the infant gut (Vieira-Silva and Rocha 2010). M3 genomes have more copies of genes involved in degrading mRNA (ATP-dependent RNA helicase; K05592), converting RNA monomers to DNA (Class I and Class III RNR genes; K00525-7), and acquiring raw materials for RNA synthesis, (guanine/hypoxanthine transporter for purine acquisition; K06901) (Supplemental Table S4). Metabolic network visualization suggests selection for generating ribose-5-phosphate, including conversion from ribose via a ribokinase and de novo synthesis from xylulose by conversion of D-xylulose to D-xylulose-5-phosphate via xylulokinase (K00854), followed by conversion to D-ribulose-5-phosphate via ribulose-phosphate 3-epimerase (K01783) (Fig. 4; Supplemental Table S4). This is of potential significance in the gut because xylose, a metabolic precursor of xylulose, is abundant in plants but not digested by human genome-encoded carbohydrate-active enzymes (Jackson and Nicolson 2002).
M3 genomes also selected genes in two parallel pathways for serine biosynthesis as defined in E. coli: a primary pathway, known as the phosphorylated pathway (K00058), and a secondary nonphosphorylated pathway (K00018) (Blatt et al. 1966). M3 species all have the enzymes necessary to make the precursor for these reactions (D-glycerate) from fructose/sucrose (Fig. 4). The M3 genomes also have an extra copy of the β subunit of tryptophan synthase (K01696), which can convert serine to tryptophan if indole is present (Fig. 4; Supplemental Fig. S6; Xie et al. 2002). Increased tryptophan synthesis may benefit M3 species, because tryptophan limitation is a human immune defense mechanism (Schaible and Kaufmann 2005). This result agrees with the selection of other genes important for M3 survival within macrophages, including the Mg2+ transporter-C.
M3 species genomes also overrepresent propionate CoA–transferase (K01026; EC2.8.3.1) (Supplemental Table S4), a key enzyme in lactate fermentation to propionate (Seeliger et al. 2002). A. caccae ferments lactate, but produces butyrate rather than propionate using butyrylCoA:acetate CoA transferase (Duncan et al. 2002). The genomic context of propionate CoA-transferase suggests that it participates in lactate fermentation to butyrate in A. caccae and C. symbiosium. Two of the three copies in A. caccae, and the single copy in C. symbiosum, may be part of an operon that contains 3-hydroxy-butyryl CoA dehydratase (K01715;EC:4.2.1.55), and in two cases butyryl-CoA dehydrogenase [EC:1.3.99.2]; both participate in a butyrate-producing pathway (Supplemental Fig. S7). Two species in M1, E. hallii and R. intestinalis, also ferment lactate to butyrate. Although these species both have 3-hydroxybutyryl CoA dehydratase in their genomes, they lack propionate CoA–transferase, indicating that initial lactate fermentation stages may differ between these M1 and M3 lactate fermenters. In all three cases, this operon contains an H+/gluconate symporter family protein (COG 2610) that can facilitate gluconic acid uptake. Approximately 70% of dietary gluconic acid reaches the large intestine, and gluconic acid intake stimulates butyrate production by colonic bacteria, although cross-feeding between lactic acid bacteria that produce lactate and bacteria that further metabolize lactate to butyrate has been implicated (Tsukahara et al. 2002; Kameue et al. 2004). Further experimental work will help verify whether this operon's expression correlates with lactate/gluconic acid metabolism in these strains.
Discussion
That species in Clostridium cluster XIVa differ dramatically in distribution patterns is consistent with known variation in their biological properties. Understanding the phylogenetic depth at which distribution patterns are conserved in different phylogenetic groups is crucial for functionally interpreting phylogenetic marker surveys. The different ecological strategies of species within Clostridium cluster XIVa, and other important groups in the gut (Supplemental Fig. S1), indicate that binning sequences by family, class, or phylum may miss significant associations with environmental parameters such as diet or disease. Informatics tools we introduced through QIIME (Caporaso et al. 2010), including scripts for evaluating differences in OTUs defined at any %ID level using both classical statistical techniques (e.g., ANOVA and Pearson correlation) and machine learning, will facilitate analyses at finer taxonomic scales. Ultimately, high-throughput whole-genome sequencing, especially from personalized culture collections of carefully phenotyped individuals (Goodman et al. 2011), are required for maximum resolution.
Understanding parallels between primary and secondary succession in the gut, or other body-habitat-associated ecosystems, has important implications for developing strategies that encourage establishment of health-promoting, stable communities. For example, breastfeeding alters the infant gut microbiota and protects against pathogens in early life (Dominguez-Bello et al. 2011; Lamberti et al. 2011). Therefore, if infant gut succession shares features with succession following disturbance, microbes or conditions that breast-feeding promotes may also foster healthier gut succession after disturbance. Adaptation of gut species to particular successional stages may also help select new probiotics: Species adapted to complex stable communities may not be competitive when introduced into a disturbed gut.
Finally, using comparative genomics to explain distribution patterns, deployed here for one specific group, may help understand ecological differentiation more broadly across the tree of life. We have shown previously that when phylogeny does not explain distribution or biological properties, we can isolate important traits against genomic backgrounds (Lozupone et al. 2001, 2008). Sequencing additional reference genomes will extend such analyses, providing insights and testable hypotheses about biological factors that drive distribution patterns in various human gut microbiota and other systems. Increasing numbers of sequences per sample, now routine with next-generation sequencing, will make these finer-scale analyses increasingly practical.
Methods
Co-occurrence
The abundance table of Qin et al. (2010) that we used for co-occurrence analysis (kindly provided by Dr. D. Ehrlich [INRA, Jouy en Josas, France]) included 155 bacterial species that had draft/complete genome sequences, were nonredundant (genomes from 932 publicly available genomes were grouped at 90% identity over 80% of their length leaving 650 groups; the largest genome represented each group), and were found with genome coverage ≥1% in at least one individual (coverage defined as the proportion of the genome matched by reads aligning to only one position among the genomes at ≥90% identity).
Bray-Curtis distance was calculated as:
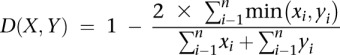 |
where x and y are two abundance profiles.
BS_FD for bootstrap with control of false discoveries
Many pairwise association tests for co-occurrence were performed, requiring multiple comparison correction [our 155 species yield (155 × 154)/2 = 11,935 tests]. We controlled for false discoveries with the BS_FD technique (Lallich et al. 2006). BS_FD first computes the distribution of differences between scores from original and bootstrapped data. The largest score difference is kept at each iteration. The adjusted threshold is the sum of the original threshold and the (1-δ)th quantile of the score difference distribution (for distances, this routine is slightly modified). Thus, BS_FD takes three parameters: the number of iterations (here, 10,000), the number of accepted false discoveries (here, 1), and the risk level (here, 0.05). We discarded species links with scores below the adjusted threshold. We applied this multiple test correction to each measure. Initial (lax) thresholds were adjusted by metric, e.g., initial thresholds for Pearson, Spearman, and Kendall were set to ±0.5, but adjusted thresholds were 0.61, 0.69, and 0.93 respectively (Supplemental Table S6).
Matching 16S rRNA sequences to co-occurrence network species
We obtained 16S rRNA sequences for most strains from a draft genome sequence of the same species using NAST (DeSantis et al. 2006). NAST identifies and aligns 16S rRNA homologs within larger sequences (here, genomic contigs). Some draft genomes lacked complete 16S rRNA sequences, often screened as repeats by assemblers. Therefore, we obtained some 16S rRNA sequences from GenBank records for targeted sequencing of the same species. Ruminococcus sp. SR1-5 was removed from the V2 analyses because no full-length 16S rRNA sequence was available, and the draft genome assembly for this gene only partially overlapped the V2 region. We calculated %ID between species from the NAST-alignments using ARB (Ludwig et al. 2004). Because NAST aligns hypervariable regions poorly, these were omitted from %ID calculations using lanemaskPH from Phil Hugenholtz's ARB database at RDP-II (Maidak et al. 2001).
16S rRNA phylogenies
16S rRNA gene sequences were aligned using NAST (DeSantis et al. 2006), then manually curated in ARB. The region covering positions 428–6723 in the NAST alignment was present for all species except Ruminococcus sp. SR1-5, which we excluded from the initial tree to use more characters. The overlapping region was filtered with lanemaskPH, excluding hypervariable regions. From this alignment, we built a neighbor-joining (NJ) tree using the F84 nucleotide substitution model in PHYLIP 3.65. We estimated bootstrap support for nodes with PHYLIP's seqboot and consense programs (1000 replicates). We imported this NJ tree into ARB, and added Ruminococcus sp. SR1-5 using parsimony insertion. Branches are colored in Figure 2 by genome size using TopiaryExplorer (Pirrung et al. 2011); genome sizes were from GOLD (Genomes Online Database: http://www.genomesonline.org).
We also made a 16S rRNA phylogeny for all species in the co-occurrence network (Supplemental Fig. S1). We aligned sequences with NAST, and built the tree by parsimony insertion into the reference tree from the RDP-II Arb database noted above. Hypervariable regions were again excluded with the lanemaskPH filter.
16S rRNA survey analysis
Supplemental Table S2 details the five 16S rRNA gut microbial community surveys we reevaluated: These used Sanger sequencing or multiplex pyrosequencing of the V2 region. We obtained sequences and metadata from GenBank (AY916135–AY916390, AY974810–AY986384 [Eckburg et al. 2005]; EF695452–EF710623 [Frank et al. 2007]) or the investigators. Community analyses of all samples were previously published, except for MLN samples collected by Frank et al. (2007), but not previously described. We reevaluated a subset of sequences from a larger set of humanized gnotobiotic mouse experiments; these evaluated colonization along the gut (Supplemental Table S2).
We quality-filtered pyrosequencing data using QIIME (Caporaso et al. 2010), excluding sequences <200 or >100 nt long, with average quality score <25, with homopolymers >6 nt long, or with primer mismatches. Sanger sequences were quality filtered by the investigators before GenBank deposition. We built a BLAST database from sequences from each study and matched Clostridium cluster XIVa 16S rRNA sequences to each database using BLASTn, determining the fraction of each sample's reads matching the near-full-length sequence at >98%ID.
Integrating infant timeseries data into the global PCoA plot
In our earlier PCoA of 464 published samples from diverse free-living and host-associated bacterial assemblages (Ley et al. 2008), we observed that human infant gut samples clustered along the second PC axis with communities likely containing “weedy” organisms, such as those that can grow in culture. To test whether successional and antibiotic-induced community changes within the gut of a human infant followed this pattern, we added five samples from the infant time-series experiment (Koenig et al. 2011). We chose OTUs at 97%ID using UCLUST, then chose the most abundant sequence from each OTU as representative using QIIME (Caporaso et al. 2010). We aligned representative sequences using NAST (DeSantis et al. 2006), then added them to a tree containing sequences from the other 464 samples, plus reference tree sequences from the ARB database of Phil Hugenholz using parsimony insertion. We excluded hypervariable regions using the lanemaskPH mask. We performed PCoA of an unweighted UniFrac distance matrix using this tree via Fast UniFrac (Lozupone and Knight 2005; Hamady et al. 2010).
Acknowledgments
This work was supported in part by the National Institutes of Health (K01DK090285, DK30292, DK70977, DK78669, HG4872, HG4866), the Crohn's and Colitis Foundation of America, and the Howard Hughes Medical Institute. K.F. and J.R. are supported by the Research Foundation, Flanders (FWO), the Flemish agency for Innovation by Science and Technology (IWT), and the Brussels Institute for Research and Innovation.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.138198.112.
Freely available online through the Genome Research Open Access option.
References
- Bartlett JG 2002. Clinical practice. Antibiotic-associated diarrhea. N Engl J Med 346: 334–339 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300 [Google Scholar]
- Biagi E, Nylund L, Candela M, Ostan R, Bucci L, Pini E, Nikkila J, Monti D, Satokari R, Franceschi C, et al. 2010. Through ageing, and beyond: Gut microbiota and inflammatory status in seniors and centenarians. PLoS ONE 5: e10667 doi: 10.1371/journal.pone.0010667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc-Potard AB, Groisman EA 1997. The Salmonella selC locus contains a pathogenicity island mediating intramacrophage survival. EMBO J 16: 5376–5385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatt L, Dorer FE, Sallach HJ 1966. Occurrence of hydroxypyruvate-L-glutamate transaminase in Escherichia coli and its separation from hydroxypyruvate-phosphate-L-glutamate transaminase. J Bacteriol 92: 668–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway RM, Walker LR 1997. Competition and facilitation: A synthetic approach to interactions in plant communities. Ecology 78: 1958–1965 [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffron S, Rehrauer H, Pernthaler J, von Mering C 2010. A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res 20: 947–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan H, Babayan V, Blyumin E, Gandhi C, Hak K, Harake D, Kumar K, Lee P, Li TT, Liu HY, et al. 2010. The P-type ATPase superfamily. J Mol Microbiol Biotechnol 19: 5–104 [DOI] [PubMed] [Google Scholar]
- Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. 2007. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2: 2366–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date SV, Marcotte EM 2003. Discovery of uncharacterized cellular systems by genome-wide analysis of functional linkages. Nat Biotechnol 21: 1055–1062 [DOI] [PubMed] [Google Scholar]
- Decousser JW, Bartizel C, Zamni M, Fadel N, Doucet-Populaire F 2007. Clostridium symbiosum as a cause of bloodstream infection in an immunocompetent patient. Anaerobe 13: 166–169 [DOI] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez-Gonzalez F, Bond DR, Jennings E, Russell JB 1999. Alternative schemes of butyrate production in Butyrivibrio fibrisolvens and their relationship to acetate utilization, lactate production, and phylogeny. Arch Microbiol 171: 324–330 [DOI] [PubMed] [Google Scholar]
- Dominguez-Bello MG, Blaser MJ, Ley RE, Knight R 2011. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology 140: 1713–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duck LW, Walter MR, Novak J, Kelly D, Tomasi M, Cong Y, Elson CO 2007. Isolation of flagellated bacteria implicated in Crohn's disease. Inflamm Bowel Dis 13: 1191–1201 [DOI] [PubMed] [Google Scholar]
- Duncan SH, Barcenilla A, Stewart CS, Pryde SE, Flint HJ 2002. Acetate utilization and butyryl coenzyme A (CoA):acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl Environ Microbiol 68: 5186–5190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA 2005. Diversity of the human intestinal microbial flora. Science 308: 1635–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsayed S, Zhang K 2004. Bacteremia caused by Clostridium symbiosum. J Clin Microbiol 42: 4390–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein W 1992. Kdp, a bacterial P-type ATPase whose expression and activity are regulated by turgor pressure. Acta Physiol Scand Suppl 607: 193–199 [PubMed] [Google Scholar]
- Fierer N, Nemergut D, Knight R, Craine JM 2010. Changes through time: Integrating microorganisms into the study of succession. Res Microbiol 161: 635–642 [DOI] [PubMed] [Google Scholar]
- Finegold SM, Song Y, Liu C, Hecht DW, Summanen P, Kononen E, Allen SD 2005. Clostridium clostridioforme: A mixture of three clinically important species. Eur J Clin Microbiol Infect Dis 24: 319–324 [DOI] [PubMed] [Google Scholar]
- Flint HJ, Duncan SH, Scott KP, Louis P 2007. Interactions and competition within the microbial community of the human colon: Links between diet and health. Environ Microbiol 9: 1101–1111 [DOI] [PubMed] [Google Scholar]
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR 2007. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci 104: 13780–13785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freilich S, Kreimer A, Meilijson I, Gophna U, Sharan R, Ruppin E 2010. The large-scale organization of the bacterial network of ecological co-occurrence interactions. Nucleic Acids Res 38: 3857–3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland JL, Cook KL, Adams JL, Kerkhof L 2001. Culturability as an indicator of succession in microbial communities. Microb Ecol 42: 150–158 [DOI] [PubMed] [Google Scholar]
- Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, Gordon JI 2011. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci 108: 6252–6257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamady M, Lozupone C, Knight R 2010. Fast UniFrac: Facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J 4: 17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Sakamoto M, Kitahara M, Benno Y 2003. Molecular analysis of fecal microbiota in elderly individuals using 16S rDNA library and T-RFLP. Microbiol Immunol 47: 557–570 [DOI] [PubMed] [Google Scholar]
- Huh HJ, Lee ST, Lee JH, Ki CS, Lee NY 2010. Clostridium symbiosum isolated from blood. Korean J Clin Microbiol 13: 90–92 [Google Scholar]
- Jackson S, Nicolson SW 2002. Xylose as a nectar sugar: From biochemistry to ecology. Comp Biochem Physiol B Biochem Mol Biol 131: 613–620 [DOI] [PubMed] [Google Scholar]
- Jean D, Briolat V, Reysset G 2004. Oxidative stress response in Clostridium perfringens. Microbiology 150: 1649–1659 [DOI] [PubMed] [Google Scholar]
- Joly F, Mayeur C, Bruneau A, Noordine ML, Meylheuc T, Langella P, Messing B, Duee PH, Cherbuy C, Thomas M 2010. Drastic changes in fecal and mucosa-associated microbiota in adult patients with short bowel syndrome. Biochimie 92: 753–761 [DOI] [PubMed] [Google Scholar]
- Kameue C, Tsukahara T, Yamada K, Koyama H, Iwasaki Y, Nakayama K, Ushida K 2004. Dietary sodium gluconate protects rats from large bowel cancer by stimulating butyrate production. J Nutr 134: 940–944 [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S 2000. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: 27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, Angenent LT, Ley RE 2011. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci 108: 4578–4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallich S, Teytaud O, Prudhomme E 2006. Statistical inference and data mining: False discoveries control. 17th COMPSTAT Symposium of the IASC, Rome, Italy, pp. 325–336 [Google Scholar]
- Lamberti LM, Fischer Walker CL, Noiman A, Victora C, Black RE 2011. Breastfeeding and the risk for diarrhea morbidity and mortality. BMC Public Health 11: S15 doi: 10.1186/1471-2458-11-S3–S15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre P, Legendre L 1998. Numerical ecology, 2nd ed. Elsevier, Amsterdam, The Netherlands [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI 2006. Microbial ecology: Human gut microbes associated with obesity. Nature 444: 1022–1023 [DOI] [PubMed] [Google Scholar]
- Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI 2008. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol 6: 776–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, Knight R 2005. UniFrac: A new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71: 8228–8235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Knight RD, Landweber LF 2001. The molecular basis of nuclear genetic code change in ciliates. Curr Biol 11: 65–74 [DOI] [PubMed] [Google Scholar]
- Lozupone CA, Hamady M, Cantarel BL, Coutinho PM, Henrissat B, Gordon JI, Knight R 2008. The convergence of carbohydrate active gene repertoires in human gut microbes. Proc Natl Acad Sci 105: 15076–15081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, Buchner A, Lai T, Steppi S, Jobb G, et al. 2004. ARB: A software environment for sequence data. Nucleic Acids Res 32: 1363–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai V, Braden CR, Heckendorf J, Pironis B, Hirshon JM 2006. Monitoring of stool microbiota in subjects with diarrhea indicates distortions in composition. J Clin Microbiol 44: 4550–4552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maidak BL, Cole JR, Lilburn TG, Parker CT Jr, Saxman PR, Farris RJ, Garrity GM, Olsen GJ, Schmidt TM, Tiedje JM 2001. The RDP-II (Ribosomal Database Project). Nucleic Acids Res 29: 173–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maukonen J, Satokari R, Matto J, Soderlund H, Mattila-Sandholm T, Saarela M 2006. Prevalence and temporal stability of selected clostridial groups in irritable bowel syndrome in relation to predominant faecal bacteria. J Med Microbiol 55: 625–633 [DOI] [PubMed] [Google Scholar]
- McCook LJ 1994. Understanding ecological community succession: Causal models and theories, a review. Plant Ecol 110: 115–147 [Google Scholar]
- Meyer PE, Lafitte F, Bontempi G 2008. minet: A R/Bioconductor package for inferring large transcriptional networks using mutual information. BMC Bioinformatics 9: 461 doi: 10.1186/1471-2105-9-461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KE, Weinstock GM, Highlander SK, Worley KC, Creasy HH, Wortman JR, Rusch DB, Mitreva M, Sodergren E, Chinwalla AT, et al. 2010. A catalog of reference genomes from the human microbiome. Science 328: 994–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, et al. 2012. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336: 489–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO 2007. Development of the human infant intestinal microbiota. PLoS Biol 5: e177 doi: 10.1371/journal.pbio.0050177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrung M, Kennedy R, Caporaso JG, Stombaugh J, Wendel D, Knight R 2011. TopiaryExplorer: Visualizing large phylogenetic trees with environmental metadata. Bioinformatics 27: 3067–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Png CW, Linden SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, McGuckin MA, Florin TH 2010. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol 105: 2420–2428 [DOI] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaible UE, Kaufmann SH 2005. A nutritive view on the host-pathogen interplay. Trends Microbiol 13: 373–380 [DOI] [PubMed] [Google Scholar]
- Schoepfer AM, Schaffer T, Seibold-Schmid B, Muller F, Seibold F 2008. Antibodies to flagellin indicate reactivity to bacterial antigens in IBS patients. Neurogastroenterol Motil 20: 1110–1118 [DOI] [PubMed] [Google Scholar]
- Schoonmaker P, Mckee A 1988. Species composition and diversity during secondary succession of coniferous forests in the western Cascade mountains of Oregon. For Sci 34: 960–979 [Google Scholar]
- Seeliger S, Janssen PH, Schink B 2002. Energetics and kinetics of lactate fermentation to acetate and propionate via methylmalonyl-CoA or acrylyl-CoA. FEMS Microbiol Lett 211: 65–70 [DOI] [PubMed] [Google Scholar]
- Sigler WV, Zeyer J 2004. Colony-forming analysis of bacterial community succession in deglaciated soils indicated pioneer stress-tolerant opportunists. Microb Ecol 48: 316–323 [DOI] [PubMed] [Google Scholar]
- Sleator RD, Hill C 2002. Bacterial osmoadaptation: The role of osmolytes in bacterial stress and virulence. FEMS Microbiol Rev 26: 49–71 [DOI] [PubMed] [Google Scholar]
- Smillie CS, Smith MB, Friedman J, Cordero OX, David LA, Alm EJ 2011. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 480: 241–244 [DOI] [PubMed] [Google Scholar]
- Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, Buhler JD, Gordon JI 2005. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 307: 1955–1959 [DOI] [PubMed] [Google Scholar]
- Stark PL, Lee A 1982. The microbial ecology of the large bowel of breast-fed and formula-fed infants during the first year of life. J Med Microbiol 15: 189–203 [DOI] [PubMed] [Google Scholar]
- Tsuda M, Hosono A, Yanagibashi T, Kihara-Fujioka M, Hachimura S, Itoh K, Hirayama K, Takahashi K, Kaminogawa S 2010. Intestinal commensal bacteria promote T cell hyporesponsiveness and down-regulate the serum antibody responses induced by dietary antigen. Immunol Lett 132: 45–52 [DOI] [PubMed] [Google Scholar]
- Tsukahara T, Koyama H, Okada M, Ushida K 2002. Stimulation of butyrate production by gluconic acid in batch culture of pig cecal digesta and identification of butyrate-producing bacteria. J Nutr 132: 2229–2234 [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. 2009a. A core gut microbiome in obese and lean twins. Nature 457: 480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI 2009b. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1: 6ra14 doi: 10.1126/scitranslmed.3000322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira-Silva S, Rocha EP 2010. The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet 6: e1000808 doi: 10.1371/journal.pgen.1000808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Jarnerot G, Tysk C, Jansson JK, Engstrand L 2010. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 139: 1844–1854.e1 [DOI] [PubMed] [Google Scholar]
- Wolin MJ, Miller TL, Collins MD, Lawson PA 2003. Formate-dependent growth and homoacetogenic fermentation by a bacterium from human feces: Description of Bryantella formatexigens gen. nov., sp. nov. Appl Environ Microbiol 69: 6321–6326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Forst C, Bonner C, Jensen RA 2002. Significance of two distinct types of tryptophan synthase beta chain in Bacteria, Archaea and higher plants. Genome Biol 3: research0004–research0004.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young VB, Schmidt TM 2004. Antibiotic-associated diarrhea accompanied by large-scale alterations in the composition of the fecal microbiota. J Clin Microbiol 42: 1203–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaneveld JR, Lozupone C, Gordon JI, Knight R 2010. Ribosomal RNA diversity predicts genome diversity in gut bacteria and their relatives. Nucleic Acids Res 38: 3869–3879 [DOI] [PMC free article] [PubMed] [Google Scholar]