

Abstract
The regulation of gene expression is mediated at the transcriptional level by enhancer regions that are bound by sequence-specific transcription factors (TFs). Recent studies have shown that the in vivo binding sites of single TFs differ between developmental or cellular contexts. How this context-specific binding is encoded in the cis-regulatory DNA sequence has, however, remained unclear. We computationally dissect context-specific TF binding sites in Drosophila, Caenorhabditis elegans, mouse, and human and find distinct combinations of sequence motifs for partner factors, which are predictive and reveal specific motif requirements of individual binding sites. We predict that TF binding in the early Drosophila embryo depends on motifs for the early zygotic TFs Vielfaltig (also known as Zelda) and Tramtrack. We validate experimentally that the activity of Twist-bound enhancers and Twist binding itself depend on Vielfaltig motifs, suggesting that Vielfaltig is more generally important for early transcription. Our finding that the motif content can predict context-specific binding and that the predictions work across different Drosophila species suggests that characteristic motif combinations are shared between sites, revealing context-specific motif codes (cis-regulatory signatures), which appear to be conserved during evolution. Taken together, this study establishes a novel approach to derive predictive cis-regulatory motif requirements for individual TF binding sites and enhancers. Importantly, the method is generally applicable across different cell types and organisms to elucidate cis-regulatory sequence determinants and the corresponding trans-acting factors from the increasing number of tissue- and cell-type-specific TF binding studies.
The precise regulation of gene expression plays an important role in development, cell differentiation, and cellular responses to the environment. The spatio-temporal expression of genes is controlled by DNA elements called enhancers (Banerji et al. 1981) or cis-regulatory modules (CRMs) (Kirchhamer et al. 1996). These elements act as docking platforms for transcription factors (TFs), and the combined regulatory cues of all bound TFs results in the activation (or repression) of gene expression (e.g., Stanojevic et al. 1991; Giese et al. 1992).
TFs bind to the DNA in a sequence-specific manner, recognizing short sequence motifs. However, the relation between TF motifs and in vivo binding sites is far from simple: First, there is typically a large discrepancy between the number of motif occurrences and in vivo binding sites and most of even the highest-scoring motif occurrences are not bound. For example, during Drosophila embryogenesis, the mesodermal TF Twist binds to only about a thousand of the roughly 1 million Twist motifs in the genome (Sandmann et al. 2007; Zeitlinger et al. 2007; Zinzen et al. 2009). Second, in vivo TF binding appears to be highly context-specific, and the same TF typically binds to different genomic binding sites in different conditions (e.g., Zeitlinger et al. 2003; Buck and Lieb 2006; Sandmann et al. 2007; Zinzen et al. 2009; Wilczynski and Furlong 2010; Palii et al. 2011). Twist-binding sites, for example, change dynamically between different time points during embryonic mesoderm development (2–8 h post-fertilization, hpf), which cannot be attributed to changes of Twist activity (e.g., by alternative splicing or post-translational modification) or concentration (Sandmann et al. 2007; Zinzen et al. 2009; Wilczynski and Furlong 2010; see also Fig. 1). Such differential binding has also been reported for several other TFs between different cell types in human and mouse (Lin et al. 2010; Palii et al. 2011), different developmental stages in C. elegans (Zhong et al. 2010), or different growth conditions in yeast (Zeitlinger et al. 2003; Buck and Lieb 2006).
Figure 1.

Context-specific Twist binding sites display differential motif content. (A) Twist binding sites identified by ChIP-chip in D. melanogaster embryos (Zinzen et al. 2009) differ between developmental time points (2–4, 4–6, and 6–8 hpf). The ‘early,’ ‘late,’ and ‘continuous’ classes of binding sites are indicated and comprise sites to which Twist binds exclusively at 2–4 (early), 6–8 hpf (late), or at all three time points (cont.). (B) The genes close to the Twist binding sites also differ between time points. (C) Genes close to the early and late binding sites are enriched in different Gene Ontology (GO) categories, as expected for respective developmental stages. Shown are hypergeometric P-values (see color legend) for selected categories that are significant for at least one time point (see Supplemental Table S2 for all categories and the GO identifiers). (D) Distribution of the Twist (TWI), Tramtrack (TTK), and ME119 motifs according to their distances to the summits of all Twi ChIP-chip peaks of the ‘early’ (purple) and ‘late’ (green) time points (shown are motif counts for each 200-bp bin averaged across all ChIP-chip peaks for each time point). While the distribution of TWI motifs does not differ between early and late binding sites, the TTK motifs are enriched and the ME119 motifs are depleted in early sites. (E) Motifs are differentially enriched between the early and late binding sites, even when corrected for different overall sequence composition using shuffled controls motifs. (Left column) Shown are fold-enrichment values for the motifs that are differentially distributed between the early and late sites (P-value ≤ 0.05; motifs from Stark et al. 2007). Interestingly, the differential motif distribution between the early and late sites stems from consistent motif enrichment and depletion with respect to the average intergenic genome (right columns), as expected for motifs that help define TF binding sites within a large genome.
The dependence of in vivo binding on the cellular context suggests that TFs might regulate different genes in different cell types. Cell-type-specific regulatory targets and functions have indeed been observed for many broadly expressed TFs. For example, although active in many tissues, Hox TFs regulate certain genes specifically in some tissues but not in others (Pearson et al. 2005). This is even more pronounced for field-specific selector genes such as scalloped (sd) (Guss et al. 2001) or TFs downstream from signaling pathways, most of which are reused throughout development and regulate different genes in different contexts (Barolo and Posakony 2002; Mullen et al. 2011; Trompouki et al. 2011).
Context-specific binding might result from other TFs facilitating or inhibiting binding either directly or via changes to the chromatin structure and DNA accessibility (e.g., Zeitlinger et al. 2003; Harbison et al. 2004; Buck and Lieb 2006; Sandmann et al. 2007; Heinz et al. 2010; He et al. 2011; John et al. 2011; Kaplan et al. 2011; Li et al. 2011). Such a combinatorial model would be an elegant way to realize many gene regulatory states with a limited number of TFs and to achieve TF binding specificity within the context of a large genome. It would also explain how TF binding can depend on both, the cellular context (because each cell type expresses a distinct combination of TFs) and the specific genomic region because each binding site has a different sequence, presumably containing a distinct combination of TF motifs. A combinatorial model requires that the binding of one TF to a given site depends on the presence or absence of motifs for other TFs in the vicinity of the site. Context-specific binding should thus be reflected in the cis-regulatory sequence around binding sites and might allow the discovery of characteristic sequence features such as motifs of partner TFs that would be predictive of binding. However, to what extent the cis-regulatory sequence is predictive of context-specific binding has remained unclear.
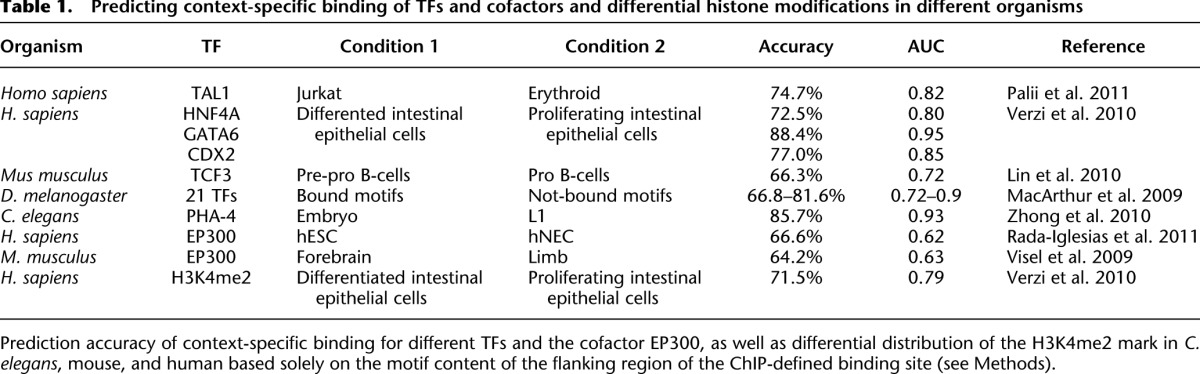
Here, we elucidate sequence determinants of context-specific TF binding in Drosophila, C. elegans, mouse, and human (Table 1; MacArthur et al. 2009; Visel et al. 2009; Zinzen et al. 2009; Lin et al. 2010; Verzi et al. 2010; Zhong et al. 2010; Palii et al. 2011; Rada-Iglesias et al. 2011), focusing mainly on a time course of Twist binding in the early Drosophila embryo (Zinzen et al. 2009). We show that context-specific Twist binding is explained by the presence of specific motif combinations for other TFs. Moving beyond bulk analyses, we determine cis-regulatory motif requirements for individual binding sites and validate them experimentally by motif-disrupting mutations. This identifies the TAGteam motif of the TF Vielfaltig (VFL; also known as Zelda) to be a key determinant of Twist binding and enhancer activity in the early Drosophila embryo. We also show that knowledge about cis-regulatory motif requirements in D. melanogaster allows the correct prediction of Twist binding across different Drosophila species, independently of the overall sequence conservation. Finally, we show that combinations of cis-regulatory motifs around in vivo binding sites are predictive of context-specific binding for other TFs and cofactors and the context-specific distribution of histone modifications in Drosophila (Zinzen et al. 2009), C. elegans (Zhong et al. 2010), mouse (Visel et al. 2009; Lin et al. 2010), and human (Verzi et al. 2010; Palii et al. 2011; Rada-Iglesias et al. 2011), for which time-, cell-type-, or tissue-specific chromatin immunoprecipitation (ChIP) data sets were available.
Table 1.
Predicting context-specific binding of TFs and cofactors and differential histone modifications in different organisms
Results
In vivo binding is context-dependent
As reported before, the binding sites of Twist differ substantially between different time points during early Drosophila embryo development, creating a picture of highly dynamic binding (Fig. 1A; Sandmann et al. 2007; Zinzen et al. 2009; Wilczynski and Furlong 2010). For example, out of 1620 Twist-binding sites at 2–4 hpf, 770 (47%) were specific for this time point (from now on called ‘early’ binding sites). Similarly, 689 out of 1576 (43.7%) were specific to 6–8 hpf (‘late’), and only 473 binding sites were bound throughout the entire time course (‘continuous’), a mere 14% compared to all 3290 sites. These observations could not be explained by altered Twist function, because Twist has not been reported to be alternatively spliced or modified post-translationally, and its expression remains constant throughout the developmental stages considered here (Sandmann et al. 2007; Zinzen et al. 2009; Wilczynski and Furlong 2010).
Importantly, the nonoverlap of binding sites between different time points appears not to be due to cutoff issues or other experimental artifacts but rather reflects changes in developmental gene expression: Genes flanking the binding sites at each time point are distinct (Fig. 1B) and are associated with functions expected for the respective developmental stages (Fig. 1C; Supplemental Table S2). Biological functions associated with genes of the early-bound set include cell division and pattern formation required during early embryonic development, whereas those associated with the late-bound set include somatic and visceral muscle development, myoblast fusion, and other processes related to later mesoderm differentiation (Wilczynski and Furlong 2010). In addition, the Twist sequence motif is conserved among closely related Drosophila species in the early and the late binding sites, suggesting that Twist binding in both contexts is under negative selection (see Supplemental Fig. S2).
These results confirm earlier observations (Wilczynski and Furlong 2010) and suggest that Twist binding depends on the cellular context and that context-specific binding is relevant for functional transcriptional regulation during development. Context-specific binding appears to be a general property of TFs and has been observed for several TFs in different species (Zeitlinger et al. 2003; Buck and Lieb 2006; Sandmann et al. 2007; Zinzen et al. 2009; Heinz et al. 2010; Lin et al. 2010; Verzi et al. 2010; Zhong et al. 2010; Palii et al. 2011). (See Supplemental Tables S1 and S5 and the Supplemental Discussion for examples in C. elegans, mouse, and human, for which time-, cell-type-, or tissue-specific ChIP data sets were available.)
Differential motif enrichment is predictive of context-specific binding
Context-specific binding might result from other TFs facilitating or inhibiting binding (e.g., Zeitlinger et al. 2003; Harbison et al. 2004; Buck and Lieb 2006; Sandmann et al. 2007; He et al. 2011). We reasoned that the motifs for such putative (activating or inhibiting) partner factors should be differentially distributed between the binding sites in different contexts. We indeed observed that the genomic sequence around early and late Twist-binding sites were enriched for different sets of TF motifs (known and predicted motifs from Stark et al. 2007), while—as expected—the Twist motif itself was found similarly enriched in both sets (Fig. 1D,E).
For example, early binding sites were enriched for motifs matching to Snail (SNA), Dorsal (DL), and Kruppel (KR), all of which are known Twist partner TFs in the early Drosophila embryo (He et al. 2011). In contrast, late binding sites were enriched for motifs matching Tinman (TIN) as observed previously (Sandmann et al. 2007) and Chorion factor 2 (CF2), which are involved in heart (Azpiazu and Frasch 1993) and muscle development (García-Zaragoza et al. 2008), respectively.
This motivated us to perform a discriminatory motif analysis of early and late binding sites (i.e., compare and contrast the sites' combined motif content) using a predictive classification framework with the aim to characterize important sequence features of each individual binding site. We decided to use an established machine learning method (a support vector machine, SVM) and reasoned that successful classification of the early and late sites solely based on sequence features (i.e., the sequences' motif content; for details, see Methods) would indicate that the respective features are relevant for TF binding.
Briefly, we excluded each binding site in turn for testing, trained the SVM on the remaining sites, and evaluated whether the test site was correctly predicted to be bound early or late (Fig. 2A; for details, see Methods). This leave-one-out cross-validation protocol works surprisingly well, correctly predicting early versus late binding for 77% of the Twist sites (area under the receiver operating characteristic [ROC] curve [AUC]: 0.84; Fig. 2B) and between 70% and 77% for other mesodermal TFs (AUCs 0.73 to 0.82; Supplemental Table S3). When we repeated the entire analysis after randomly shuffling the sites' assignments to the early and late classes, the predictions are ∼50% in all cases (Fig. 2B; Supplemental Table S3), as expected for random binary classifications. This confirms our cross-validation protocol and suggests that the successful predictions do not result from the computational procedure per se but rather from sequence differences (i.e., motif content) between early and late binding sites. The 35 motifs that appear to be important for the SVM predictions overlap the differentially distributed motifs above and included 10 motifs enriched in early and 25 in late binding sites (Supplemental Fig. S1).
Figure 2.

Cis-regulatory motif content predicts TF binding. (A) Schematic overview of the approach. To computationally classify context-specific TF binding sites based on their motif content, we counted all instances of known cis-regulatory motifs (denoted here as M1, M2, M3, etc.) in 401-nt-long windows centered on each ChIP-chip peak's summit (left; purple and green denote early and late bound sites, respectively). We used the motif counts (M1, M2, M3, etc.) for all binding sites as feature vectors for binary classification with a support vector machine (SVM) using leave-one-out cross-validation (LOOCV). Briefly, we excluded each binding site in turn for testing (blue), trained the SVM on the remaining sites, and predicted the test site (right). (B,C) Prediction (binary classification) of early versus late Twist binding sites (B) or continuously bound versus not bound Twist motifs (C) based solely on the motif content. (Orange) Prediction accuracies (percent of correctly classified sites; left) and receiver-operating-characteristic (ROC) curves (right; area under the curve [AUC] values are indicated). We repeated the entire procedure after randomizing the sites' class assignments (controls; gray), which yields random classifications (∼50%; AUC∼0.5) as expected (see Supplemental Discussion). The accuracy and AUC values for all other mesodermal TFs can be found in Supplemental Table S3; and for TFs, cofactors, and histone modifications in C. elegans, mouse, and human in Table 1.
Using the same approach, we were able to predict context-specific binding for all other TFs in Drosophila, C. elegans, mouse, and human for which ChIP data from different time points, cell types, or tissues were available (Visel et al. 2009; Zinzen et al. 2009; Lin et al. 2010; Verzi et al. 2010; Zhong et al. 2010; Palii et al. 2011; Rada-Iglesias et al. 2011). The prediction success varied between the data sets from an accuracy of 66.3% (AUC = 0.72) for TCF3 (also known as E2A; mouse) (Lin et al. 2010) to 88.4% (AUC = 0.95) for GATA6 (human) (Table 1; Verzi et al. 2010). Remarkably, we could also distinguish binding of the transcriptional cofactor EP300 between human embryonic stem cells and neuroectodermal spheres (Rada-Iglesias et al. 2011) and between mouse forebrain and limb (Visel et al. 2009) (67%; AUC = 0.62 and 64.2%; AUC = 0.63 respectively) (Table 1). Similarly, differential genomic distribution of histone-3-lysine-4 dimethylation (H3K4me2) between differentiated and proliferating intestinal epithelial cells (Verzi et al. 2010) could be predicted successfully (71.5%; AUC = 0.79) (Table 1). This shows that the TF and cofactor binding site sequences, as well as genomic regions with specific chromatin marks, contain distinct TF motif combinations that are indicative for the particular contexts. The successful classification of unseen sites during cross-validation further argues that sufficiently many binding sites share similar motif combinations, thereby enabling the identification of general rules (cross-validated classification would not be successful if each binding site were bound by entirely independent means).
Because our results suggest that partner motifs might determine context-specific TF binding, we wondered if they could also influence more generally whether a genomic instance of a TF motif was bound in vivo or not. Indeed, occurrences of Twist motifs in the genome sequence that were bound continuously and occurrences (of the same motif score) that were not bound according to ChIP data (Zinzen et al. 2009) could be classified successfully with an accuracy of 86% (AUC = 0.93) using leave-one-out cross-validation solely based on the motif content of the flanking sequences (Fig. 2C). Similar results were obtained for all other pairwise comparison of bound versus nonbound motif occurrences for all main mesodermal TFs in Drosophila embryos (Supplemental Table S3; Zinzen et al. 2009) and for several TFs important for early embryonic anterior/posterior (AP) patterning (accuracy of up to 81.6%; AUC = 0.9) (Table 1; MacArthur et al. 2009). While this performance is not enough to predict TF binding in the entire genome, the 86% correctly classified sites (vs. 50% expected by chance) suggest that partner TF motifs in the vicinity of binding sites carry information indicative of TF binding that is independent of the bound TF's motif itself.
Scoring the classification of individual sites
Successful classification indicates that cis-regulatory requirements are shared between different early and late binding sites, respectively (see above). However, such bulk analyses do not assess the robustness of each site's classification, i.e., to what extent repeated classification with different training sets would lead to the same outcome. For example, sites that share important features with many other sites might be more robustly predicted, while the prediction of sites with more unique features might more strongly depend on the individual training sets. To obtain a score that assesses the robustness of the prediction for each individual site, we classified each site 100 times using 100 different training sets that each did not include the test site (Fig. 3A) (for details, see Methods). This bootstrapping protocol yields a score between 0 and 100 for each site, which indicates the number of correct predictions.
Figure 3.

A prediction score for individual TF binding sites. (A) Schematic overview of the bootstrapping approach used to calculate a prediction score for each individual binding site. We excluded a site for testing (blue) and subsampled the remaining sites (left) 100 times to obtain 100 different training sets (middle), each of which we used to predict the test site (right) (see Fig. 2 for details). The prediction score represents the number of correct predictions and ranges from 0 to 100. (B) Distribution of the prediction scores for the classification of early versus late Twist binding sites (orange) and for controls for which we repeated the entire procedure after randomizing the sites' assignments to the early and late classes (gray). (C) Predictability of binding might be underestimated because sites with low prediction scores are less often reproducible by ChIP: Twist binding sites with high prediction scores (red; score-range 85–100) are significantly more often detected in an independent ChIP-seq data set (from He et al. 2011) than sites with low scores (blue; score range 0–15). Shown is the number of total and confirmed binding sites for sites in both score ranges and the binomial P-value.
We found that the majority of sites (59%) were confidently predicted with scores above 75 and 43% had scores above 90 (Fig. 3B). In contrast, if we repeated the protocol after shuffling the site assignments to the early and late classes, no single site reached a score above 75 (two out of 1043 sites [0.2%] scored exactly 75) (Fig. 3B).
The classification score for each individual site also allows us to assess the difference between sites with high and sites with low scores, potentially obtaining an explanation for the failure to predict some sites. We assessed the experimental reproducibility of the early Twist-binding sites by comparing the ChIP-chip data used here (Zinzen et al. 2009) to recent ChIP-seq data for Twist at the same time point (2–4 hpf) (Fig. 3C; He et al. 2011). Interestingly, 64% of the well-predicted sites but only 30% of the poorly predicted sites were found in the independent ChIP data set. This highly significant difference (2.1-fold; P ≤ 10−6) suggests that some of the low-scoring sites might be false positives of the ChIP approach and that the overall prediction performance might have been underestimated. Indeed, when we repeated the initial classification on binding sites detected by both ChIP approaches, the fraction of correctly predicted sites increased from 77% to 81%; and to near 100% when only peaks that scored 75 or above were used.
Identifying the cis-regulatory requirements of individual binding sites
The method above provides a prediction score for each binding site and thereby the means to determine the combinatorial motif requirements for each individual site (i.e., the site's cis-regulatory requirements). For this, we rescored each well-predicted site (score ≥75) after deleting all occurrences of a specific TF motif in silico (Fig. 4A) (for details, see Methods). These in silico mutations resulted in a substantial drop for 186 (30%) of the 612 well-predicted sites. Simultaneous mutations of all the occurrences of two TF motifs impaired the predictions of an additional 43% of sites, with a remaining 26% of robust sites (Fig. 4B).
Figure 4.

Prediction and validation of early Twist-bound enhancers' cis-regulatory requirements: Vielfaltig's TAGteam motif is a key component of early transcriptional regulation. (A) Schematic overview of the in silico mutations approach. To assess the importance of each particular TF motif (denoted here as M1, M2, M3, etc.), we set the number of its occurrences in the SVM feature table to zero. The differences in the prediction scores after these in silico motif deletions reflect the importance of the motif for the correct prediction of a particular binding site. In the example given, motif M2 (but not M1 or M3) is important for correctly classifying the binding site. (B) Fraction of well-predicted Twist early and late bound sites (prediction score ≥75) that drop substantially (≥20 points) when motifs of one (blue) or two (red) TFs are mutated in silico (the remaining sites are robust; gray). (C) Fraction of sites that are affected by a given motif (from Fig. 4B, blue sector). The most frequent motifs are the TTK-motif and Vielfaltig's TAGteam motif (VFL/ME38) for early (purple) and ME17 for late bound sites (green). Note that the number of sites affected by each motif's ablation is a conservative (i.e., low) estimate due to the overlap of motif instances in the sites' sequences. (D) The activity of early Twist-bound enhancers depends on Vielfaltig's TAGteam (VFL/ME38) motif. The wild-type (wt; left) and TAGteam mutated (right) enhancers of four different genes (btl, wntD, ths, and cact) were placed upstream of a Gal4 reporter gene (see cartoon on top), the expression of which was visualized by in situ hybridization in early Drosophila embryos (bottom). In all cases, the enhancer activity was abolished or strongly reduced when all (between three and five) instances of the TAGteam motif were mutated (all embryos are oriented laterally, dorsal site up, anterior to the left; stages 5 and 8 correspond to 2 and 3 hpf, respectively). Note that we took great care to ensure that comparisons between wild-type and mutant constructs are valid despite the generally semiquantitative nature of enzymatic in situ hybridization: Reporter constructs differed only by the indicated motif-disrupting sequence changes and were inserted into the identical genomic positions (landing site), the identical probe was used in all cases, and hybridizations were performed in parallel. (E) Twist binding depends on Vielfaltig's TAGteam motif. (Top) Schematic cartoons illustrate the genotype of the transgenic embryos, which carry a Gal4 reporter construct for the Twist-binding site in the cact locus (purple) with either the wild-type sequence (Enh-wt) or a sequence in which all four TAGteam motifs are mutated (Enh-mut). The inward pointing arrowheads indicate the primer pairs used to distinguish the endogenous cact locus (left) and the reporter (right) during qPCR (note that they are offset from the ChIP-chip peak summit to discriminate between the different sequence constructs). (Bottom) Twist ChIP-qPCR results from stage 5–7 embryos (2–4 hpf) that carry either the wild-type (wt) or the TAGteam mutant (mut) reporter. Shown is Twist ChIP enrichment (percent recovery compared with an unrelated genomic region) at the reporter (dark bars) and the endogenous cact locus (light bars). Error bars indicate standard deviations of three biological replicates from independent embryo collections (replicates are shown individually in Supplemental Fig. S4); P-values from t-tests are shown.
Interestingly, motifs for certain TFs appeared to be required for many of the 186 Twist-binding sites (Fig. 4C): the Tramtrack (TTK) motif (early; 52%), the motif ME38 (early; 38%; ME [for motif-in-enhancers] refers to motifs discovered in Stark et al. 2007), and the ME17 motif (late; 58%) were the most frequent ones. Interestingly, Tramtrack is known to repress zygotic expression of fushi tarazu (ftz) prior to the maternal-to-zygotic transition (Pritchard and Schubiger 1996), and ME38 matches the TAGteam motif that is bound by Vielfaltig, an essential activator of zygotic transcription (ten Bosch et al. 2006; Liang et al. 2008). Both tramtrack and vielfaltig transcripts are maternally deposited to the egg and present at very early stages of Drosophila embryo development. ME17 is a novel motif for which the corresponding TF is unknown (Stark et al. 2007), but which has been reported to associate with nucleosome-depleted open chromatin in the Drosophila embryo (Mavrich et al. 2008). The importance of the identified motifs is further supported by their increased evolutionary conservation in the regions that appear affected by the motifs' in silico ablations (see Supplemental Fig. S3).
The in silico mutations approach also identified cis-regulatory requirements for Twist binding in other contexts (see the Supplemental Discussion and Supplemental Fig. S5). For example, continuously bound Twist sites differed from early and late Twist-binding sites by an increased number of Twist motifs and the presence of motifs for Trithorax-like (TRL; also known as GAGA or GAF) and Zeste (Z). We also found an increased number of Twist motifs around individual instances of the Twist sequence motif that were bound compared with motif instances that were not bound (up to twofold enrichment, p ≤ 2.032 × 10−127), suggesting that TF binding sites often contain several copies of the TF's motif (Berman et al. 2002). Additionally, motifs for Snail—an important Twist partner TF (Zeitlinger et al. 2007)—were also enriched close to bound Twist motifs as observed previously (Sandmann et al. 2007; Zeitlinger et al. 2007).
Early embryonic enhancers depend on Vielfaltig's TAGteam motif
Several Twist-bound regions that we predict to depend on the TAGteam motif of the TF Vielfaltig (when classifying both early vs. late sites and early vs. nonbound sites) function as early enhancers with diverse patterns in the Drosophila embryo (Ohshiro and Saigo 1997; Markstein et al. 2004; Zeitlinger et al. 2007). In addition, the TAGteam motif is well-defined and largely nondegenerate, making it an ideal candidate for experimental validation by motif-disrupting mutations. We therefore chose four early Twist-bound embryonic enhancers close to the genes breathless (btl) (Ohshiro and Saigo 1997), wnt inhibitor of Dorsal (wntD) (Zeitlinger et al. 2007), thisbe (ths) (Markstein et al. 2004), and cactus (cact) to experimentally validate the dependence on the TAGteam motif (Fig. 4D; for details, see the Supplemental Material).
For each of these regions, we cloned wild-type and mutant sequences in which we disrupted all TAGteam motifs by point mutations upstream of a Gal4 reporter gene and inserted them into the Drosophila melanogaster genome by site-specific integration (Pfeiffer et al. 2008). We found that all four wild-type sequences drove reporter expression in diverse dorso-ventral patterns in the early embryo (Fig. 4D). In contrast, all four mutant sequences in which the TAGteam motif had been disrupted were nonfunctional or had severely reduced enhancer activity, suggesting that the TAGteam motif is required for enhancer activity (Fig. 4E).
The experiments above show that the TAGteam motif is required for the activity of early Twist-bound enhancers. To directly test if the TAGteam motif is also required for Twist binding (which was the basis of the computational predictions), we selected the Twist-binding site in the cact locus for chromatin-immunoprecipitation (ChIP) experiments. We performed ChIP with an antibody against Twist (Zinzen et al. 2009) from stage 5–7 embryos (2–4 hpf) carrying either the wild-type or the TAGteam motif mutant cact Gal4 reporter (Fig. 4E; Supplemental Fig. S4). For both transgenic lines, we assessed Twist binding to the reporter construct and to the endogenous cact locus using quantitative PCR (qPCR) with primer pairs that could distinguish between both sequence contexts. Twist binding to the mutant reporter construct was indeed strongly reduced (3.4-fold decrease; t-test P-value ≤ 0.003), while binding to the endogenous cact locus, which serves as an internal control, was comparable (t-test p = 0.29). This indicates that Twist binding in the early Drosophila embryo depends on the TAGteam motif and thereby suggests that Vielfaltig facilitates or enables the binding of Twist and potentially other TFs in this context.
Predicting Twist binding across six Drosophila species
We were wondering if the motif combinations that predict context-specific Twist binding in D. melanogaster would allow predictions across closely related Drosophila species. This would test if the function of combinatorial motif patterns is evolutionarily conserved and also independently assesses our predictions based on motif-disrupting evolutionary sequence changes and their effect on Twist binding.
We trained an SVM in D. melanogaster and predicted each Twist-bound site in D. melanogaster and its corresponding orthologous sequence in Drosophila simulans, Drosophila erecta, Drosophila yakuba, Drosophila ananassae, and Drosophila pseudoobscura using leave-one-out cross-validation (i.e., we trained only in D. melanogaster and when we predicted a site across species, the corresponding D. melanogaster site was held out and not used for training). We then compared the results of these predictions with recent ChIP-seq data across species (He et al. 2011) and found that high-scoring orthologous sites across the different species were significantly more often bound by Twist (He et al. 2011) than orthologous sites with low scores (Fig. 5A). For example, the prediction scores for a binding site near the genes Distal-less (Dll) and CG3650 correlate well with binding across all six species (Fig. 5B) and are random for the D. erecta sequence and below random for the D. pseudoobscura sequence, which are indeed bound at low or nondetectable levels, respectively.
Figure 5.

Predicting Twist binding across Drosophila species. (A) Sequences orthologous to D. melanogaster early Twist binding sites in five Drosophila species are significantly more often bound by Twist if they had high prediction scores (red; low-scoring sites are blue). For each of the 407 early Twist binding sites in D. melanogaster found by two independent ChIP studies (Zinzen et al. 2009; He et al. 2011), we classified the orthologous sequences from Drosophila simulans, Drosophila erecta, Drosophila yakuba, Drosophila ananassae, or Drosophila pseudoobscura with a SVM trained in D. melanogaster only (for details, see Methods). We then assessed whether the sites were bound in vivo using data from He et al. (2011) (note that the majority of D. melanogaster binding sites are bound across species as reported by He et al. 2011, leading to a high overall binding rate). Shown is the fraction of bound sites for the best and worst scoring sites (score ranges: 0–15 versus 85–100). (B) Prediction scores (orange) and ChIP-seq signals (normalized read density; black density track) correlate well across six different Drosophila species for a Twist binding site (black bar) close to the genes Dll and CG3650. (C) Examples of loss (left) or conservation (right) of a Twist binding site between D. melanogaster and D. simulans that had been correctly predicted despite largely similar (left) and different (right) overall motif content, respectively. The motif content is shown as a heatmap in which gray represents motifs with identical counts in both species (14 for the left vs. 10 for the right example, respectively) and shades of red and green represent smaller or higher motif counts in D. simulans, respectively. The UCSC phastCons track indicates sequence conservation across 14 insect species (Siepel et al. 2005). Consistent with the motif content heatmaps, the binding site sequence on the left is overall more highly conserved across species: 38.3% (left) versus 8.7% (right) of all nucleotides have a perfect phastCons score of 1.0, and the overall nucleotide identity between D. melanogaster and D. simulans was 86.2% (left) and 84.8% (right), respectively.
Interestingly, the predictions were indicative of binding independent of the sites' overall sequence similarity or their overall TF motif content (Fig. 5C). This suggests that the trained SVM was able to assess motif presence and absence patterns in a functionally meaningful way, e.g., by weighting motifs according to their importance during training. For example, Twist binding to a site on chromosome 2L (Fig. 5C, left) was lost in D. simulans as predicted and likely due to the disruption of Tramtrack and Vielfaltig motifs, which were both important for the successful classification of the D. melanogaster sequence. In contrast, Twist binding to a site on chromosome X with lower overall sequence conservation and more divergent motif content (Fig. 5C, right) was conserved as predicted. The correct prediction is expected because successful classification of the D. melanogaster sequence was robust against all in silico mutations except for the combined ablation of both Tramtrack and Kruppel motifs, the latter being present in both species. Overall, these results suggest that rules about Twist binding learned in D. melanogaster apply across different Drosophila species. The confirmation of predicted conservation or loss of binding across species also constitutes an independent validation of our predictions, based on naturally occurring sequence changes across species (instead of the targeted motif disruptions above; Fig. 4D).
Discussion
Recent ChIP experiments revealed that in vivo TF binding sites differ between different cell types (or more generally cellular contexts) (Zeitlinger et al. 2003; Buck and Lieb 2006; Sandmann et al. 2007; Zinzen et al. 2009; Heinz et al. 2010; Lin et al. 2010; Zhong et al. 2010; John et al. 2011; Palii et al. 2011), consistent with the frequent reuse of TFs in different cellular or developmental contexts (Fujioka et al. 2003; Blanchard et al. 2010; Tran et al. 2010) and their context-specific functions. However, whether and how context-specific TF binding is encoded in the cis-regulatory sequences and the relation between the DNA sequence and in vivo binding has remained unclear.
Here, we use binding sites of a single TF in different contexts as pivots to study the sequence determinants of in vivo binding. By systematically comparing the binding site sequences, we show that they contain motifs for other TFs that are characteristic for each context and allow the prediction of context-specific binding. The motif-based predictions were sufficiently strong to pinpoint cis-regulatory requirements for individual binding sites, providing specific testable hypotheses, which we validated experimentally.
This finding has important implications for transcriptional regulation: First, it argues that context-dependent TF binding is determined by the cis-regulatory sequence, consistent with the sufficiency of enhancer sequences to recapitulate their endogenous chromatin state (i.e., histone modifications and DNA methylation) (e.g., Lienert et al. 2011) and activity (e.g., Banerji et al. 1983; Doyle et al. 1989; Visel et al. 2009) in different contexts. Second, in vivo binding appears to be determined by combinations of TF motifs rather than a single TF's motif, therefore substantially increasing the information content and specificity of in vivo binding. Individual motifs are often only 4–6 nt long and would therefore occur every 256–4096 nt by chance (i.e., in random DNA sequences—even when motif degeneracy is not taken into account). Second, because different motif combinations are functional, a single TF can have context-specific binding sites and target genes depending on both, the cis-regulatory sequence that contains a certain combination of motifs and the cell type that expresses the corresponding TFs. For example, Twist motifs in the vicinity of motifs for Snail, Dorsal, or Vielfaltig are preferentially bound early, while those near motifs for Tinman (TIN) or Chorion factor 2 (CF2) are preferentially bound late, when these TFs are present, respectively.
We observe that Twist binding correlates with the binding of other mesodermal TFs (e.g., early with Myocyte enhancer factor 2 [MEF2] and TIN with Pearson correlation coefficients of 0.2 and 0.4, respectively) and that ChIP-chip data (from Zinzen et al. 2009) for other mesodermal TFs are predictive of Twist binding using cross-validation (83%; AUC = 0.88), suggesting that partner TFs might assist each other's binding in a correlated fashion.
In general, the action of partner TFs might be direct, e.g., mediated by direct protein–protein interactions (suggested, e.g., for ‘condition-altered binding’) (Harbison et al. 2004), or passive, e.g., by opening or otherwise preparing chromatin for TF binding (suggested, e.g., in yeast) (Buck and Lieb 2006) or for ‘assisted loading’ (Voss et al. 2011). Some of the uncharacterized motifs (from Stark et al. 2007) might, for example, recruit chromatin-remodeling factors, and one of them (ME17) indeed correlates with nucleosome-depleted open chromatin (Mavrich et al. 2008). It is conceivable that chromatin-mediated functions might be temporally decoupled such that partner TFs could act sequentially rather than simultaneously.
Vielfaltig's TAGteam motif is a key regulator of early TF binding and enhancer function
The TF Vielfaltig's TAGteam motif appears to be a key determinant of early Twist binding: We found it enriched in early binding sites, required to successfully classify them in a predictive framework, required for function of four early enhancers with diverse activity patterns, and necessary for Twist binding. Similarly, the early binding sites of other factors are enriched in TAGteam motifs (e.g., early MEF2 binding sites; 1.97-fold; P ≤ 4 × 10−6), suggesting that it is a general determinant of early binding and enhancer function. Interestingly, Vielfaltig is maternally deposited and has been shown to bind to TAGteam motifs, a set of motifs that are enriched in regulatory regions of early blastodermal genes (ten Bosch et al. 2006; Liang et al. 2008) and in TF binding sites in early D. melanogaster and D. yakuba embryos (Li et al. 2008; Bradley et al. 2010). Vielfaltig is sufficient to activate enhancers that contain TAGteam motifs and required for early gene expression and cellularization (Liang et al. 2008). While this work was under revision, it has further been shown that Vielfaltig binds to ∼60% of all genomic instances of the TAGteam motif (Harrison et al. 2011; Nien et al. 2011). Our demonstration that Vielfaltig is a key determinant of early binding is intriguing and suggests that Vielfaltig might help to open (or keep open) chromatin and allow TFs to access their binding sites on DNA thereby defining early enhancers.
We also find the motif for Tramtrack to be important for early Twist binding. Maternal Tramtrack has been proposed to repress zygotic transcription of early patterning genes in a concentration-dependent manner, thereby explaining the timing of zygotic activation (Pritchard and Schubiger 1996). Due to the overlap of different motifs, the 38% and 52% early Twist-binding sites that depend on Vielfaltig and Tramtrack are conservative estimates, and both factors are likely important for additional binding sites. Our study suggests that Vielfaltig and Tramtrack play an important regulatory role in the early embryo, preparing and/or regulating enhancers of a broad set of genes.
Context-specific codes are shared among binding sites
Our finding that context-specific TF binding can be predicted using cross-validation indicates that the motif combinations extracted from training sequences were sufficiently general to correctly predict previously unseen test sequences. This means that different sites share characteristic sequence features and might function by similar means. In fact, we find similar patterns of motifs enriched in binding sites for different TFs in the same context (e.g., the early Drosophila mesoderm), suggesting that different cell types have specific “codes” that are indicative of binding for different TFs. We found this to be generally true for all data sets we studied in species as diverse as human, mouse, C. elegans, and Drosophila. In its ability to discover which cis-regulatory motifs (and the corresponding TFs) are relevant for different functionally defined sets of sequences (e.g., those active or bound in defined cellular contexts), our approach is similar to recent k-mer-based enhancer predictions in mammals (Lee et al. 2011). It is complementary to thermodynamic models of gene expression in the early Drosophila embryo (Jaeger et al. 2004; Janssens et al. 2006; Segal et al. 2008; Kazemian et al. 2010). Here, all relevant TFs, their motifs, and their cellular protein concentrations are known, and the models predict enhancer activity for selected DNA sequences in order to gain insights into mechanistic aspects of transcriptional regulation, e.g., the importance of weak binding or homotypic and heterotypic TF–TF interactions.
Here, we show that motif analyses of context-specific binding sites can identify the precise cis-regulatory sequence requirements and the trans-acting factors for individual genomic sites. This has important implications for the many TFs such as Hox factors (Pearson et al. 2005) or TFs downstream from signaling pathways (Barolo and Posakony 2002), which are broadly expressed but regulate certain genes specifically in some tissues but not in others: we foresee that the recent increase in cell-type-specific ChIP analyses will reveal specific cis-regulatory requirements and the corresponding trans-acting factors that define the regulatory state for many cell types. Because TF binding has been shown to be predictive of cell-type-specific enhancer activity (Zinzen et al. 2009; see also Stark 2009; He and Sinha 2010), this will bridge the gap between the sequence, TF binding, and enhancer/CRM function and will ultimately reveal how cell-type-specific regulatory information is encoded in the DNA sequence.
Methods
Definition of context-specific binding sites
We obtained ChIP-chip peaks and the raw ChIP-chip data for Twist (TWI), Tinman (TIN), Binou (BIN), and Myocyte enhancer factor 2 (MEF2) from Zinzen et al. (2009). We defined stage-specific peaks as those present (i.e., called as peaks by Zinzen et al. 2009) only at one stage for: Twist early (2–4 hpf), Twist late (6–8 hpf), Tinman early (2–4 hpf), Tinman late (6–8 hpf), Binou early (6–8 hpf), Binou late (10–12 hpf), MEF2 early1 (2–4 hpf), MEF2 early2 (4–6 hpf), MEF2 late1 (8–10 hpf), and MEF2 late2 (10–12 hpf). We also required that the raw ChIP-chip signals at the respective stage were higher than at any other stages for the same TF. We further defined continuously bound peaks as peaks present (i.e., called as peaks by Zinzen et al. 2009) during all stages for which ChIP-chip data were available, i.e., Binou continuous (6–8, 8–10, 10–12 hpf), MEF2 continous (2–4, 4–6, 6–8, 8–10, 10–12 hpf), Twist continuous (2–4, 4–6, 6–8 hpf), and Tinman continuous (2–4, 4–6, 6–8 hpf).
For non-Drosophila ChIP data sets (i.e., mouse, human and C. elegans), we used the raw ChIP-seq reads from Lin et al. (2010), Verzi et al. (2010), Zhong et al. (2010), and Palii et al. (2011). We mapped the reads to the genomes of their respective organisms (genome sequence releases hg18, mm9, ce6; all obtained from the UCSC Genome Browser; http://genome.ucsc.edu/) using Bowtie (Langmead et al. 2009) and called ChIP-seq peaks using MACS (Zhang et al. 2008) with default parameters. We considered the best 1000 peaks for Lin et al. (2010), Verzi et al. (2010), and Palii et al. (2011) and the best 500 peaks for Zhong et al. (2010) according to the peaks' P-values. For EP300 we used ‘class_I’ and ‘class II-I’ regions as defined by Rada-Iglesias et al. (2011) and the Forebrain- and Limb-specific EP300 binding sites as defined by Visel et al. (2009).
For the TFs BIN, MEF2, TIN, and TWI (Supplemental Table S1, upper part), we considered two peaks overlapping if their summits were located in the same CRM defined by Zinzen et al. (2009). For TCF3, TAL1, PHA-4, HNF4A, H3K4me2, GATA6, CDX2, and EP300 (Supplemental Table S1, lower part), we considered peaks as overlapping if their summits peak were closer than 300 bp.
For the motif analysis and the SVM predictions, we needed to ensure that the flanking regions (a 401-bp window centered on each peak summit, i.e., a 200-bp flanking sequence on each side) did not overlap. We therefore excluded peaks of different contexts if their summits were closer than 400 bp and kept only one of two or more such overlapping peak regions if they belonged to the same context. To account for the more spread-out nature of histone modifications, we extended the flanking regions to 300 bp when analyzing H3K4me2 in differentiated and proliferating intestinal epithelial cells.
Definition of bound and nonbound motif instances
We defined ‘not bound’ motif instances for mesodermal TFs as motif matches to the D. melanogaster intergenic and nonrepeat genome (PWM-cutoff p = 1/1024) that were within 50 bp of a microarray probe (from the raw Zinzen et al. ChIP-chip data) with a ChIP-chip signal among the lowest 10%. For comparisons of these ‘not bound’ motifs with bound motifs of the continuous or stage-specific classes above, we selected only those regions with at least one TF motif match within 50 bp of a ChIP-chip peak summit of the respective class.
Statistical analyses and computations
All statistical computations to obtain binomial or hypergeometric P-values and to correct them for multiple testing were done in R (version 2.14). To intersect coordinates and identify overlapping regions, we used the tool intersectBed, and to obtain random genomic regions, we used the tool shuffleBed from bedtools (Quinlan and Hall 2010).
Conservation of Twist binding across species
To define confident binding sites that are confirmed by two independent ChIP experiments, we intersected the ChIP-chip peaks above with the 3488 D. melanogaster ChIP-seq peaks by He et al. (2011). To validate the predictions of Twist binding across species, we intersected the predictions with ChIP-seq peaks of He et al. (2011) in each species (D. simulans, D. erecta, D. yakuba, D. ananasae, and D. pseudoobscura; P-value ≤ 10−10).
Peak-to-gene assignment and GO analysis
We obtained the genome annotation, i.e., genes and their genomic coordinates, from FlyBase (Drosophila; release 5.22); Ensembl (human; Ensembl Genes 64 for NCBI36); and mouse (Ensembl Genes 65 for NCBIm37); and WormBase (C. elegans; release WS190) and the Gene Ontologies (downloaded on January 2012). We assigned each ChIP peak summit to the gene with the closest transcription start site (TSS; 5′ end of gene). We then calculated the enrichment (hypergeometric P-values) of functional categories from Gene Ontology (Ashburner et al. 2000) in each set of binding regions by comparing the number of genes in each category obtained in each ChIP experiment with the corresponding number among all genes. We corrected the hypergeometric P-values obtained for each category for multiple testing using the R-function p.adjust with the method ‘FDR’ and applied a cutoff of ≤0.05.
Motif analysis
We used known and predicted motifs from Stark et al. (2007) (Supplemental Table S4), UniProbe (January 2009) (Newburger and Bulyk 2009), JASPAR (release 2009) (Bryne et al. 2008), and TRANSFAC (release 2009.3) (Matys et al. 2003). We converted IUPAC motif consensus sequences to position-specific-weight matrices (PWMs) by using a pseudocount of 0.001 and assuming an equal background distribution of 0.25 for all four nucleotides. To account for the redundancy of all motif data sets, we clustered the motifs into groups (see below) and used one motif as representative.
For each ChIP-chip data set, we scanned the respective species' genome as in Stark et al. (2007) within a 401-bp window centered on each peak's summit for occurrences of regulatory motifs with a PWM-cutoff 2.44 × 10−4 (1/4096). For the SVM predictions in Drosophila TFs, mouse TCF3, EP300, C. elegans PHA-4, and human CDX data, we used a PWM-cutoff of 3.9 × 10−3 (1/256) to increase sensitivity.
We assessed the differential distribution of each motif between the context-specific binding sites by calculating a fold-enrichment value as the ratio of the number of motif occurrences, corrected for differences in average sequence composition by the number of occurrences of shuffled control motifs as in Kheradpour et al. (2007) and similar to Frith et al. (2004), Ho Sui et al. (2005), and Chang et al. (2006). We assessed the statistical significance of the differential enrichment by a hypergeometric P-value and report only significant motifs (P-value ≤ 0.05).
Feature selection
To reduce redundancy from highly similar motifs, we first clustered motifs by the number of co-occurrences in randomly chosen genomic sequences (K-means clustering using the implementation in R [function: kmeans]; N = 73), and used one motif per cluster as representative. We then performed feature selection by backward elimination (Guyon 2003) to remove motifs with low predictive value for the respective classification tasks: We first ranked the features by their individual contribution (i.e., the difference in prediction accuracy when using all 73 features vs. leaving out that feature individually). We then repeated the predictions with all 73 features, eliminated increasingly many low-ranking features (i.e., we first eliminated feature 73, then 73 and 72, then 73 and 72 and 71, etc.), and chose to cut the feature list at the global maximum of the resulting curve (e.g., keeping the top 35 features for Twist early vs. late and 31 for Twist continuous vs. not bound). Because feature selection can improve prediction performance even on random data, we repeated the entire analysis after shuffling the class assignments.
SVM prediction
To assess global classification performance, we used a binary SVM classifier (Boser et al. 1992) under the leave-one-out cross-validation (LOOCV) framework implemented by the R package e1071 with a radial basis function as kernel (default parameters). As features we used the motifs as described above and as attributes the number of motif instances (= PWM matches; see above) in each region (Fig. 2A). The performance (accuracy and ‘Area Under the Curve’ [AUC]) of all SVM predictions was computed by the R package ROCR (Sing et al. 2005) as was the average of 10 times bootstrapping the larger of both binding site classes to obtain two sets of equal sizes. We also used negative controls by randomly shuffling the class assignments of all binding sites and performing identical computations (see the Supplemental Discussion).
Calculation of prediction scores
We computed a prediction score for each binding site by bootstrapping SVM predictions using SVM-light (Joachims 1999) with a radial basis function as kernel and default parameters and a manually implemented leave-one-out cross-validation (LOOCV). For this, we excluded each binding site in turn for testing, trained 100 SVMs by randomly subsampling the remaining regions 100 times (i.e., choosing 10% of the sites in each class at random), and predicted the test site with all 100 SVMs; the score corresponds to the fraction of correct predictions (in percent).
In silico mutations
It is typically difficult to interpret the results of SVM-based predictions, especially with respect to the relevant features or feature combinations, i.e., TF motifs. To investigate the importance of a motif for the classification success of an individual region, we manually deleted all occurrences of that motif by setting the respective count in the region's feature vector to zero (thus in silico mutations) and recomputed the prediction score as described above (Fig. 4A). We considered drops of at least 20 points as substantial, because 20 points correspond to the difference of well-predicted enhancers and the best random scores (see Fig. 3B). This step is computationally intense because SVM predictions with LOOCV have to be run 100 times for each motif (37 motifs for the early vs. late comparison) and for each pair of motifs (37 × 36/2 = 666 pairwise combinations for the early vs. late comparison). Depending on the data set, the computations took typically between 500 and 1000 CPU-hours and were generally finished within a few hours on a cluster of 16 Sun Microsystem servers, each equipped with 2 AMD Opteron 2427 CPUs, 2.2 GHz (64 GB main memory, 300 GB local harddisk) and running Debian Linux (lenny) and a Sun Gridengine scheduling system. Despite the multiple testing situation for all TF motif pairs, the results remain specific because the majority of all affected binding sites were affected by the mutation of motif pairs that included the TTK (85%), KR (69%), and VFL/ME38 motifs (63%) for early Twist-binding sites, and BRK (59%) and ME17 (82%) for late Twist-binding sites, consistent with the results for individual motif in silico mutations (Fig. 4C).
Predictions across different Drosophila species
To predict binding sites across different Drosophila species (D. melanogaster, D. simulans, D. erecta, D. yakuba, D. ananasae, and D. pseudoobscura), we first retrieved the orthologous sequences for all D. melanogaster binding sites from a UCSC whole-genome alignment as in Stark et al. (2007) and scanned them for motifs as described above. Then, we excluded each D. melanogaster site in turn for testing, trained 100 SVMs by subsampling the remaining D. melanogaster regions as described above, and scored the test site in D. melanogaster and the orthologous regions in each of the other Drosophila species. We evaluated the across-species predictions by intersecting high- and low-scoring predictions with ChIP data from He et al. (2011) (using the set of peaks called with p ≤ 10−10). We evaluated the overall sequence conservation for Figure 5C in the 401-bp regions around the peak summits that we used for the predictions. phastCons scores were obtained from UCSC (Fujita et al. 2011), and percent nucleotide identity was calculated according to the UCSC BLASTZ/MultiZ multiple genome alignment of 14 insect species (Siepel et al. 2005).
Comparing prediction score to experimental ChIP reproducibility
To test the prediction scores, we assessed the number of high- and low-scoring early-binding sites that were reproduced by a recent ChIP-seq study (He et al. 2011) and evaluated the difference by a hypergeometric P-value.
Experimental validation
Cloning and transgenesis
Wild-type enhancers were PCR-amplified from genomic DNA, and mutagenized enhancers were chemically synthesized (Invitrogen) (sequences in the Supplemental Material). All were cloned into a pBPGUw vector, verified by Sanger sequencing, and integrated into an attP2 landing site on chromosome 3 (Pfeiffer et al. 2008). Finally, transgenic flies were confirmed by PCR and Sanger sequencing.
Whole-mount in situ hybridization
An antisense Gal4 RNA probe generated using the primers described in Pfeiffer et al. (2008) was used to visualize the expression of the Gal4 reporter gene in transgenic embryos. Briefly, embryos were collected, fixed, and hybridized with the digoxigenin-labeled antisense RNA probe followed by staining with anti-DIG FAB fragments coupled to alkaline phosphatase (Roche cat. no. 11093274910) as described in Lécuyer et al. (2008). Gal4 reporter RNA was visualized by addition of NBT/BCIP substrate (NBT, Roche cat. no. 11383213001; BCIP, Roche cat. no. 11383221001) to embryos. While AP staining is generally semiquantitative, we took great care that comparisons across different transgenic lines are valid: Reporter constructs differed only by the indicated motif-disrupting sequence changes and were inserted into the identical genomic positions (landing site), the identical probe was used in all cases, and hybridizations of control and embryos with mutated fragments were performed identically and in parallel.
Twist ChIP-qPCR
We performed three biological replicates ChIP-qPCR experiments from independent embryo collections. For each, we collected and snap-froze embryos at 2–4 hpf (stages 5–7) from two transgenic Drosophila lines (see above), which either carried the wild-type or the TAGteam mutant cact Gal4 reporter (cact enh-wt or cact enh-mut) (see above and the Supplemental Material). We prepared and fixed the embryos and extracted chromatin as described (Sandmann et al. 2006) from ∼0.5–0.8 g of embryos. We sonicated the chromatin to a product size of ∼400–800 bp using Bioruptor Standard (Diagenode) for two cycles (30 sec on/30 sec off) for biological replicates 1 and 3 and with a Tip sonicator (Omni Sonic Ruptor 250 Watt Ultrasonic Homogenizer) with four cycles (1 min on [Duty cycle 30%, Output 20%], 2 min off) for biological replicate 2. Approximately 30–50 μg of chromatin was incubated with 5 μL of rabbit anti-twist antibody (DRK2 bleed #1, a gift from Eileen Furlong, EMBL, Heidelberg) and precipitated with 50 μL of 50% protein A-Sepharose CL-4B beads suspension (GE Healthcare Lot # 10021997) according to Sandmann et al. (2006). The precipitated chromatin was assessed by quantitative PCR (qPCR) using three different primer pairs to probe the reporter construct, the endogenous cactus enhancer, and an unrelated genomic region (see the Supplemental Material and the Supplemental Discussion). We used an MyiQ single color RT-PCR detection system and SsoFast EvaGreen Supermix (Bio-Rad laboratories, Inc. cat no. 172-5204; qPCR conditions were: 3 min at 95°C; 20 sec at 95°C, 20 sec at 64°C, 20 sec at 72°C [50 cycles]; melting curve: 30 sec at 95°C, 1 min at 55°C; ramp from 55°C to 95°C with 1°C increment for 30 sec [41 cycles]). We first calculated the percent recovery of each of the three loci during the IP (IP vs. input) and then the enrichments compared with the endogenous cact locus or an unrelated genomic region as indicated (this controls for varying IP efficiencies).
Data access
The list of transcription factor (TF) motifs, genomic coordinates of TF binding sites, the motif matches used as SVM input, and results can be downloaded from http://www.starklab.org/data.
Acknowledgments
We thank Wolfgang Lugmayr (IMP) for administering the IMP/IMBA compute-cluster, Michaela Pagani and Katharina Schernhuber (IMP) for excellent technical support, and Gerald Stampfel (IMP) for help with image processing. We are grateful to Eileen Furlong (EMBL) for sharing the anti-Twist antibody, and to her and Robert Zinzen (EMBL) for help with the ChIP-qPCR experiments. J.O.Y-C. was supported by the Austrian Ministry for Science and Research through the GEN-AU Bioinformatics Integration Network III. H.Q.D. is supported by a GMI grant to Ortrun Mittelsten-Scheid and a WWTF grant to Arndt von Haeseler. D.S. is supported by an ERC Starting Grant from the European Community's Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement no. 242922 awarded to A.S. Basic research at the IMP is supported by Boehringer Ingelheim GmbH.
Author contributions: J.O.Y-C. and H.Q.D. performed the computational analyses and predictions. E.Z.K. cloned the reporter constructs and performed the in situ experiments. J.O.Y-C. and E.Z.K. established the transgenic Drosophila stocks. D.S. performed the ChIP-qPCR experiments. A.S. conceived, planned, and supervised the project. J.O.Y-C. and A.S. analyzed the data and wrote the paper.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.132811.111.
Freely available online through the Genome Research Open Access option.
References
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. 2000. Gene Ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azpiazu N, Frasch M 1993. tinman and bagpipe: Two homeo box genes that determine cell fates in the dorsal mesoderm of Drosophila. Genes Dev 7: 1325–1340 [DOI] [PubMed] [Google Scholar]
- Banerji J, Rusconi S, Schaffner W 1981. Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell 27: 299–308 [DOI] [PubMed] [Google Scholar]
- Banerji J, Olson L, Schaffner W 1983. A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy chain genes. Cell 33: 729–740 [DOI] [PubMed] [Google Scholar]
- Barolo S, Posakony JW 2002. Three habits of highly effective signaling pathways: Principles of transcriptional control by developmental cell signaling. Genes Dev 16: 1167–1181 [DOI] [PubMed] [Google Scholar]
- Berman BP, Nibu Y, Pfeiffer BD, Tomancak P, Celniker SE, Levine M, Rubin GM, Eisen MB 2002. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc Natl Acad Sci 99: 757–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard FJ, Collins B, Cyran SA, Hancock DH, Taylor MV, Blau J 2010. The transcription factor Mef2 is required for normal circadian behavior in Drosophila. J Neurosci 30: 5855–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boser BE, Guyon I, Vapnik VN 1992. A training algorithm for optimal margin classifiers. In Proceedings of the Fifth Annual Workshop on Computational Learning Theory, pp. 144–152. http://dl.acm.org/citation.cfm?id=130385 [Google Scholar]
- Bradley RK, Li X-Y, Trapnell C, Davidson S, Pachter L, Chu HC, Tonkin LA, Biggin MD, Eisen MB 2010. Binding site turnover produces pervasive quantitative changes in transcription factor binding between closely related Drosophila species. PLoS Biol 8: e1000343. doi: 10.1371/journal.pbio.1000343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryne JC, Valen E, Tang M-HE, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A 2008. JASPAR, the open access database of transcription factor-binding profiles: New content and tools in the 2008 update. Nucleic Acids Res 36: D102–D106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MJ, Lieb JD 2006. A chromatin-mediated mechanism for specification of conditional transcription factor targets. Nat Genet 38: 1446–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L-W, Nagarajan R, Magee JA, Milbrandt J, Stormo GD 2006. A systematic model to predict transcriptional regulatory mechanisms based on overrepresentation of transcription factor binding profiles. Genome Res 16: 405–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle HJ, Kraut R, Levine M 1989. Spatial regulation of zerknüllt: A dorsal–ventral patterning gene in Drosophila. Genes Dev 3: 1518–1533 [DOI] [PubMed] [Google Scholar]
- Frith MC, Fu Y, Yu L, Chen J-F, Hansen U, Weng Z 2004. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res 32: 1372–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka M, Lear BC, Landgraf M, Yusibova GL, Zhou J, Riley KM, Patel NH, Jaynes JB 2003. Even-skipped, acting as a repressor, regulates axonal projections in Drosophila. Development 130: 5385–5400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, et al. 2011. The UCSC Genome Browser database: Update 2011. Nucleic Acids Res 39: D876–D882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Zaragoza E, Mas JA, Vivar J, Arredondo JJ, Cervera M 2008. CF2 activity and enhancer integration are required for proper muscle gene expression in Drosophila. Mech Dev 125: 617–630 [DOI] [PubMed] [Google Scholar]
- Giese K, Cox J, Grosschedl R 1992. The HMG domain of lymphoid enhancer factor 1 bends DNA and facilitates assembly of functional nucleoprotein structures. Cell 69: 185–195 [DOI] [PubMed] [Google Scholar]
- Guss KA, Nelson CE, Hudson A, Kraus ME, Carroll SB 2001. Control of a genetic regulatory network by a selector gene. Science Magazine 292: 1164–1167 [DOI] [PubMed] [Google Scholar]
- Guyon I 2003. An introduction to variable and feature selection. J Mach Learn Res 3: 1157–1182 [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, MacIsaac KD, Danford TW, Hannett NM, Tagne J-B, Reynolds DB, Yoo J, et al. 2004. Transcriptional regulatory code of a eukaryotic genome. Nature 431: 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison MM, Li X-Y, Kaplan T, Botchan MR, Eisen MB 2011. Zelda binding in the early Drosophila melanogaster embryo marks regions subsequently activated at the maternal-to-zygotic transition. PLoS Genet 7: e1002266 doi: 10.1371/journal.pgen.1002266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Sinha S 2010. ChIPs and regulatory bits. Nat Biotechnol 28: 142–143 [DOI] [PubMed] [Google Scholar]
- He Q, Bardet AF, Patton B, Purvis J, Johnston J, Paulson A, Gogol M, Stark A, Zeitlinger J 2011. High conservation of transcription factor binding and evidence for combinatorial regulation across six Drosophila species. Nat Genet 43: 414–420 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Sui SJ, Mortimer JR, Arenillas DJ, Brumm J, Walsh CJ, Kennedy BP, Wasserman WW 2005. oPOSSUM: Identification of over-represented transcription factor binding sites in co-expressed genes. Nucleic Acids Res 33: 3154–3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger J, Surkova S, Blagov M, Janssens H, Kosman D, Kozlov KN, Manu, Myasnikova E, Vanario-Alonso CE, Samsonova M, et al. 2004. Dynamic control of positional information in the early Drosophila embryo. Nature 430: 368–371 [DOI] [PubMed] [Google Scholar]
- Janssens H, Hou S, Jaeger J, Kim A-R, Myasnikova E, Sharp D, Reinitz J 2006. Quantitative and predictive model of transcriptional control of the Drosophila melanogaster even skipped gene. Nature 38: 1159–1165 [DOI] [PubMed] [Google Scholar]
- Joachims T. 1999 Making large-scale SVM learning practical. In Advances in kernel methods—support vector learning (ed. B Schölkopf et al.). MIT Press, Boston. [Google Scholar]
- John S, Sabo PJ, Thurman RE, Sung M-H, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA 2011. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet 43: 264–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan T, Li X-Y, Sabo PJ, Thomas S, Stamatoyannopoulos JA, Biggin MD, Eisen MB 2011. Quantitative models of the mechanisms that control genome-wide patterns of transcription factor binding during early Drosophila development. PLoS Genet 7: e1001290 doi: 10.1371/journal.pgen.1001290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemian M, Blatti C, Richards A, McCutchan M, Wakabayashi-Ito N, Hammonds AS, Celniker SE, Kumar S, Wolfe SA, Brodsky MH, et al. 2010. Quantitative analysis of the Drosophila segmentation regulatory network using pattern generating potentials. PLoS Biol 8: e1000456 doi: 10.1371/journal.pbio.1000456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradpour P, Stark A, Roy S, Kellis M 2007. Reliable prediction of regulator targets using 12 Drosophila genomes. Genome Res 17: 1919–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhamer CV, Yuh CH, Davidson EH 1996. Modular cis-regulatory organization of developmentally expressed genes: Two genes transcribed territorially in the sea urchin embryo, and additional examples. Proc Natl Acad Sci 93: 9322–9328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 doi: 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lécuyer E, Parthasarathy N, Krause HM 2008. Fluorescent in situ hybridization protocols in Drosophila embryos and tissues. Methods Mol Biol 420: 289–302 [DOI] [PubMed] [Google Scholar]
- Lee D, Karchin R, Beer MA 2011. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res 21: 2167–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-Y, MacArthur S, Bourgon R, Nix D, Pollard DA, Iyer VN, Hechmer A, Simirenko L, Stapleton M, Luengo Hendriks CL, et al. 2008. Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS Biol 6: e27 doi:10.1371/journal.pbio.0060027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-Y, Thomas S, Sabo PJ, Eisen MB, Stamatoyannopoulos JA, Biggin MD 2011. The role of chromatin accessibility in directing the widespread, overlapping patterns of Drosophila transcription factor binding. Genome Biol 12: R34 doi: 10.1186/gb-2011-12-4-r34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H-L, Nien C-Y, Liu H-Y, Metzstein MM, Kirov N, Rushlow C 2008. The zinc-finger protein Zelda is a key activator of the early zygotic genome in Drosophila. Nature 456: 400–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienert F, Wirbelauer C, Som I, Dean A, Mohn F, Schübeler D 2011. Identification of genetic elements that autonomously determine DNA methylation states. Nat Genet 43: 1091–1097 [DOI] [PubMed] [Google Scholar]
- Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, et al. 2010. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol 11: 635–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur S, Li X-Y, Li J, Brown JB, Chu HC, Zeng L, Grondona BP, Hechmer A, Simirenko L, Keränen SVE, et al. 2009. Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions. Genome Biol 10: R80 doi: 10.1186/gb-2009-10-7-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markstein M, Zinzen R, Markstein P, Yee K-P, Erives A, Stathopoulos A, Levine M 2004. A regulatory code for neurogenic gene expression in the Drosophila embryo. Development 131: 2387–2394 [DOI] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gössling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. 2003. TRANSFAC: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res 31: 374–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, Tomsho LP, Qi J, Glaser RL, Schuster SC, et al. 2008. Nucleosome organization in the Drosophila genome. Nature 453: 358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AC, Orlando DA, Newman JJ, Lovén J, Kumar RM, Bilodeau S, Reddy J, Guenther MG, DeKoter RP, Young RA 2011. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 147: 565–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newburger DE, Bulyk ML 2009. UniPROBE: An online database of protein binding microarray data on protein–DNA interactions. Nucleic Acids Res 37: D77–D82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nien C-Y, Liang H-L, Butcher S, Sun Y, Fu S, Gocha T, Kirov N, Manak JR, Rushlow C 2011. Temporal coordination of gene networks by Zelda in the early Drosophila embryo. PLoS Genet 7: e1002339 doi: 10.1371/journal.pgen.1002339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshiro T, Saigo K 1997. Transcriptional regulation of breathless FGF receptor gene by binding of TRACHEALESS/dARNT heterodimers to three central midline elements in Drosophila developing trachea. Development 124: 3975–3986 [DOI] [PubMed] [Google Scholar]
- Palii CG, Perez-Iratxeta C, Yao Z, Cao Y, Dai F, Davison J, Atkins H, Allan D, Dilworth FJ, Gentleman R, et al. 2011. Differential genomic targeting of the transcription factor TAL1 in alternate haematopoietic lineages. EMBO J 30: 494–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson JC, Lemons D, McGinnis W 2005. Modulating Hox gene functions during animal body patterning. Nat Rev Genet 6: 893–904 [DOI] [PubMed] [Google Scholar]
- Pfeiffer BD, Jenett A, Hammonds AS, Ngo T-TB, Misra S, Murphy C, Scully A, Carlson JW, Wan KH, Laverty TR, et al. 2008. Tools for neuroanatomy and neurogenetics in Drosophila. Proc Natl Acad Sci 105: 9715–9720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard DK, Schubiger G 1996. Activation of transcription in Drosophila embryos is a gradual process mediated by the nucleocytoplasmic ratio. Genes Dev 10: 1131–1142 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470: 279–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmann T, Jensen LJ, Jakobsen JS, Karzynski MM, Eichenlaub MP, Bork P, Furlong EEM 2006. A temporal map of transcription factor activity: Mef2 directly regulates target genes at all stages of muscle development. Dev Cell 10: 797–807 [DOI] [PubMed] [Google Scholar]
- Sandmann T, Girardot C, Brehme M, Tongprasit W, Stolc V, Furlong EEM 2007. A core transcriptional network for early mesoderm development in Drosophila melanogaster. Genes Dev 21: 436–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Raveh-Sadka T, Schroeder M, Unnerstall U, Gaul U 2008. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature 451: 535–540 [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing T, Sander O, Beerenwinkel N, Lengauer T 2005. ROCR: Visualizing classifier performance in R. Bioinformatics 21: 3940–3941 [DOI] [PubMed] [Google Scholar]
- Stanojevic D, Small S, Levine M 1991. Regulation of a segmentation stripe by overlapping activators and repressors in the Drosophila embryo. Science 254: 1385–1387 [DOI] [PubMed] [Google Scholar]
- Stark A 2009. Learning the transcriptional regulatory code. Mol Syst Biol 5: 1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, Lin MF, Kheradpour P, Pedersen JS, Parts L, Carlson JW, Crosby MA, Rasmussen MD, Roy S, Deoras AN, et al. 2007. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature 450: 219–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Bosch JR, Benavides JA, Cline TW 2006. The TAGteam DNA motif controls the timing of Drosophila pre-blastoderm transcription. Development 133: 1967–1977 [DOI] [PubMed] [Google Scholar]
- Tran KD, Miller MR, Doe CQ 2010. Recombineering Hunchback identifies two conserved domains required to maintain neuroblast competence and specify early-born neuronal identity. Development 137: 1421–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trompouki E, Bowman TV, Lawton LN, Fan ZP, Wu D-C, DiBiase A, Martin CS, Cech JN, Sessa AK, Leblanc JL, et al. 2011. Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell 147: 577–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzi MP, Shin H, He HH, Sulahian R, Meyer CA, Montgomery RK, Fleet JC, Brown M, Liu XS, Shivdasani RA 2010. Differentiation-specific histone modifications reveal dynamic chromatin interactions and partners for the intestinal transcription factor CDX2. Dev Cell 19: 713–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457: 854–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss TC, Schiltz RL, Sung M-H, Yen PM, Stamatoyannopoulos JA, Biddie SC, Johnson TA, Miranda TB, John S, Hager GL 2011. Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell 146: 544–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilczynski B, Furlong EEM 2010. Dynamic CRM occupancy reflects a temporal map of developmental progression. Mol Syst Biol 6: 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger J, Simon I, Harbison CT, Hannett NM, Volkert TL, Fink GR, Young RA 2003. Program-specific distribution of a transcription factor dependent on partner transcription factor and MAPK signaling. Cell 113: 395–404 [DOI] [PubMed] [Google Scholar]
- Zeitlinger J, Zinzen RP, Stark A, Kellis M, Zhang H, Young RA, Levine M 2007. Whole-genome ChIP-chip analysis of Dorsal, Twist, and Snail suggests integration of diverse patterning processes in the Drosophila embryo. Genes Dev 21: 385–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137 doi: 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M, Niu W, Lu ZJ, Sarov M, Murray JI, Janette J, Raha D, Sheaffer KL, Lam HYK, Preston E, et al. 2010. Genome-wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA-4/FOXA in development and environmental response. PLoS Genet 6: e1000848 doi: 10.1371/journal.pgen.1000848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzen RP, Girardot C, Gagneur J, Braun M, Furlong EEM 2009. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature 462: 65–70 [DOI] [PubMed] [Google Scholar]