

Abstract
Death receptor 3 (DR3, TNFRSF25), the closest family relative to tumor necrosis factor receptor 1, promotes CD4+ T-cell-driven inflammatory disease. We investigated the in vivo role of DR3 and its ligand TL1A in viral infection, by challenging DR3-deficient (DR3KO) mice and their DR3WT littermates with the β-herpesvirus murine cytomegalovirus or the poxvirus vaccinia virus. The phenotype and function of splenic T-cells were analyzed using flow cytometry and molecular biological techniques. We report surface expression of DR3 by naive CD8+ T cells, with TCR activation increasing its levels 4-fold and altering the ratio of DR3 splice variants. T-cell responses were reduced up to 90% in DR3KO mice during acute infection. Adoptive transfer experiments indicated this was dependent on T-cell-restricted expression of DR3. DR3-dependent CD8+ T-cell expansion was NK and CD4 independent and due to proliferation, not decreased cell death. Notably, impaired immunity in DR3KO hosts on a C57BL/6 background was associated with 4- to 7-fold increases in viral loads during the acute phase of infection, and in mice with suboptimal NK responses was essential for survival (37.5%). This is the first description of DR3 regulating virus-specific T-cell function in vivo and uncovers a critical role for DR3 in mediating antiviral immunity.
Keywords: murine cytomegalovirus, vaccinia virus
The tumor necrosis factor receptor superfamily (TNFRSF) is a group of structurally related cytokine receptors, through which ligand-induced signaling is critical for orchestrating the formation, maintenance and function of the peripheral immune system (1–4). Death receptor 3 (DR3, TRAMP, LARD, WSL-1, Apo-3, TR3, TNFRSF25) is one of the more recently discovered TNFRSF members and belongs to a subdivision that contains a death domain, an intracellular region that can recruit TNFR1-associated death domain (TRADD) and Fas-associated death domain (FADD) proteins as downstream effectors of apoptosis following receptor overexpression (5–10) or binding of its ligand, TNF-like protein 1A (TL1A, TNFSF15; refs. 11, 12). However, DR3 can also recruit TRAF2 via TRADD, thus activating the transcription factor NFκB, which, in turn, induces multiple immune activation and cell survival genes (11, 13). This contextual multifunctionality has been borne out in in vivo studies of mice genetically deficient for DR3 expression or overexpressing TL1A. DR3 regulates T-cell receptor (TCR)-triggered death of thymocytes (14) but is also expressed by natural killer T (NKT), CD4+ T and myeloid cells (15–20), through which DR3 controls the induction and immunopathology of several autoimmune and inflammatory diseases, including inflammatory arthritis, experimental autoimmune encephalomyelitis, colitis, ileitis, allergic lung inflammation, and experimental autoimmune uveoretinitis (16, 19–27). DR3 expression can also be triggered on tubular epithelial cells, through which this axis controls responses to renal inflammation and injury (28, 29). To date, essential in vivo roles for DR3 in immunity to viruses have not been reported.
Murine cytomegalovirus (MCMV) is a double-stranded β-herpesvirus endemic in most rodent populations, which has been used extensively to examine host-pathogen relationships and adaptive immunity (30–32). During acute MCMV infection, the dominant cellular antiviral response consists of the development and expansion of a large pool of CD8+ T cells directed against several virus antigens (33–36). Notably, sustained cellular immune control by antiviral CD8+ T cells is required for active suppression of latent MCMV reactivation (33). In addition, MCMV-specific CD8+ T-cell lines and MCMV-primed polyclonal CD8+ T cells protect against MCMV infection on adoptive transfer into immune-deficient hosts (31). Thus, CD8+ T cells play an important role in the provision of long-term antiviral immunity against cytomegalovirus infection (31, 37).
We examined the role of DR3 during acute MCMV infection in DR3-deficient DR3-knockout (DR3KO) mice, paying particular attention to DR3-dependent effects on CD8+ T cells. Here we report constitutive surface expression of DR3 on CD8+ T cells that is tightly regulated following activation. DR3KO mice exhibited compromised CD4+ and CD8+ T-cell expansion following MCMV challenge that was associated with impaired control of virus replication. These findings reveal that DR3 can orchestrate immune responses elicited by evolutionarily divergent viruses and suggest that manipulation of this pathway can promote T-cell immunity to pathogens.
MATERIALS AND METHODS
Mice
All MCMV experiments were undertaken on 8- to 10-wk-old Thy1.1+ or Thy1.2+ DR3 wild type (DR3WT) and DR3KO mice (obtained from Cancer Research UK; ref. 14) on a C57BL/6 or DBA-1 background (7 backcrosses) bred in-house, as described previously (38). All procedures were performed in strict accordance with UK Home Office project licenses 30/2580 and 30/2442.
Cellular staining and flow cytometry
Splenocytes were isolated, treated with red blood cell lysis buffer, incubated with Fc block (BD Pharmingen, San Diego, CA, USA) and stained with combinations of preconjugated α-mouse CD44, NK1.1, CD8, CD4, β-TCR (BD Pharmingen), CD25 (Invitrogen, Carlsbad, CA, USA) and CD69 (eBioscience, San Diego, CA, USA) mAbs. In some experiments, MCMV-specific CD4 and CD8 T cells were identified following ex vivo peptide stimulation, as described previously (39). Cell proliferation and apoptosis were detected using the Ki67 set and annexin-V apoptosis detection kit (both BD Pharmingen), respectively. In some experiments, after 30 min blocking with 5% normal goat serum (Vector, Burlingame, CA, USA), splenocytes were stained with polyclonal α-mouse DR3-biotin (BAF 2437; R&D Systems, Minneapolis, MN, USA) followed by SA-APC (Molecular Probes, Eugene, OR, USA). To evaluate the effects of TL1A on proliferation and TCR-induced DR3 expression, whole splenocytes, or splenic CD8+ T cells isolated to >98% purity using magnetic selection (Miltenyi Biotec, Bergisch Gladbach, Germany), were incubated in 96-well plates (1×105 cells/well) with combinations of murine sTL1A (50 ng/ml; R&D Systems), anti-mouse CD3 (10 μg/ml; clone 145-2C11, plate bound) and anti-CD28 (0.5 μg/ml; clone 37.51, plate bound; BD Pharmingen) for up to 4 d before anti-DR3, CD4, CD8, CD25, CD44, and CD69 staining. Isolated T cells were prelabeled with 0.25 μM carboxy-fluorescein succinyl ester (CFSE) as described previously (15). Data were acquired either on a FACSCalibur (BD Biosciences, San Jose, CA, USA) or a CyAn ADP flow cytometer (Beckman Coulter, Fullerton, CA, USA) and analyzed with CellQuest Pro (BD Biosciences), Summit (Beckman Coulter) or FlowJo (Tree Star, Ashland, OR , USA) software. Cell numbers were determined using a BD Accuri C6 flow cytometer and CFlow software (BD Biosciences).
Splice variant analysis
CD8+ T cells were isolated by positive magnetic selection (Miltenyi Biotec) from OT-1-derived spleen and lymph node cells with or without activation using 100 pM SIINFEKL peptide for 24h. RNA was extracted using the RNeasy mini kit, cDNA synthesized from 2.5 μg of total RNA using the Superscript III first strand synthesis system (Invitrogen, Carlsbad, CA, USA) and DR3 transcripts amplified using the following primers (5′–3′): for Fig. 1C, forward TGACCCTTGAGAACTGCTCGG and reverse GGATAGCCCCAAAAAGGAACGC; for cloning and sequencing, a nested PCR using forward F1F-CAGAGCCGCACTCACAAGGG, forward F1N-GGTACACACCGCAATGGAGG, reverse R1F-CAGAGCCGCACTCACAAGGG, and forward R1N-GATGGACCTTCCATCACGGGC.
Figure 1.

Expression of DR3 on splenocytes. A–C) DR3 expression on NK and NKT (A), CD4+ T (B), and CD8+ T (C) cells. Representative flow cytometric dotplots of splenic NK1.1+ lymphocytes stained for TCRβ, and CD4+ or CD8+ T cells stained for CD44, DR3, or control Ig from DR3WT and DR3KO mice. D) Preferential expression of DR3 on naive CD44lo rather than antigen-experienced CD44hi CD4+ and CD8+ T cells. Each symbol represents a different DR3WT mouse.
Virus, mouse infections, and treatments
C57BL/6 and DBA mice were infected i.p. with 5 × 104 to 2 × 105 or 1–4 × 104 plaque-forming units (PFU) of MCMV Smith strain [American Type Culture Collection (ATCC), Manassas, VA, USA], respectively, isolated and purified from the salivary glands of infected Balb/C mice as described previously (40). Mice were sacrificed between d 0 and 32 postinfection, and MCMV tissue titers (spleen, kidney, liver, lungs, and salivary glands) were determined by titration on 3T3 cells (40). In some cases, mice were treated on d −2 and 0 with anti-CD4 mAbs (clones YTA3.1.2 and YTA 191.1.2, 100 μg of each). In other experiments, Thy1.2+ DR3WT or DR3KO T cells were isolated using MACS (Miltenyi Biotec) purification kits, and 4 × 106 cells were adoptively transferred into congenic Thy1.1+ mice before MCMV challenge. For recombinant vaccinia virus (rVV) challenge, mice were challenged i.p. with 5 × 106 PFU rVV, tissues were removed on d 4, 8, and 12, and rVV titers were determined as described previously (41).
Statistics
All statistical analysis was performed using GraphPad Prism software (GraphPad, San Diego, CA, USA). Values of P < 0.05 were considered statistically significant and are included in the figures.
RESULTS
DR3 is expressed by CD8+ T cells and regulated following activation
Cell surface expression of DR3 was investigated on subpopulations of immune cells using an anti-DR3 polyclonal Ab and DR3KO splenocytes as a control for specificity. In agreement with previous reports (20), cell surface DR3 was found on NK1.1+TCRβ+ NKT (Fig. 1A) and CD4+ T cells (Fig. 1B) in the spleen but was absent from NK1.1+TCRβ− NKT cells (Fig. 1A). DR3 expression on CD4+ T cells was found on the majority of both CD44lo naive (mean±se; 87±0.6%) and CD44hi (55±1.5%) antigen-experienced cells (Fig. 1B, D). In contrast, preferential DR3 surface expression was observed on naive CD44loCD25lo CD8+ T cells (66±1.5%), with considerably less on the antigen-experienced CD44hiCD25hi subset (20±1.5%) from DR3WT mice (Fig. 1C, D). Overall expression of DR3 relative to control staining was higher on CD4+ T cells compared to CD8+ T cells (Fig. 1). DR3 surface expression on T cells was also monitored in whole-splenocyte and purified T-cell cultures after stimulation with agonistic anti-CD3 mAb. Following activation, DR3 was up-regulated in distinct patterns depending on the T-cell subset. For CD4+ T cells, DR3 expression increased ~4-fold to peak at 72 h poststimulation there-after. In comparison, CD8+ T cells up-regulated DR3 more rapidly, peaking at 24 h before declining to reach baseline levels by 72 h (Fig. 2A).
Figure 2.

Kinetics of DR3 expression by T cells. A) Kinetics of DR3 expression by CD4+ and CD8+ T cells following stimulation through the TCR. Values represent relative mean fluorescence, calculated as the ratio of geometric mean fluorescence intensity of the sample above negative control staining, following stimulation without (○), or with anti-CD3 mAb (●) on TCRβ+ lymphocytes stained using anti-DR3 and analyzed by flow cytometry. B, C) Expression of DR3 splice variants from CD8+ T cells following TCR stimulation. RNA was isolated from naive and activated OT-1 CD8+ DR3WT T cells; DR3 transcripts were amplified, cloned, and sequenced as described in Materials and Methods. B) RT-PCR for DR3 and β-actin before and after activation. C) Splice variant analysis and frequency of sequenced clones from CD8+ T cells after activation. Arrows mark the position of primers. CRR, cysteine-rich region; TM, transmembrane; DD, death domain. Numbers and boxes indicate coding exons. Plus symbol denotes position of ATG. Asterisk denotes position of stop codon.
Several alternative DR3 mRNAs have been isolated from peripheral immune cells in mice and humans (5, 10, 42, 43) and alterations in splice variant expression have been described for T cells following activation in both species (10, 21). Considering the paucity of information on DR3 and murine CD8+ T cells, we examined splice variant expression in this subset, using RT-PCR, cloning, and sequencing for DR3 transcripts. To ensure specific activation, naive CD8+ T cells from OT-1 transgenic DR3WT mice were purified and stimulated with the OT-1 TCR-specific SIINFEKL peptide. Naive OT-1 CD8+ T cells expressed low levels of both the truncated (variant III) and full-length (variant I) transcripts of membrane-bound DR3 (Fig. 2B, C). In contrast, OT-1 CD8+ T cells activated for 24 h expressed more abundant levels of the full-length DR3 mRNA transcript, as well as significant levels of variant II encoding for a predicted soluble form of DR3 (Fig. 2B, C).
DR3 is required for optimal adaptive antiviral T-cell responses
To evaluate the role of DR3 during virus infection in vivo, we challenged DR3WT and DR3KO mice with MCMV and followed the dynamics of immune responses early during MCMV infection. The mouse spleen represents a site of robust virus-specific T- and NK cell responses during early infection by MCMV (44, 45). Kinetics of splenic NK cell expansion were similar between DR3WT and DR3KO mice following MCMV challenge (Fig. 3Ai). Critically, however, large alterations were observed in CD4+ and CD8+ T-cell kinetics. CD4+ T-cell expansion in DR3WT mice was significantly larger than in DR3KO mice 6 d after challenge (Fig. 3Aii). In contrast, significant differences in CD8+ T-cell expansion were observed throughout the time course of early MCMV infection (Fig. 3Aiii). At its peak on d 6, CD4+ T-cell numbers were 2.4-fold higher in MCMV-challenged DR3WT mice compared to unchallenged DR3WT controls, while expansion in CD4+ DR3KO mice was virtually absent (1.1-fold higher). This equated to a 90% reduction in CD4+ T-cell expansion and 86% decrease in numbers in DR3KO mice following MCMV challenge (Fig. 3B). A similar pattern was observed in CD8+ T cells, with MCMV-challenged DR3WT mice showing 3.3-fold higher numbers than unchallenged DR3WT controls, compared to 2.4-fold in their DR3KO littermates. This was equivalent to a 37% reduction in CD8+ T-cell expansion and 57% decrease in numbers (Fig. 3B). This DR3-dependent defect in T-cell expansion following viral challenge was not restricted to MCMV, as it was also observed after rVV infection. To assess whether DR3 regulated T-cell immunity to other virus infection, we challenged DR3-deficient mice with rVV. At 8 d after rVV infection, CD4+ and CD8+ T-cell numbers had increased 2.9- and 5.7-fold in DR3WT mice, respectively, compared to 1.7- and 3.3-fold in DR3KO littermates (Fig. 3C, D), thus demonstrating that DR3 is a critical regulator of T-cell responses elicited by evolutionarily diverse viruses.
Figure 3.

Regulation of T-cell responses by DR3 following viral infection. A) Time courses showing numbers of NK (i), CD4+ T (ii), and CD8+ T (iii) cells in the spleens from DR3WT and DR3KO mice following MCMV challenge. DR3WT receiving PBS (●) or MCMV (▲); DR3KO receiving PBS (○) or MCMV (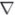 ). Each data point represents mean ± se from n ≥ 5 mice and is representative of 4 separate experiments. *P < 0.05 at indicated time points; Student’s t test. B) Relative expansion of CD4+ and CD8+ T cells compared to unchallenged controls following MCMV challenge at peak (d 6 for CD4+, d 7 for CD8+). Bars show means ± se from n ≥ 8 mice. P values were determined by Mann-Whitney U test. C) Time courses showing numbers of total splenocytes (i), CD4+ T cells (ii), and CD8+ T cells (iii) in the spleens from DR3WT and DR3KO mice following rVV challenge. DR3WT receiving PBS (●) or rVV (▲); DR3KO receiving PBS (○) or rVV (). Each data point represents mean ± se from n ≥ 6 mice. * P < 0.05 at indicated time points; Student’s t test. D) Relative expansion of CD4+ and CD8+ T cells compared to unchallenged controls following rVV challenge at d 8. Bars show means ± se from n ≥ 6 mice. P values were determined by Mann-Whitney U test.
). Each data point represents mean ± se from n ≥ 5 mice and is representative of 4 separate experiments. *P < 0.05 at indicated time points; Student’s t test. B) Relative expansion of CD4+ and CD8+ T cells compared to unchallenged controls following MCMV challenge at peak (d 6 for CD4+, d 7 for CD8+). Bars show means ± se from n ≥ 8 mice. P values were determined by Mann-Whitney U test. C) Time courses showing numbers of total splenocytes (i), CD4+ T cells (ii), and CD8+ T cells (iii) in the spleens from DR3WT and DR3KO mice following rVV challenge. DR3WT receiving PBS (●) or rVV (▲); DR3KO receiving PBS (○) or rVV (). Each data point represents mean ± se from n ≥ 6 mice. * P < 0.05 at indicated time points; Student’s t test. D) Relative expansion of CD4+ and CD8+ T cells compared to unchallenged controls following rVV challenge at d 8. Bars show means ± se from n ≥ 6 mice. P values were determined by Mann-Whitney U test.
As an indication of function, cytokine release of antiviral CD8+ and CD4+ T cells was also measured against select viral proteins directly ex vivo, including the dominant MHC I-restricted responses to M38, M45, and m139 and the MHC II-restricted response to M25 (44, 46–48). The proportion of T cells producing IFNγ in response to any of these peptides was not significantly different between DR3WT and DR3KO mice in any direct comparison (Fig. 4A), but DR3KO mice recorded lower responses in 11 of 13 data points across 3 separate experiments, and 2-way ANOVA indicated a significant contribution by genotype (Supplemental Table S1), suggesting overall that DR3KO responses were marginally smaller. Other readouts, including the proportion of T cells releasing TNF-α or degranulating as measured by recirculation of surface CD107, also showed no significant differences (Fig. 4B), suggesting that priming was intact in the absence of DR3. However, DR3KO mice generated fewer virus-specific CD4+ and CD8+ T cells during the peak of the primary T-cell response to MCMV when compared to their DR3WT counterparts, as revealed by quantification of virus-specific IFNγ-expressing T-cell numbers (Fig. 4C). Intriguingly, this impairment in development of early antiviral immunity was not sustained into late immune responses. When secondary memory responses were studied 32 d after infection, numbers of cytokine-producing CD4+ and CD8+ T cells were similar in MCMV-infected DR3WT and DR3KO mice (Fig. 4D).
Figure 4.

Antigen-specific responses following MCMV infection in the absence of DR3. A) IFN response of T cells in DR3WT and DR3KO mice following MCMV challenge. Representative flow cytometric dotplots of T cells showing CD8 and intracellular IFNγ staining in spleens from DR3WT and DR3KO mice, 7 d after MCMV challenge, to the M45 peptide. B) Intracellular staining for TNF-α and circulation of CD107a on splenic CD8+ T cells in response to M45 and m139 peptides following MCMV challenge. Values are means ± se of n ≥ 6 mice. C, D) Numbers of IFNγ-producing CD4+ T cells specific for the indicated peptide (left panel) and CD8+ T cells to IE3, M38, M45 and m139 in DR3WT (▲) and DR3KO () mice (right panel) either 7 d (C) or 32 d (D) following MCMV challenge. Bars indicate means. Each symbol represents a mouse. One of 3 experiments is shown. P values were determined by Student’s t test.
DR3/TL1A pathway influences the control of viral infection in vivo
To study the importance of DR3 in the control of viral infection, the kinetics of MCMV growth in the peripheral tissues of DR3WT and DR3KO mice was determined by plaque titration ex vivo. In C57BL/6 mice, control of early MCMV infection (before d 4) is facilitated by expression of the Ly49H receptor on NK cells, which recognizes the MCMV-encoded protein m157 (49). The absence of DR3 on NK cells (Fig. 1A), the finding that there were no significant differences in splenic NK cell numbers between DR3WT and DR3KO mice 4 d after infection (Fig. 3A), and the fact that low-dose (5×104 PFU/mouse) MCMV challenge resulted in very low levels of detectable plaque-forming virus up to 7 d after infection in both DR3WT and DR3KO mice (data not shown), led us to perform high-dose (2×105 PFU/mouse) challenge experiments to allow easier comparison of plaque-forming virus titers. High-titer challenge resulted in accelerated weight loss in DR3KO compared to DR3WT mice at early time points prior to 4 d, though the animals had recovered by d 6, and the overall curve was not significantly different when analyzed using 2-way ANOVA (Fig. 5A). On d 7 at the peak of the CD8+ T-cell response, increased MCMV titers were observed in 4 of the 5 tissues tested (liver, lung, spleen, and salivary glands) from DR3KO mice compared to their DR3WT littermates. Between 4- and 7-fold more replicating virus was recovered in DR3KO mice, depending on the organ studied (Fig. 5B). A similar increase in viral titer was observed in the ovaries of DR3KO mice following rVV infection (Fig. 5C). These data are consistent with the hypothesis that DR3/TL1A signaling is essential for an optimal T-cell response to either MCMV or rVV, disruption of which impairs control of these viruses.
Figure 5.

Weight loss and viral titers in the absence of DR3. A) Weight loss in DR3WT or DR3KO mice on a C57Bl/6 background following infection with MCMV. No significant difference observed using 2-way ANOVA. B, C) MCMV (B) and rVV (C) titers (PFU/g tissue) in the indicated tissues of C57Bl/6 DR3WT or DR3KO mice at d 7 following MCMV or rVV challenge, respectively. Bars represent median and interquartile range of n ≥ 6 mice. D) Weight loss in DR3WT or DR3KO mice on a DBA-1 background following infection with a low dose (1×104 PFU/mouse) of MCMV. P value determined by 2-way ANOVA. E) MCMV titers (PFU/g tissue) in the salivary glands of DBA DR3WT or DR3KO mice at d 7 following low-dose challenge. Bars represent median and interquartile range of n ≥ 6 mice. P values were determined by Mann-Whitney U test. One of 2 experiments is shown. F) Weight loss (left panel) and survival curves (right panel) of DR3WT or DR3KO mice on a DBA-1 background following infection with a high dose (4×104 PFU/mouse) of MCMV. P value determined by Fisher’s exact test. One of 2 experiments is shown.
We tested this further by infecting mice on a DBA-1 background, which genetically lack Ly49H and therefore cannot mount efficient NK-dependent resistance to MCMV. At low doses (104 PFU/mouse), DR3KO mice again showed significantly greater weight loss compared to their DR3WT littermates, which was more sustained than in C57B/6 mice and had only partially recovered by d 6 (Fig. 5D). MCMV titers in the salivary glands were 4-fold higher at d 7 (Fig. 5E). At high doses (4×104 PFU/mouse), weight loss became critical, with 62.5% of DR3KO mice, but no DR3WT animals, reaching the endpoint weight (80% of initial body weight) at which animals had to be killed according to our license procedures (Fig. 5F). The biological implication is that in the absence of fully protective NK responses, DR3 is critical for survival during MCMV challenge.
MCMV-driven CD8+ T-cell expansions are independent of CD4+ T cells and Ly49H+ NK cells, but require intrinsic expression of DR3
To further explore the underlying mechanisms behind DR3-dependent T-cell expansion, depletion and adoptive transfer experiments were performed. Splenocyte expansions were measured in DBA-1 mice, thus investigating the dependence on Ly49H-driven NK responses. DR3KO mice still displayed impaired CD8+ T-cell expansion compared to DR3WT controls on a DBA-1 background (Fig. 6A), thus demonstrating that Ly49H-mediated control of viral load and innate immune activation does not influence DR3 regulation of CD8+ T-cell responses. The current literature on DR3 primarily describes essential roles for the accumulation of effector CD4+ T cells in different models of disease. To prove that DR3-mediated regulation of CD4+ T cells did not influence CD8+ T-cell responses, MCMV-infected C57BL/6 mice were depleted of CD4+ T cells with mAb. CD4+ T-cell depletion prior to MCMVchallenge had no significant effect on CD8+ T-cell expansion or virus-specific CD8+ T-cell numbers in DR3WT mice, while both remained significantly reduced in the absence of DR3 (Fig. 6B; data not shown). The former is in keeping with reports that MCMV-driven primary CD8 expansion is unaffected in CD4−/− and MHC II−/− mice (39, 50, 51). Finally, to study whether the impairments in T-cell expansion were dependent on intrinsic expression of DR3, we adoptively transferred DR3WT and DR3KO T cells into congenic C57BL/6 mice identical except for the Thy1 locus, thus allowing us to identify and count transferred cells. Following MCMV challenge, only DR3WT, and not DR3KO, CD4+ and CD8+ T cells expanded, showing that intrinsic DR3 expression on T cells is required for this effect (Fig. 6C).
Figure 6.
Depletion and adoptive transfer experiments prior to MCMV challenge. A) CD8+CD44hi T-cell expansion in DR3WT or DR3KO mice on a DBA-1 background at 7 d following infection with a low dose (1×104 PFU/mouse) of MCMV. Each symbol represents an individual animal. B) MCMV-driven CD8+ T-cell expansion following CD4+ depletion. CD4+ T cells were depleted from mice prior to MCMV challenge. Bars indicate means. Each symbol represents an individual animal. C) Numbers of CD4+ (left panel) and CD8+ (right panel) DR3WT or DR3KO T cells following adoptive transfer and challenge with MCMV. Data represent means + se of n ≥ 5 mice. One of 5 experiments is shown. All P values were determined by Student’s t test.
DR3 regulates proliferation, not cell death, of CD8+ T cells after MCMV challenge
A number of hypotheses could explain the impaired T, cell response of DR3KO T cells. Signaling by TNFRSF members alters T-cell life span by promoting cell division or cell death by competing pathways (52, 53). As DR3 mediates TCR-triggered apoptosis of thymocytes (14), we first addressed whether the failure of CD4+ and CD8+ T-cell subset expansion in DR3KO mice during virus infection was due to altered cell death. Splenocytes from MCMV-challenged DR3WT and DR3KO mice were stained for expression of surface annexin-V to visualize apoptotic cells, with total uptake of the exclusion dye, 7AAD, as a marker for dead cells. While MCMV infection generally increased splenic T-cell death, similar proportions of apoptotic and dead cells were observed in both CD4+ and CD8+ T cells from DR3WT and DR3KO MCMV-infected mice (Fig. 7), suggesting that DR3’s primary function on T cells after MCMV challenge was not related to cell death.
Figure 7.
T-cell apoptosis following MCMV infection in the absence of DR3. DR3WT or DR3KO mice were challenged with MCMV, and T-cell apoptosis was measured using mAbs to CD4, CD8, and TCR and staining with annexin V and 7AAD at d 7. A) Representative flow cytometric dotplots for CD8+ T cells. B) Summary for CD4+ and CD8+ T cells following MCMV challenge. Analysis for different groups was carried out using the plots shown in A: live, annexin Vlo7AAD−; apopototic, annexin Vhi7AADlo; dead, annexin Vhi7AADhi. Data represent means ± se for n ≥ 5 mice. One experiment of 3 is shown. No significant differences were observed.
We therefore tested the effect of TL1A on the numbers of purified CD8+ T cells recovered after anti-CD3 and anti-CD28 stimulation in vitro. TCR activation induced expansion of CD8+ T-cell numbers from both DR3WT and DR3KO mice to an equivalent level. TL1A by itself had no effect, but in the presence of anti-CD3 boosted CD8+ T-cell numbers in a DR3-dependent fashion (Fig. 8A). The increase in TL1A-induced numbers was equivalent to that provided by giving additional costimulation through CD28 (Fig. 8A). CFSE plots indicated that TL1A induced more cell division from DR3WT T cells (Fig. 8B), but the defect in DR3KO T cells was not associated with any impairment in activation as measured by expression of CD25, CD44, or CD69 (Fig. 8C).
Figure 8.

In vitro T cell proliferation in the absence of DR3. A) Effect of TL1A on purified naive CD8+ T cells. CD8+ T cells were purified from spleens and treated as shown. Cells were counted at the indicated time points. Data are means ± se from n ≥ 5 mice. B) Proliferation of CD8+ T cells stimulated with anti-CD3 and TL1A. DR3WT (left panel) or DR3KO T cells (right panel) were loaded with CFSE and stimulated as described in Materials and Methods. Plots are histograms demonstrating loss of CFSE signal after 4 d stimulation. C) Density plots showing staining of CD8+ T cells for CD25, CD44, and CD69, 4 d after stimulation with anti-CD3 and TL1A.
Investigating expression of nuclear Ki-67 in T cells as a marker of proliferation in vivo, MCMV infection induced extensive CD8+ T-cell proliferation in DR3WT mice, with 75% of CD8+ cells expressing Ki-67. In comparison, only ~50% of DR3KO CD8+ T cells were Ki-67 positive during the peak (d 7) of the acute-phase antiviral response (Fig. 9A). Time courses counting Ki-67+ T cells reflected the patterns observed in overall numbers of T cells following MCMV challenge (Fig. 3). Thus, numbers of DR3WT CD4+ Ki-67+ T cells were significantly increased in the spleen compared to their DR3KO counterparts after MCMV infection, peaking at d 6. Meanwhile, numbers of DR3WT CD8+ Ki-67+ T cells peaked at d 7 and were significantly greater in quantity than DR3KO CD8+ Ki-67+ T cells on d 4, 7, and 9 (Fig. 9B). There was a significant correlation between increasing numbers of CD8+ T cells and proportion of Ki-67+ cells, with a clear division between DR3WT and DR3KO populations (Fig. 9C). These increases in Ki-67+ cells were also reflected in the antigen-specific response, with significantly greater numbers of CD8+ T cells in DR3WT mice responding to M38 and M45 peptide than from DR3KO mice (Fig. 9D). Therefore, we conclude that impaired antiviral T-cell generation and associated protection in DR3KO mice is caused by intrinsic defects in the capacity of CD4+ and CD8+ T cells to divide in response to cognate antigen.
Figure 9.

T-cell proliferation following MCMV infection in the absence of DR3. DR3WT or DR3KO mice were challenged with MCMV, and T-cell proliferation was measured using mAbs to CD4, CD8, TCR, and Ki-67 at d 7. A) Representative flow cytometric dotplots of Ki-67 expression in unchallenged CD8+ T cells (i) and in CD8+ T cells following MCMV challenge (ii). B) Numbers of CD4+ T cells and CD8+ T cells expressing Ki-67 from uninfected DR3WT (●) and DR3KO (○) or MCMV-challenged DR3WT (■) and DR3KO (□) mice. Data represent means ± se of n ≥ 5 mice. One of 3 experiments is shown. *P < 0.05 at indicated time points; Student’s t test. C) Correlation between increasing numbers of CD8+ T cells, expression of Ki-67, and DR3 genotype. Increase in CD8+ T-cell numbers was calculated by subtracting the average number of splenic CD8+ T cells recovered from uninfected animals from the number at 7 d after MCMV infection. DR3WT (●) and DR3KO (○) mice are shown. Line represents best-fit curve; dotted lines indicate 95% confidence limits. D) Numbers of CD8+, Ki-67+, and IFNγ+ T cells responding to M38, M45, and m139 peptide challenge in DR3WT (▲) and DR3KO () mice 7 d following MCMV challenge. Bars indicate means. Each symbol represents a mouse. One experiment is shown. P values were determined by Student’s t test.
DISCUSSION
In recent years, DR3 has emerged as a major regulator of CD4+ T-cell function, affecting multiple T-cell-dependent autoimmune and inflammatory diseases. However, in vivo functional data on the role of DR3 in models of infection have been limited, with only very recent data showing a reduction in the control of Salmonella in DR3KO mice that is mediated by CD4+ T cells (54). For the first time, we have shown, using DR3KO mice and the MCMV and rVV challenge models, that DR3 also plays an essential role in the generation of an efficient antiviral T-cell response, influencing both CD4+ and CD8+ T-cell subsets. In the absence of protective DR3-independent NK responses, the absence of DR3 can prove critical for survival.
Prior to this report, cell surface DR3 protein by naive CD8+ T cells was not well characterized. Naive CD8+ T cells were reported to express very low levels (55), or potential absence of surface DR3 (20), though murine DR3 mRNAs for transmembrane forms have been described (9). These differences could be explained either by low receptor expression and/or preferential binding of some mAbs to specific DR3 isoforms, as has been recently suggested (56). Irrespective, our data highlight a requirement for careful consideration of the function of DR3 splice variants, which are differentially expressed following T-cell activation in both mice and humans (10, 21). In humans, resting T cells only express significant levels of mRNAs encoding for soluble forms of DR3 (10), which is distinct from the constitutive expression of a truncated transmembrane form of DR3 (variant III) in both murine CD4+ and CD8+ T cells reported here (Fig. 2C) and elsewhere (21). Intriguingly, variant III did not confer in vitro proliferative responsiveness to TL1A in the absence of TCR signaling, even though the truncation removes a cysteine-rich domain not involved in ligand binding (42). The possibility remains that variant III has additional, undiscovered functions. What is evident is that, in agreement with others (16, 55), TL1A-induced CD4+ or CD8+ T-cell expansion in vitro requires TCR activation, and, at least for CD8+ T cells, this correlates with an increase in signal for full-length DR3 mRNA (Fig. 2). It is tempting to speculate that selective DR3 splice variant expression may act to limit DR3/TL1A signaling and prevent unrestrained naive cytotoxic T-cell responsiveness. However, the level to which variant III and full-length DR3 contribute to TL1A effects on activated CD8+ T cells is unclear. Reduced DR3 expression on CD44hiCD8+ T cells (Fig. 1A) may be significant, but a more detailed study of DR3 splice variant expression and function throughout CD8+ T-cell differentiation is needed before further conclusions can be drawn.
In the first week following MCMV infection, DR3-deficient mice exhibited reduced numbers of virus-specific T cells, although the decreases in percentages of peptide-specific responses were not as dramatic. Whether differences in proportions vs. total numbers reflect the presence of another DR3-regulated MCMV-derived peptide-specific T-cell population not examined, reduced proportions of the studied peptide-specific T cells at earlier time points, and/or impaired concurrent expansion of non-virus-specific T cells in DR3 deficient mice, is unclear. Irrespective, our data clearly demonstrate that DR3 orchestrates expansion of virus-specific T-cell numbers during acute MCMV and rVV infection.
In contrast, gene deletion of the TNRSF members OX40 or 4-1BB has distinct effects depending on the viral challenge. OX40 has no effect on primary MCMV, but drives accumulation of vaccinia-specific, CD8+ T-cell responses (39, 57). 4-1BB has little effect on vaccinia (58), but inhibits primary CD8 T-cell responses, yet paradoxically enhances MCMV-specific CD8+ T-cell memory accumulation (44). Therefore, fundamental differences exist between these molecules in the regulation of T-cell immunity to different acute virus infection. Intriguingly, impaired acute T-cell responses in DR3-deficient mice were comparable to those observed in mice lacking CD28 ligands (59). Enhanced TCR-induced proliferation following stimulation with TL1A suggests that DR3 is a regulator of CD28-independent early effector T-cell responses, though it is evident that while TL1A can compensate for CD28 costimulation in terms of proliferation (Fig. 8A), it cannot compensate for efficient CD4+ T-cell cytokine production (16). Direct TL1A-driven naive CD8+ T-cell expansion may explain the long-term accumulation of CD8+ T cells in the spleen and mesenteric lymph nodes of transgenic mice engineered with TL1A overexpression by myeloid cells and T cells (25) and the promotion of antitumor CD8+ T cells by TL1A-expressing plasmacytomas (55). In the case of TL1A-driven MCMV-specific CD8+ T-cell expansion, this process was generally resistant to CD4+ depletion, which is consistent with previous reports that acute CD8+ T-cell expansion to MCMV is independent of CD4+ T cells (39, 50). This CD4 independence also indicates that CD4+ T cells are not the major source of TL1A that drives early CD8+ T-cell responses to MCMV. Endothelial cells (60, 61), macrophages (62–64), monocytes (62) and dendritic cells (16, 62) have all been reported as sources of TL1A, and further work will be required to identify which of these are important during MCMV infection.
DR3 also promoted early CD4+ T-cell expansion following MCMV challenge. This is in keeping with the current literature, which has demonstrated a role for DR3 in the accumulation of pathological effector CD4+ T cells in autoimmune, inflammatory, and bacterial disease, whether they belong to Th1, Th2, or Th17 subsets (16, 20, 21, 23, 54). Agonistic anti-DR3 mAbs or overexpression of TL1A has also been shown to preferentially expand CD4+ regulatory T cells (18, 19, 25). Even in this respect, DR3 function cannot be considered simply as an amplifier of CD4+ numbers, as TL1A can inhibit Th17 differentiation or maintain Th17 response depending on the differentiation state of the T cells that receive the DR3 signal (15). Regardless, DR3’s regulation of CD4+ T-cell function is likely to be different from CD8+ T cells, as the majority of CD44hi cells of the former, but a minority of the latter, express surface DR3 (Fig. 1B, D).
Our data reveal a critical cell-intrinsic role for DR3 in regulating T-cell proliferation that is independent of overall TCR-induced cellular activation, as measured by surface expression of activation markers (Fig. 8C). This finding is consistent with studies in organ cultures indicating that the absence of DR3 only affects NFκB activation induced by TL1A and not other NFκB activation pathways, such as TNF (28). The mechanisms through which DR3 controls CD4+ or CD8+ T-cell expansion seem distinct. TL1A-driven CD4+ T-cell proliferation is largely dependent on IL-2 (16), but in keeping with our observation that impaired DR3-dependent CD8+ T-cell expansion following MCMV challenge is CD4 independent, TL1A-driven CD8+ T-cell proliferation has been recently reported to be very resistant to treatment with antagonistic anti-IL-2 mAbs (55). However, a contribution by DR3 in splenic T-cell accumulation and/or recruitment cannot be excluded. In addition, DR3 is not just expressed on T cells, and its presence on endothelia and bone marrow-derived macrophages could further influence the response to MCMV. Both cell types are targets for MCMV replication in vivo (65), but we have found no direct antiviral effect of TL1A on the in vitro growth of MCMV in 3T3 cell lines (Supplemental Fig. S1). TL1A may also influence the way immunity develops via downstream soluble effector molecules. Indeed, MMP-9 and IL-8 have been reportedly released following treatment of human macrophages with TL1A (64, 66). Furthermore, DR3 can regulate macrophage differentiation in vitro into more specialist subsets, such as osteoclasts (22) or foam cells (67). In addition, TL1A has distinct isoforms generated by differential ectodomain shedding that have a number of effects on endothelial cells in vitro, including inducing growth arrest, triggering apoptosis, and promoting senescence (61). How these effects apply in vivo is another interesting area of further research.
Finally, our data show that the absence of DR3 results in dysregulated control of viral replication in vivo, leading to significant increases in acute MCMV replication in the liver, spleen, and a site of virus persistence and shedding, the salivary glands (45, 68). Critically, DR3 deficiency on a DBA background that lacks Ly49H-dependent NK cell activation (49) can be lethal. Weight recovery after initial MCMV challenge in DR3WT mice began after d 4 and correlated with the acute expansion of splenic T cells that peaked at d 7. While this is consistent with the T-cell response protecting from MCMV challenge, further adoptive transfer experiments into immunodeficient mice would be required to formally prove this and the possibility remains that there are other DR3-dependent innate mechanisms that contribute to the reduced control of MCMV observed in DR3KO mice.
Intriguingly, the impaired early T-cell expansion in DR3KO mice and concomitant increase in MCMV titers did not result in defective memory (Fig. 4D), indicating that DR3’s primary function is in acute and not long-lived antiviral T-cell immunity. These data have several interesting implications; one is that DR3 may control the contraction of the acute T-cell response, without influencing differentiation to memory. The underlying mechanism is unclear and requires further study, but it could be speculated that TL1A/DR3 signaling may directly (or indirectly through other downstream effector molecules) trigger the contraction process. Another possibility in the MCMV system is that continuous antigenic stimulation overcomes the observed DR3 defects in T-cell proliferation, coinciding with the decrease in DR3 expression on CD44hi antigen-experienced T-cells (Fig. 1D). This contrasts with 4-1BB and OX40, which both contribute significantly to the development of the MCMV-specific CD8+ memory T-cell response (39, 44) and highlights the different contributions of TNFRSF members to immunity against persistent viral infections. These findings identify DR3 as critical for the orchestration of immunity to acute virus infection and identify the DR3/TL1A pathway as a potential target for boosting antiviral immunity.
Supplementary Material
Acknowledgments
The authors thank the Central Biotechnology Service (School of Medicine, Cardiff University) for supply and access to flow cytometers. This work was supported by grant funding from the British Medical Research Council (MRC) and the Wellcome Trust. J.P.T. is funded by MRC grant G0901119 awarded to E.C.Y.W. and I.R.H. I.R.H. holds a Wellcome Trust Career Development Fellowship. S.M.C. is funded by a Biotechnology and Biological Sciences Research Council project grant.
Abbreviations
- CFSE
carboxy-fluorescein succinyl ester
- DR3
death receptor 3
- DR3KO
DR3-knockout
- DR3WT
DR3 wild type
- MCMV
murine cytomegalovirus
- NKT
natural killer T
- PFU
plaque-forming unit
- rVV
recombinant vaccinia virus
- TCR
T-cell receptor
- TL1A
TNF-like protein 1A
- TNFRSF
tumor necrosis factor receptor superfamily
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat. Rev. Immunol. 2003;3:609–620. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 2.Hehlgans T, Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology. 2005;115:1–20. doi: 10.1111/j.1365-2567.2005.02143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfeffer K. Biological functions of tumor necrosis factor cytokines and their receptors. Cytokine Growth Factor Rev. 2003;14:185–191. doi: 10.1016/s1359-6101(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 4.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu. Rev. Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 5.Bodmer JL, Burns K, Schneider P, Hofmann K, Steiner V, Thome M, Bornand T, Hahne M, Schroter M, Becker K, Wilson A, French LE, Browning JL, MacDonald HR, Tschopp J. TRAMP, a novel apoptosis-mediating receptor with sequence homology to tumor necrosis factor receptor 1 and Fas(Apo-1/CD95) Immunity. 1997;6:79–88. doi: 10.1016/s1074-7613(00)80244-7. [DOI] [PubMed] [Google Scholar]
- 6.Chinnaiyan AM, O’Rourke K, Yu GL, Lyons RH, Garg M, Duan DR, Xing L, Gentz R, Ni J, Dixit VM. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science. 1996;274:990–992. doi: 10.1126/science.274.5289.990. [DOI] [PubMed] [Google Scholar]
- 7.Kitson J, Raven T, Jiang YP, Goeddel DV, Giles KM, Pun KT, Grinham CJ, Brown R, Farrow SN. A death-domain-containing receptor that mediates apoptosis. Nature. 1996;384:372–375. doi: 10.1038/384372a0. [DOI] [PubMed] [Google Scholar]
- 8.Marsters SA, Sheridan JP, Pitti RM, Huang A, Skubatch M, Baldwin D, Yuan J, Gurney A, Goddard AD, Godowski P, Ashkenazi A. A novel receptor for Apo2L/ TRAIL contains a truncated death domain. Curr. Biol. 1997;7:1003–1006. doi: 10.1016/s0960-9822(06)00422-2. [DOI] [PubMed] [Google Scholar]
- 9.Tan KB, Harrop J, Reddy M, Young P, Terrett J, Emery J, Moore G, Truneh A. Characterization of a novel TNF-like ligand and recently described TNF ligand and TNF receptor superfamily genes and their constitutive and inducible expression in hematopoietic and non-hematopoietic cells. Gene. 1997;204:35–46. doi: 10.1016/s0378-1119(97)00509-x. [DOI] [PubMed] [Google Scholar]
- 10.Screaton GR, Xu XN, Olsen AL, Cowper AE, Tan R, McMichael AJ, Bell JI. LARD: a new lymphoid-specific death domain containing receptor regulated by alternative pre-mRNA splicing. Proc. Natl. Acad. Sci. U. S. A. 1997;94:4615–4619. doi: 10.1073/pnas.94.9.4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wen L, Zhuang L, Luo X, Wei P. TL1A-induced NF-kappaB activation and c-IAP2 production prevent DR3-mediated apoptosis in TF-1 cells. J. Biol. Chem. 2003;278:39251–39258. doi: 10.1074/jbc.M305833200. [DOI] [PubMed] [Google Scholar]
- 12.Migone TS, Zhang J, Luo X, Zhuang L, Chen C, Hu B, Hong JS, Perry JW, Chen SF, Zhou JX, Cho YH, Ullrich S, Kanakaraj P, Carrell J, Boyd E, Olsen HS, Hu G, Pukac L, Liu D, Ni J, Kim S, Gentz R, Feng P, Moore PA, Ruben SM, Wei P. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16:479–492. doi: 10.1016/s1074-7613(02)00283-2. [DOI] [PubMed] [Google Scholar]
- 13.Pobezinskaya YL, Choksi S, Morgan MJ, Cao X, Liu ZG. The adaptor protein TRADD is essential for TNF-like ligand 1A/death receptor 3 signaling. J. Immunol. 2011;186:5212–5216. doi: 10.4049/jimmunol.1002374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang EC, Thern A, Denzel A, Kitson J, Farrow SN, Owen MJ. DR3 regulates negative selection during thymocyte development. Mol. Cell. Biol. 2001;21:3451–3461. doi: 10.1128/MCB.21.10.3451-3461.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones GW, Stumhofer JS, Foster T, Twohig JP, Hertzog P, Topley N, Williams AS, Hunter CA, Jenkins BJ, Wang EC, Jones SA. Naive and activated T cells display differential responsiveness to TL1A that affects Th17 generation, maintenance, and proliferation. FASEB J. 2011;25:409–419. doi: 10.1096/fj.10-166843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meylan F, Davidson TS, Kahle E, Kinder M, Acharya K, Jankovic D, Bundoc V, Hodges M, Shevach EM, Keane-Myers A, Wang EC, Siegel RM. The TNF-family receptor DR3 is essential for diverse T cell-mediated inflammatory diseases. Immunity. 2008;29:79–89. doi: 10.1016/j.immuni.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papadakis KA, Zhu D, Prehn JL, Landers C, Avanesyan A, Lafkas G, Targan SR. Dominant role for TL1A/DR3 pathway in IL-12 plus IL-18-induced IFN-gamma production by peripheral blood and mucosal CCR9+ T lymphocytes. J. Immunol. 2005;174:4985–4990. doi: 10.4049/jimmunol.174.8.4985. [DOI] [PubMed] [Google Scholar]
- 18.Schreiber TH, Wolf D, Tsai MS, Chirinos J, Deyev VV, Gonzalez L, Malek TR, Levy RB, Podack ER. Therapeutic Treg expansion in mice by TNFRSF25 prevents allergic lung inflammation. J. Clin. Invest. 2010;120:3629–3640. doi: 10.1172/JCI42933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taraban VY, Slebioda TJ, Willoughby JE, Buchan SL, James S, Sheth B, Smyth NR, Thomas GJ, Wang EC, Al-Shamkhani A. Sustained TL1A expression modulates effector and regulatory T-cell responses and drives intestinal goblet cell hyperplasia. Mucosal Immunol. 2011;4:186–196. doi: 10.1038/mi.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang L, Adkins B, Deyev V, Podack ER. Essential role of TNF receptor superfamily 25 (TNFRSF25) in the development of allergic lung inflammation. J. Exp. Med. 2008;205:1037–1048. doi: 10.1084/jem.20072528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bamias G, Mishina M, Nyce M, Ross WG, Kollias G, Rivera-Nieves J, Pizarro TT, Cominelli F. Role of TL1A and its receptor DR3 in two models of chronic murine ileitis. Proc. Natl. Acad. Sci. U. S. A. 2006;103:8441–8446. doi: 10.1073/pnas.0510903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bull MJ, Williams AS, Mecklenburgh Z, Calder CJ, Twohig JP, Elford C, Evans BA, Rowley TF, Slebioda TJ, Taraban VY, Al-Shamkhani A, Wang EC. The death receptor 3-TNF-like protein 1A pathway drives adverse bone pathology in inflammatory arthritis. J. Exp. Med. 2008;205:2457–2464. doi: 10.1084/jem.20072378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pappu BP, Borodovsky A, Zheng TS, Yang X, Wu P, Dong X, Weng S, Browning B, Scott ML, Ma L, Su L, Tian Q, Schneider P, Flavell RA, Dong C, Burkly LC. TL1A-DR3 interaction regulates Th17 cell function and Th17-mediated autoimmune disease. J. Exp. Med. 2008;205:1049–1062. doi: 10.1084/jem.20071364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takedatsu H, Michelsen KS, Wei B, Landers CJ, Thomas LS, Dhall D, Braun J, Targan SR. TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T-helper 1 and T-helper 17 activation. Gastroenterology. 2008;135:552–567. doi: 10.1053/j.gastro.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meylan F, Song YJ, Fuss I, Villarreal S, Kahle E, Malm IJ, Acharya K, Ramos HL, Lo L, Mentink-Kane MM, Wynn TA, Migone TS, Strober W, Siegel RM. The TNF-family cytokine TL1A drives IL-13-dependent small intestinal inflammation. Mucosal Immunol. 2011;4:172–185. doi: 10.1038/mi.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shih DQ, Barrett R, Zhang X, Yeager N, Koon HW, Phaosawasdi P, Song Y, Ko B, Wong MH, Michelsen KS, Martins G, Pothoulakis C, Targan SR. Constitutive TL1A (TNFSF15) expression on lymphoid or myeloid cells leads to mild intestinal inflammation and fibrosis. PLoS One. 2011;6:e16090. doi: 10.1371/journal.pone.0016090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calder CJ, Wang EC. An essential role for death receptor 3 in experimental autoimmune uveoretinitis. Ocul. Immunol. Inflamm. 2012;20:212–214. doi: 10.3109/09273948.2012.658135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Lamki RS, Wang J, Tolkovsky AM, Bradley JA, Griffin JL, Thiru S, Wang EC, Bolton E, Min W, Moore P, Pober JS, Bradley JR. TL1A both promotes and protects from renal inflammation and injury. J. Am. Soc. Nephrol. 2008;19:953–960. doi: 10.1681/ASN.2007060706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Al-Lamki RS, Lu W, Finlay S, Twohig JP, Wang EC, Tolkovsky AM, Bradley JR. DR3 signaling protects against cisplatin nephrotoxicity mediated by TNF. Am J Pathol. 2012;180:1454–1464. doi: 10.1016/j.ajpath.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Booth TW, Scalzo AA, Carrello C, Lyons PA, Farrell HE, Singleton GR, Shellam GR. Molecular and biological characterization of new strains of murine cytomegalovirus isolated from wild mice. Arch. Virol. 1993;132:209–220. doi: 10.1007/BF01309855. [DOI] [PubMed] [Google Scholar]
- 31.Reddehase MJ, Podlech J, Grzimek NK. Mouse models of cytomegalovirus latency: overview. J. Clin. Virol. 2002;25(Suppl. 2):S23–S36. doi: 10.1016/s1386-6532(02)00087-2. [DOI] [PubMed] [Google Scholar]
- 32.Smith LM, McWhorter AR, Masters LL, Shellam GR, Redwood AJ. Laboratory strains of murine cytomegalovirus are genetically similar to but phenotypically distinct from wild strains of virus. J. Virol. 2008;82:6689–6696. doi: 10.1128/JVI.00160-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polic B, Hengel H, Krmpotic A, Trgovcich J, Pavic I, Luccaronin P, Jonjic S, Koszinowski UH. Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J. Exp. Med. 1998;188:1047–1054. doi: 10.1084/jem.188.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reddehase MJ, Keil GM, Koszinowski UH. The cytolytic T lymphocyte response to the murine cytomegalovirus. I. Distinct maturation stages of cytolytic T lymphocytes constitute the cellular immune response during acute infection of mice with the murine cytomegalovirus. J. Immunol. 1984;132:482–489. [PubMed] [Google Scholar]
- 35.Reddehase MJ, Keil GM, Koszinowski UH. The cytolytic T lymphocyte response to the murine cytomegalovirus. II. Detection of virus replication stage-specific antigens by separate populations of in vivo active cytolytic T lymphocyte precursors. Eur. J. Immunol. 1984;14:56–61. doi: 10.1002/eji.1830140111. [DOI] [PubMed] [Google Scholar]
- 36.Simon CO, Holtappels R, Tervo HM, Bohm V, Daubner T, Oehrlein-Karpi SA, Kuhnapfel B, Renzaho A, Strand D, Podlech J, Reddehase MJ, Grzimek NK. CD8 T cells control cytomegalovirus latency by epitope-specific sensing of transcriptional reactivation. J. Virol. 2006;80:10436–10456. doi: 10.1128/JVI.01248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moss P, Khan N. CD8(+) T-cell immunity to cytomegalovirus. Hum. Immunol. 2004;65:456–464. doi: 10.1016/j.humimm.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 38.Twohig JP, Roberts MI, Gavalda N, Rees-Taylor EL, Giralt A, Adams D, Brooks SP, Bull MJ, Calder CJ, Cuff S, Yong AA, Alberch J, Davies A, Dunnett SB, Tolkovsky AM, Wang EC. Age-dependent maintenance of motor control and corticostriatal innervation by death receptor 3. J. Neurosci. 2010;30:3782–3792. doi: 10.1523/JNEUROSCI.1928-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Humphreys IR, Loewendorf A, de Trez C, Schneider K, Benedict CA, Munks MW, Ware CF, Croft M. OX40 costimulation promotes persistence of cytomegalovirus-specific CD8 T Cells: A CD4-dependent mechanism. J. Immunol. 2007;179:2195–2202. doi: 10.4049/jimmunol.179.4.2195. [DOI] [PubMed] [Google Scholar]
- 40.Reddehase MJ, Weiland F, Munch K, Jonjic S, Luske A, Koszinowski UH. Interstitial murine cytomegalovirus pneumonia after irradiation: characterization of cells that limit viral replication during established infection of the lungs. J. Virol. 1985;55:264–273. doi: 10.1128/jvi.55.2.264-273.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Richards H, Longhi MP, Wright K, Gallimore A, Ager A. CD62L (L-selectin) down-regulation does not affect memory T cell distribution but failure to shed compromises anti-viral immunity. J. Immunol. 2008;180:198–206. doi: 10.4049/jimmunol.180.1.198. [DOI] [PubMed] [Google Scholar]
- 42.Wang EC, Kitson J, Thern A, Williamson J, Farrow SN, Owen MJ. Genomic structure, expression, and chromosome mapping of the mouse homologue for the WSL-1 (DR3, Apo3, TRAMP, LARD, TR3, TNFRSF12) gene. Immunogenetics. 2001;53:59–63. doi: 10.1007/s002510000290. [DOI] [PubMed] [Google Scholar]
- 43.Bamias G, Martin C, 3rd, Marini M, Hoang S, Mishina M, Ross WG, Sachedina MA, Friel CM, Mize J, Bickston SJ, Pizarro TT, Wei P, Cominelli F. Expression, localization, and functional activity of TL1A, a novel Th1-polarizing cytokine in inflammatory bowel disease. J. Immunol. 2003;171:4868–4874. doi: 10.4049/jimmunol.171.9.4868. [DOI] [PubMed] [Google Scholar]
- 44.Humphreys IR, Lee SW, Jones M, Loewendorf A, Gostick E, Price DA, Benedict CA, Ware CF, Croft M. Biphasic role of 4-1BB in the regulation of mouse cytomegalovirus-specific CD8(+) T cells. Eur. J. Immunol. 2010;40:2762–2768. doi: 10.1002/eji.200940256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lathbury LJ, Allan JE, Shellam GR, Scalzo AA. Effect of host genotype in determining the relative roles of natural killer cells and T cells in mediating protection against murine cytomegalovirus infection. J. Gen. Virol. 1996;77(Pt. 10):2605–2613. doi: 10.1099/0022-1317-77-10-2605. [DOI] [PubMed] [Google Scholar]
- 46.Holtappels R, Simon CO, Munks MW, Thomas D, Deegen P, Kuhnapfel B, Daubner T, Emde SF, Podlech J, Grzimek NK, Oehrlein-Karpi SA, Hill AB, Reddehase MJ. Subdominant CD8 T-cell epitopes account for protection against cytomegalovirus independent of immunodomination. J. Virol. 2008;82:5781–5796. doi: 10.1128/JVI.00155-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Munks MW, Cho KS, Pinto AK, Sierro S, Klenerman P, Hill AB. Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J. Immunol. 2006;177:450–458. doi: 10.4049/jimmunol.177.1.450. [DOI] [PubMed] [Google Scholar]
- 48.Walton SM, Wyrsch P, Munks MW, Zimmermann A, Hengel H, Hill AB, Oxenius A. The dynamics of mouse cytomegalovirus-specific CD4 T cell responses during acute and latent infection. J. Immunol. 2008;181:1128–1134. doi: 10.4049/jimmunol.181.2.1128. [DOI] [PubMed] [Google Scholar]
- 49.Corbett AJ, Coudert JD, Forbes CA, Scalzo AA. Functional consequences of natural sequence variation of murine cytomegalovirus m157 for Ly49 receptor specificity and NK cell activation. J. Immunol. 2011;186:1713–1722. doi: 10.4049/jimmunol.1003308. [DOI] [PubMed] [Google Scholar]
- 50.Snyder CM, Loewendorf A, Bonnett EL, Croft M, Benedict CA, Hill AB. CD4+ T cell help has an epitope-dependent impact on CD8+ T cell memory inflation during murine cytomegalovirus infection. J. Immunol. 2009;183:3932–3941. doi: 10.4049/jimmunol.0900227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walton SM, Torti N, Mandaric S, Oxenius A. T-cell help permits memory CD8(+) T-cell inflation during cytomegalovirus latency. Eur. J. Immunol. 2011;41:2248–2259. doi: 10.1002/eji.201141575. [DOI] [PubMed] [Google Scholar]
- 52.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 53.Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J. 2009;23:1625–1637. doi: 10.1096/fj.08-111005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buchan SL, Taraban VY, Slebioda TJ, James S, Cunningham AF, Al-Shamkhani A. Death receptor 3 is essential for generating optimal protective CD4(+) T-cell immunity against Salmonella. Eur. J. Immunol. 2012;42:580–588. doi: 10.1002/eji.201041950. [DOI] [PubMed] [Google Scholar]
- 55.Slebioda TJ, Rowley TF, Ferdinand JR, Willoughby JE, Buchan SL, Taraban VY, Al-Shamkhani A. Triggering of TNFRSF25 promotes CD8 T-cell responses and anti-tumor immunity. Eur. J. Immunol. 2011;41:2606–2611. doi: 10.1002/eji.201141477. [DOI] [PubMed] [Google Scholar]
- 56.Taraban VY, Ferdinand JR, Al-Shamkhani A. Expression of TNFRSF25 on conventional T cells and Tregs. J. Clin. Invest. 2011;121:463–464. doi: 10.1172/JCI45832. author reply 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salek-Ardakani S, Moutaftsi M, Crotty S, Sette A, Croft M. OX40 drives protective vaccinia virus-specific CD8 T cells. J. Immunol. 2008;181:7969–7976. doi: 10.4049/jimmunol.181.11.7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao Y, Croft M. Dispensable role for 4-1BB and 4-1BBL in development of vaccinia virus-specific CD8 T cells. Immunol. Lett. 2012;141:220–226. doi: 10.1016/j.imlet.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arens R, Loewendorf A, Redeker A, Sierro S, Boon L, Klenerman P, Benedict CA, Schoenberger SP. Differential B7-CD28 costimulatory requirements for stable and inflationary mouse cytomegalovirus-specific memory CD8 T cell populations. J. Immunol. 2011;186:3874–3881. doi: 10.4049/jimmunol.1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim S, Zhang L. Identification of naturally secreted soluble form of TL1A, a TNF-like cytokine. J. Immunol. Methods. 2005;298:1–8. doi: 10.1016/j.jim.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 61.Muck C, Herndler-Brandstetter D, Micutkova L, Grubeck-Loebenstein B, Jansen-Durr P. Two functionally distinct isoforms of TL1A (TNFSF15) generated by differential ectodomain shedding. J. Gerontol. A Biol. Sci. Med. Sci. 2010;65:1165–1180. doi: 10.1093/gerona/glq129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Prehn JL, Thomas LS, Landers CJ, Yu QT, Michelsen KS, Targan SR. The T cell costimulator TL1A is induced by FcgammaR signaling in human monocytes and dendritic cells. J. Immunol. 2007;178:4033–4038. doi: 10.4049/jimmunol.178.7.4033. [DOI] [PubMed] [Google Scholar]
- 63.Kamada N, Hisamatsu T, Honda H, Kobayashi T, Chinen H, Takayama T, Kitazume MT, Okamoto S, Koganei K, Sugita A, Kanai T, Hibi T. TL1A produced by lamina propria macrophages induces Th1 and Th17 immune responses in cooperation with IL-23 in patients with Crohn’s disease. Inflamm. Bowel Dis. 2010;16:568–575. doi: 10.1002/ibd.21124. [DOI] [PubMed] [Google Scholar]
- 64.Kang YJ, Kim WJ, Bae HU, Kim DI, Park YB, Park JE, Kwon BS, Lee WH. Involvement of TL1A and DR3 in induction of pro-inflammatory cytokines and matrix metalloproteinase-9 in atherogenesis. Cytokine. 2005;29:229–235. doi: 10.1016/j.cyto.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 65.Hsu KM, Pratt JR, Akers WJ, Achilefu SI, Yokoyama WM. Murine cytomegalovirus displays selective infection of cells within hours after systemic administration. J. Gen. Virol. 2009;90:33–43. doi: 10.1099/vir.0.006668-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Su WB, Chang YH, Lin WW, Hsieh SL. Differential regulation of interleukin-8 gene transcription by death receptor 3 (DR3) and type I TNF receptor (TNFRI) Exp. Cell Res. 2006;312:266–277. doi: 10.1016/j.yexcr.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 67.McLaren JE, Calder CJ, McSharry BP, Sexton K, Salter RC, Singh NN, Wilkinson GW, Wang EC, Ramji DP. The TNF-like protein 1A-death receptor 3 pathway promotes macrophage foam cell formation in vitro. J. Immunol. 2010;184:5827–5834. doi: 10.4049/jimmunol.0903782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cavanaugh VJ, Deng Y, Birkenbach MP, Slater JS, Campbell AE. Vigorous innate and virus-specific cytotoxic T-lymphocyte responses to murine cytomegalovirus in the submaxillary salivary gland. J. Virol. 2003;77:1703–1717. doi: 10.1128/JVI.77.3.1703-1717.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.