

Abstract
CD4+CD25+FoxP3+ regulatory T cells (Tregs) are understood to maintain peripheral tolerance to self-antigens and inhibit antitumor immune responses. However, compelling evidence suggests that, Tregs provide no anti-inflammatory protection in the tumor microenvironment, but rather contribute to a T helper 17 (Th17)-driven pro-carcinogenic process. Using three-color flow cytometry, we evaluated the percentage of circulating CD4+CD25+FoxP3+ Tregs in the peripheral blood of pancreatic carcinoma patients prior to and after chemotherapy [gemcitabine (GEM) alone, or GEM+oxaliplatin (GEMOX) or bevacizumab+capecitabine+radiotherapy (BEV+CAPE+RT)]. Correlations were sought between Treg counts and plasma levels of cytokines relevant to controlling the Treg/Th17 balance, i.e., interleukin (IL)-23, IL-17A, IL-6 and transforming growth factor β 1 (TGF-β1), as measured by ELISA and the clinical features of pancreatic cancer. Treg, IL-6 and TGF-β1 levels were higher in locally advanced and metastatic pancreatic carcinoma patients compared to controls. No parameter was correlated with disease stage except IL-6. IL-17A and TGF-β1 were significantly associated with increased risk of poor prognosis. IL-17A was positively correlated with IL-23. Treg and IL-6 levels decreased following GEM monochemotherapy, IL-17A levels decreased after GEMOX, and IL-6 levels were reduced subsequent to BEV+CAPE+RT treatment. IL-23, IL-17A and TGF-β1 levels were significantly lower in patients responding to chemotherapy (partial remission/stable disease) than in nonresponders to chemotherapy (progressive disease). These results suggest an impact of the Treg/Th17-balance in pancreatic carcinoma, highlighting the significance of TGF-β1 and IL-17A as potential prognostic and predictive indicators. Immunological changes induced by mono and/or combined chemotherapy indicate specific windows of opportunity for introducing integrative interventions on a new target in pancreatic cancer, i.e. IL-17A, possibly improving survival in this highly lethal disease.
Keywords: T regulatory cells, T helper 17, interleukin-17, interleukin-23, interleukin-6, transforming growth factor β 1, pancreatic carcinoma
Introduction
The typical late presentation of pancreatic adenocarcinoma, its early systemic dissemination and its poor response to chemoor radiation therapy, render prognosis and chance of a cure under present management poor. The dearth of conventional therapeutic options motivates the search for novel immuno-therapeutic approaches to pancreatic cancer, including the development of monoclonal antibodies, cytokines, cellular immunotherapy and vaccines (1).
The risk of developing cancer may be increased by poorly regulated local inflammatory responses to pathogenic challenge. Although the etiology of pancreatic adenocarcinoma is not entirely clear, in vivo and in vitro studies show that inflammation is important in its carcinogenesis (2). Whereas acute inflammatory responses may be beneficial to the host, as it eliminates cancer cells, if this response becomes chronic it may hinder antitumor immune responses, enhancing tumor growth, angiogenesis, invasion and metastasis.
Natural CD25+CD4+ regulatory T-cells (Tregs) in the human peripheral bloodstream (identified by high CD25 expression and the IL-2 receptor α chain), alongside transcriptional factor forkhead box P3 (FoxP3) (3), have been implicated in regulating immune responses affecting tumor growth. Normally, Tregs maintain self-tolerance by suppressing immune responses against autoantigens and restore immune homeostasis during chronic inflammatory disorders (4). Although Tregs may reduce the risk of inflammation-associated cancer by downregulating inflammation, it is believed that, in cancer, they mainly function by suppressing the antitumor immune response. However, under poorly regulated pro-inflammatory conditions, Tregs fail to inhibit, and may stimulate, a pro-inflammatory T helper 17 (Th17) response, driving the pro-carcinogenic process (5).
Tregs, which promote tolerance, and Th17 cells, which induce inflammation (6), appear to arise from common naïve CD4+ T precursor cells. During activation of human precursor CD4+ T cells, interleukin-6 (IL-6) and transforming growth factor β (TGF-β) promote differentiation toward Th17 cells, whereas TGF-β alone promotes differentiation of Tregs and suppresses Th17 (7). Furthermore, IL-23, a heterodimer produced by dendritic cells (DC) comprising the unique p19 subunit plus the p40 subunit in common with IL-12, also contributes to differentiation and maintenance of Th17 cells (8). Moreover, recent data indicate late-stage plasticity of a subpopulation of Tregs, which can be selectively induced to adopt a Th17 phenotype, suggesting that Tregs may do more to modulate cancer than simply block constructive anticancer responses (5).
Tregs and Th17 are involved in opposing immune responses critically involved in modulating inflammation in several diseases, including cancer. IL-6, TGF-β1, IL-23 and IL-17A are key contributors to the Th17/Treg balance; the present study concentrated on these factors.
Materials and methods
Patients
The cohort comprised 62 patients (39 males, 23 females; median age 64.5 years, range 31–80) diagnosed with locally advanced or metastatic pancreatic adenocarcinoma at Centro Oncologico Ematologico Subalpino (COES), San Giovanni Battista Hospital, Turin, Italy, between March 2007 and March 2010. None of the patients had undergone anticancer treatment and all provided informed consent prior to entering the study. Study procedures complied with the Helsinki Declaration. Tumors were classified according to the International Union Against Cancer (UICC) Staging System at the start of chemotherapy; Table I presents the clinicopathological features of the patients. Of the 62 patients, 15 underwent radical surgery, only entering chemotherapy at disease progression and seven underwent palliative surgery, entering chemotherapy immediately afterwards. Four patients received no chemotherapy due to rapid progression. Protocols were: gemcitabine (GEM, 1250 mg/m2 on days 1 and 8 every 21 days; 28 patients); GEM (1000 mg/m2 on day 1) plus oxaliplatin (OX, 100 mg/m2 on day 2 every 14 days; 23 patients); bevacizumab (BEV, 5 mg/kg every 14 days) plus capecitabine (CAPE, 825 mg/m2/b.i.d.) and concomitant radiotherapy (RT, 50.4 Gy in 28 cycles, following the study protocol; six patients); 5-fluorouracil (5-FU, 500 mg/m2) plus levofolinate calcium (250 mg/m2 on days 1, 8, and 15 every 28 days; one patient). After the first restaging, responses (by RECIST criteria) were: no complete responses, one partial response (patient with locally advanced carcinoma treated with chemo-radiotherapy in combination with BEV followed by radical surgery), 30 stabilizations and 26 disease progressions (clinical progression in eight cases, not documented radiologically). In two cases the response was not evaluable: one patient succumbed to unrelated causes after a single dose of chemotherapy and one patient discontinued therapy after two cycles due to toxicity and poor compliance. Follow-up continued until death or May 2010. A total of 44 patients died and the median survival time was 7.2 months. Seventeen patients survived to the end of the study: with four being in follow-up and 13 being in chemotherapy. Survival was 34.4% at one year after diagnosis, 11.5% at two years.
Table I.
Clinicopathological features of the patients.
| Feature | Group A (n=27) | Group B (n=35) | Total (n=62) |
|---|---|---|---|
| Gender [n (%)] | |||
| Female | 11 (40.74) | 12 (34.29) | 23 (37.1) |
| Male | 16 (59.26) | 23 (65.71) | 39 (62.9) |
| Age [mean (median)], in years | 63.81 (66) | 64.69 (62) | 64.30 (64.5) |
| Disease stage at start of chemotherapy [n (%)] | |||
| II | 2 (7.41) | 5 (14.29) | 7 (11.29) |
| III | 3 (11.11) | 12 (34.29) | 15 (24.19) |
| IV | 22 (81.48) | 18 (51.43) | 40 (64.52) |
| Surgery [n (%)] | |||
| None | 20 (74.07) | 20 (57.14) | 40 (64.52) |
| Palliative | 1 (3.7) | 6 (17.14) | 7 (11.29) |
| Radical | 6 (22.22) | 9 (25.71) | 15 (24.19) |
| Metastases [n (%)] | |||
| No | 5 (18.52) | 17 (48.57) | 22 (35.48) |
| Yes | 22 (81.48) | 18 (51.43) | 40 (64.52) |
| Metastatic site [n (%)] | |||
| Liver | 13 (59.09) | 7 (38.89) | 20 (50) |
| Peritoneum | 4 (18.18) | 4 (22.22) | 8 (20) |
| Lung | 2 (9.09) | 2 (11.11) | 4 (10) |
| Liver, peritoneum | 1 (4.55) | 4 (22.22) | 5 (12.5) |
| Liver, lung | 1 (4.55) | 0 (0) | 1 (12.5) |
| Lung, peritoneum | 1 (4.55) | 1 (5.56) | 2 (5) |
| Chemotherapy [n (%)] | |||
| No | 4 (14.81) | 0 (0) | 4 (6.45) |
| Yes | 23 (85.19) | 35 (100) | 58 (93.55) |
| Type of chemotherapy [n (%)] | |||
| GEM | 13 (56.52) | 15 (42.86) | 28 (48.28) |
| GEMOX | 9 (39.13) | 14 (40) | 23 (39.66) |
| BEV+CAPE+RT | 0 (0) | 6 (17.14) | 6 (10.34) |
| 5-FU+levofolinate calcium | 1 (4.35) | 0 (0) | 1 (1.72) |
| Response [n (%)] | |||
| Complete remission (CR) | 0 (0) | 0 (0) | 0 (0) |
| Partial remission (PR) | 0 (0) | 1 (2.86) | 1 (1.72) |
| Stable disease (SD) | 8 (34.78) | 22 (62.86) | 30 (51.72) |
| Disease progression (DP) | 14 (60.87) | 12 (34.29) | 26 (44.83) |
Group A, patients evaluated only on the day of admission prior to chemotherapy (time 0); group B, patients evaluated on the day of admission (time 0) (prior to chemotherapy) and at first restaging (after an interval of 2–3 months depending on the chemotherapy regime) in parallel with evaluation of the clinical course. GEM, gemcitabine; OX, oxaliplatin; BEV, bevacizumab; CAPE, capecitabine; RT, radiotherapy; 5-FU, 5-fluorouracil.
Cell isolation and plasma collection
Peripheral blood was collected from the patients in heparinized tubes at admission (time 0, prior to chemotherapy) and at first restaging (after 2–3 months, depending on chemotherapy regime) in parallel with evaluation of clinical course, and from the controls (20 age- and gender-matched volunteers). Peripheral blood mononuclear cells (PBMCs) and plasma were collected at interface and upper phase, respectively, after centrifugation of blood over a Ficoll-Hypaque density gradient. Plasma was stored at −20°C until use.
Flow cytometry
Circulating CD4+/CD25high/FoxP3+ Tregs were enumerated by three-color flow cytometry. PBMCs were incubated with anti-CD4-FITC (Caltag Laboratories, Burlingame, CA, USA) and anti-CD25-PC5 (Beckman Coulter, Immunotech, France) mAb for 30 min at 4°C. Subsequent to washing with PBS, PBMCs were fixed and permeabilized with fixation/permeabilization buffer for 30 min at 4°C, washed twice with permeabilization buffer and stained with anti-human FoxP3-PE mAb, following the manufacturer’s instructions (eBioscience, San Diego, CA, USA). Following a 30-min incubation at 4°C, cells were washed and analyzed by flow cytometry in a Coulter Epics IV Cytometer (Beckman Coulter, Inc, Fullerton, CA, USA) employing Expo32 Software (Beckman Coulter). Cells were gated on viable lymphocytes, following standard forward and sideways scattering parameters. Among cells included in this gate, we evaluated Treg subpopulations as FoxP3+ within the CD4+/CD25bright subset. The results are expressed as percentages of triple-positive cell values, calculated subtracting the autofluorescence of unstained cells (morphology) from the fluorescence-intensity of antibody-labeled cells. Statistical analyses involved ≥30,000 events gated on the population of interest.
ELISA determination of IL-23, IL-17A, IL-6 and TGF-β1 plasma levels
IL-23, IL-17A, IL-6 and TGF-β1 plasma concentrations were determined using commercially available ELISA kits. For TGF-β1, plasma samples were tested after transient acidification (reducing pH to 1.5 by adding 1 N HCl for 10 min at room temperature and neutralizing with 1.2 N NaOH in 0.5 M HEPES). All evaluations were carried out in duplicate. The lower detection limits of the assays were: 4 pg/ml for IL-17A (eBioscience), 44.7 pg/ml for IL-23 and 4.61 pg/ml for TGF-β1 (R&D, Minneapolis, MN, USA) and 7 pg/ml for IL-6 (Orgenium Laboratories-Ani Biotech Oy, Vantaa, Finland).
Statistics
Patient features were presented using mean and median for continuous variables and absolute and percentage frequency for categorical variables. To compare the Treg count and cytokine levels of patients with those of the healthy donors, we used the non-parametric Mann-Whitney two-sample statistic. Correlations among Tregs, IL-23, IL-17A, IL-6 and TGF-β1 were assessed by Spearman’s correlation coefficient. Overall survival was defined as the interval from diagnosis to death or last follow-up. Hazard ratios were estimated using the semi-parametric Cox model. The Wilcoxon matched-pairs signed-ranks test was used to compare Treg counts and cytokine levels pre- and post-treatment. Statistical analysis was carried out with Stata 9.2 (StataCorp LP, College Station, TX, USA) and SigmaStat 3.1 (Jandel Scientific, San Rafael, CA, USA].
Results
Pre-chemotherapy circulating Treg counts in pancreatic cancer patients versus healthy donors
Tregs were characterized by flow cytometry, based on coexpression of CD4/CD25high/FoxP3, in PBMCs from 62 patients with locally advanced or metastatic pancreatic cancer prior to chemotherapy, and from 20 healthy donors. Patients had a statistically significantly higher percentage of Tregs than controls [median (range): 4.72 (0.34–16.7) vs. 2.13 (0.2–7.4), p=0.004] (Fig. 1).
Figure 1.
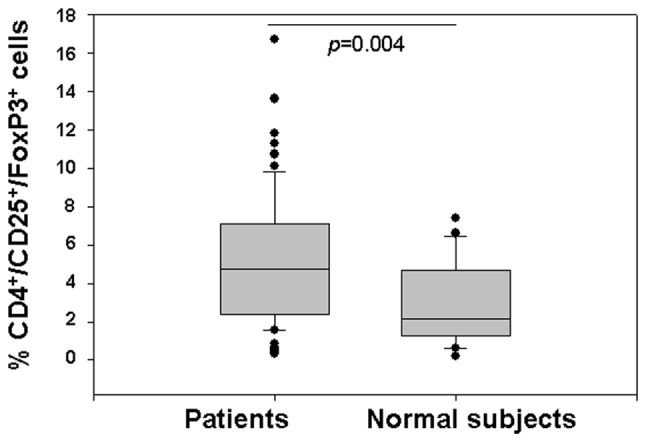
Basal Treg counts determined by flow cytometry in the peripheral blood of patients with locally advanced or metastatic pancreatic carcinoma and that of normal donors. p-values were obtained using the Mann-Whitney U test.
Pre-chemotherapy plasma levels of IL-23, IL-17A, IL-6 and TGF-β1 in pancreatic cancer patients versus healthy donors
To investigate whether the tumor microenvironment perturbs Treg generation or function via soluble factors, for the first time for pancreatic cancer, we simultaneously analyzed the profile of all the circulating cytokines involved in these complex regulatory mechanisms. Patient plasma concentrations of IL-23, IL-17A, IL-6 and TGF-β1 were measured by ELISA prior to chemotherapy and compared with corresponding healthy levels. There was no significant difference between patients and controls for levels of IL-23 [median (range) pg/ml: 18.72 (0–154.4) vs. 29.54 (0–75.67), p=0.340]. Levels of IL-6 [median (range) pg/ml: 43.42 (4.54–470.58) vs. 20.41 (1.34–48.1), p<0.001] and TGF-β1 [median (range) pg/ml: 1762.90 (907.60–5986.40) vs. 1247.70 (847.20–2290.00), p=0.003] were higher in cancer patients than those in the controls (Fig. 2). The patient-control difference in IL-17A was at the limit of statistical significance [median (range) pg/ml: 3.60 (0–130.61) vs. 0 (0–15.94), p=0.075].
Figure 2.
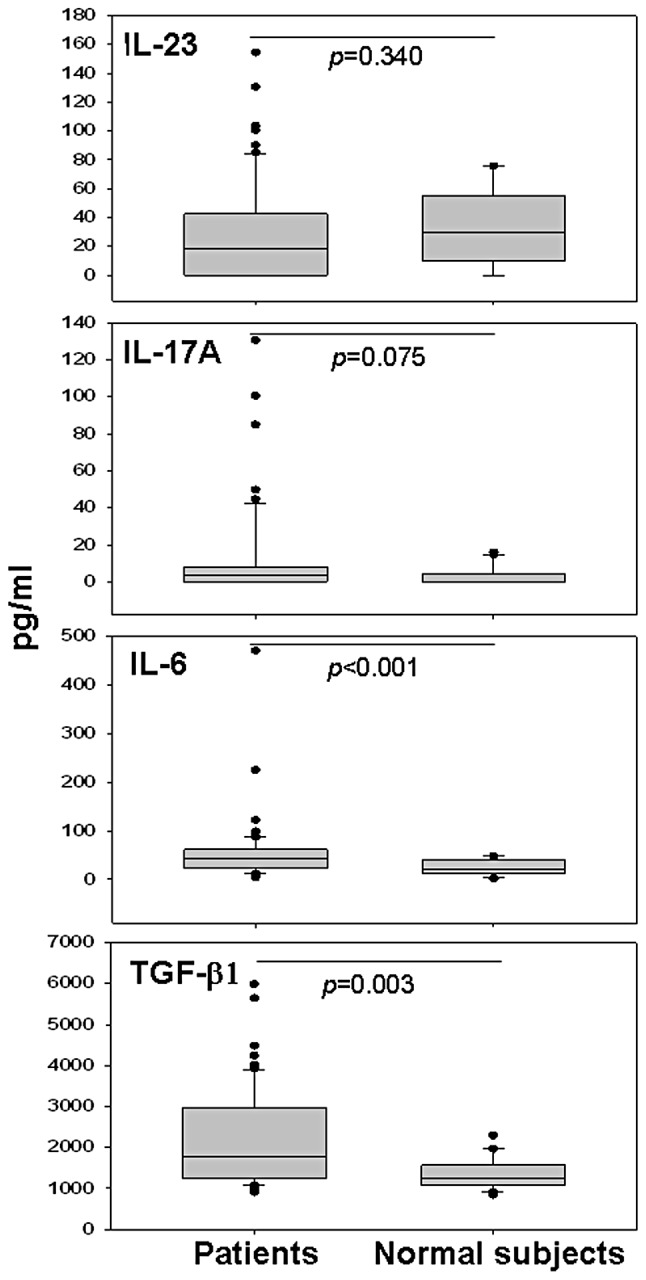
IL-23, IL-17A, IL-6 and TGF-β1 levels determined by ELISA in the plasma of patients with locally advanced or metastatic pancreatic carcinoma compared to normal donors. p-values were obtained using the Mann-Whitney U test.
Correlation among Tregs, IL-23, IL-17A, IL-6 and TGF-β1
To assess any correlation between Tregs and cytokines, we looked at the relationship between pre-chemotherapy Treg counts and the plasma concentrations of IL-23, IL-17A, IL-6 and TGF-β1. There was no correlation between Treg counts and levels of IL-23, IL-17A, IL-6 or TGF-β1 (r=0.128, p=0.322; r=0.099, p=0.444; r=−0.066, p=0.609; r=0.072, p=0.578, respectively). Concerning relationships among cytokines, there was no correlation between IL-23 and IL-6 levels (r= −0.031, p=0.809) nor between IL-23 and TGF-β1 (r=0.232, p=0.070), IL-17A and IL-6 (r=0.242, p=0.058), IL-17A and TGF-β1 (r=0.211, p=0.099) nor IL-6 and TGF-β1 (r=0.023, p=0.856). However, there was a statistically-significant positive correlation between IL-23 and IL-17A (r=0.261, p=0.041).
Correlation between disease stage, Treg count and levels of plasma IL-23, IL-17A, IL-6 and TGF-β1
To clarify the clinical significance of these findings in locally advanced or metastatic pancreatic cancer, pre-chemotherapy levels of circulating Tregs and cytokines were compared between patients categorized as being at stages II/III (n=22) and stage IV (n=40). Levels of peripheral Tregs did not differ significantly between early (II/III) and late (IV) stage disease [median (range) %: 5.13 (0.41–16.7) vs. 3.86 (0.34–13.60), p=0.162]. Furthermore, circulating levels of IL-23, IL-17A or TGF-β1 did not differ significantly between early and late stage disease [median (range] IL-23 pg/ml: 15.44 (0–130.61) vs. 18.72 (0–154.40), p=0.881; median (range) IL-17A pg/ml: 0.40 (0–130.61) vs. 4.35 (0–49.76), p=0.465; median (range) TGF-β1 pg/ml: 1580.80 (943.60–5643.50) vs. 1842.30 (907.60–5986.40), p=0.431]. Conversely, early-stage patients had significantly lower median levels of IL-6 vs. late-stage patients [median (range) pg/ml: 23.72 (10.68–225.07) vs. 52.61 (4.54–470.58), p=0.008].
Overall survival
The Cox model, adjusting for age, radical surgery, metastasis and gender, showed that the Treg counts, IL-23, and IL-6 plasma levels were not prognostic for overall survival (p=0.75, p=0.133, p=0.246, respectively). However, IL-17A, and especially TGF-β1, were significantly associated with increased risk of poor prognosis (p=0.050 and p=0.001, respectively) (Table II).
Table II.
Hazard ratio estimated by Cox model.
| Variables | Hazard ratioa | 95% CI | P-value |
|---|---|---|---|
| Tregs | 1.015 | 0.320–1.110 | 0.75 |
| IL-23 | 1.007 | 0.998–1.015 | 0.133 |
| IL-17A | 1.019 | 0.998–1.040 | 0.050 |
| IL-6 | 1.002 | 0.998–1.007 | 0.246 |
| TGF-β1 | 1.050 | 1.021–1.079 | 0.001 |
Adjusted for age, radical surgery, metastasis and gender.
Effect of different treatment protocols on the percentage of Tregs and plasma levels of IL-23, IL-17A, IL-6 and TGF-β1
Percentages of circulating Tregs pre- (Time 0) and post-chemotherapy were analyzed comparatively at first restaging (Time 1) in 15 patients treated with GEM, 14 with GEMOX and 6 treated with BEV+CAPE+RT. As Table III shows, following the stratification of patients by treatment protocol, there was no significant pre- or post-treatment difference in the percentages of peripheral Tregs in those who received GEMOX and BEV+CAPE+RT treatment. However, those receiving GEM alone showed a statistically significant reduction in Tregs (p=0.021).
Table III.
Treg and plasma levels of IL-23, IL-17A, IL-6 and TGF-β1 pre- and post-treatment.
| Chemotherapy | % Trega | IL-23 | IL-17A | IL-6 | TGF-β1 |
|---|---|---|---|---|---|
|
|
|
||||
| median (range) | pg/mlb median (range) | ||||
| GEM (n=15) | |||||
| Time 0c | 4.90 (0.87–16.7) | 34.21 (0–85) | 0 (0–85) | 40.70 (10.68–225.07) | 1691.80 (907.6–5643.5) |
| Time 1d | 2.80 (0.33–7.9) | 24.88 (0–120.24) | 0 (0–6.2) | 17.19 (3.24–160.21) | 1469.80 (690.3–3256.3) |
| p-value | 0.021 | 0.842 | 0.161 | 0.001 | 0.100 |
| GEMOX (n=14) | |||||
| Time 0 | 3.95 (0.57–11.8) | 31.10 (0–103.61) | 0 (0–130.61) | 29.67 (4.54–470.58) | 1502.90 (1127.4–5986.4) |
| Time 1 | 3.6 (0.4–10) | 21.77 (0–80.85) | 0 (0–50.7) | 20.58 (3.24–230.91) | 1855.65(1074.8–3753.3) |
| p-value | 0.638 | 0.059 | 0.046 | 0.088 | 0.683 |
| BEV+CAPE+RT (n=6) | |||||
| Time 0 | 5.10 (0.41–11.3) | 4.15 (0–81.89) | 0 (0–2.07) | 24.08 (20.27–37.62) | 1548.85 (1069.4–3269.2) |
| Time 1 | 6.36 (1.5–13) | 23.32 (0–40.43) | 0 (0–0) | 12.70 (4.54–25.60) | 1964.95 (1056–3134.8) |
| p-value | 0.753 | 0.518 | 0.159 | 0.028 | 0.463 |
Percentage of CD4+/CD25+/FoxP3+ cells obtained by flow cytometry;
plasma levels measured by ELISA.
Pre-chemotherapy;
post-chemotherapy (first restaging).
Pre- and post-therapy plasma levels of IL-23 and TGF-β1 did not differ significantly with GEM or BEV+CAPE+RT. Only GEMOX significantly reduced levels of IL-17A (p= 0.046) and IL-23 levels but only at the limit of statistical significance (p=0.059). IL-6 was significantly reduced by treatment with GEM or BEV+CAPE+RT (p=0.001, p=0.028, respectively).
Among the patients who passed first restaging after pharmacological treatment (n=57), 55.6% of those receiving GEM had stable disease (SD) with median (range) survival 457 (106–2005) days, whereas 44.4% had disease progression (DP) with median (range) survival 158.5 (43–601) days; of patients receiving GEMOX, 43.5% showed SD with median (range) survival 375 (112–836) days, whereas 56.5% had DP, with median (range) survival 118 (73–957) days; 83.4% of patients receiving BEV+CAPE+RT (n=5) had SD with median (range) survival 406 (126–770) days and 16.6% (n=1) were in partial remission (PR) with median survival of 728 days.
Objective tumor response rate, percentage of Tregs, plasma levels of IL-23, IL-17A, IL-6 and TGF-β1
Overall chemotherapy response rate was 54.3% (31 responders comprising 1 PR and 30 SD, and 26 non-responders with DP). No statistically significant responder/non-responder difference was found between pre-treatment Treg percentages and IL-6 plasma levels [median (range) % Treg: 5.06 (0.34–16.70) vs. 4.10 (1.60–13.60), p=0.773; median (range) IL-6 pg/ml: 40.7 (4.54–225.07) vs. 50.73 (4.54–470.57), p=0.178]. However, IL-23, IL-17A and TGF-β1 levels were significantly lower in responders than non-responders [median (range) IL-23 pg/ml: 4.68 (0–103.66) vs. 33.17 (0–130.61), p=0.030; median (range) IL-17A pg/ml: 0 (0–85) vs. 5.65 (0–130.61), p=0.040; median (range) TGF-β1 pg/ml: 1469.8 (907.6–5643.5) vs. 1912.85 (934.7–5986.4), p=0.032].
Discussion
Tregs are significant in tumor immune evasion, operating both by blocking generation of immunity to tumor antigens in the periphery and by neutralizing tumor-infiltrating effector T cells. They are therefore a significant obstacle to successful tumor immunotherapy (9). However, it has recently emerged that, in the tumor microenvironment, Treg cells can undergo aberrant conversion, producing the pro-inflammatory IL-17 instead of the immunosuppressive IL-10 (5). Thus, the role of Tregs may be dual, helping the tumor to survive and expand – Tregs fail to inhibit, and may contribute to, a Th17-driven pro-carcinogenic process.
Concurrently, alongside DC activation, local and systemic alterations of the effector T cell response have been reported in pancreatic cancer patients (10,11). However, in pancreatic cancer little is known regarding the effects of host Treg switch from protecting to suppressing the antitumor immune response.
This study analyzed the frequency of circulating Tregs and, for the first time, concomitant levels of IL-23, IL-17A, IL-6 and TGF-β1, cytokines implicated in the complex mutual regulation mechanism of Treg/Th17 subpopulations. Patients with locally advanced or metastatic pancreatic carcinoma were evaluated prior and subsequent to palliative chemo-therapy. Three-color flow cytometry was employed for Tregs, and ELISA was carried out to assess levels of cytokines. The results were compared with those of matched controls, investigating correlation with clinical features.
Treg levels were approximately twice the normal level in pancreatic carcinoma patients, consistent with other studies reporting increased Treg levels in various cancer patients (12–15), including pancreatic carcinoma (16).
According to the physiological-steady-state theory, Tregs in tumor patients may increase to the highest level compatible with prolonged tumor-host interaction, allowing the tumor to escape immune-mediated rejection, while protecting the host from excessive and indiscriminate immunosuppression. However, this equilibrium is temporary, and the host eventually surrenders to the overwhelming tumor burden. Animal studies have demonstrated that during tumor progression, the initial concomitant immunity is progressively lost as suppressor CD4+ T cells are generated (17). In vitro studies have revealed that suppressor T cells generated in tumor-draining lymph nodes to abrogate antitumor reactivity possess the same functional properties and FoxP3 expression levels as Tregs naturally present in the thymus in order to maintain self-tolerance (18). Tumor cells may actively recruit, activate, and expand Tregs by directly or indirectly presenting self-antigenic peptides for their recognition. Although they are refractory to T cell receptor stimulation in vitro, Tregs are able to proliferate in response to self-antigens in vivo in the presence of IL-2 (19). Moreover, suppressive cytokines such as IL-10 and TGF-β and chemokines such as CCL22, all secreted by tumor cells, tumor-infiltrating macrophages, myeloid suppressor cells and by DC, not only recruit Tregs to tumor sites, but also favor the conversion of non-suppressive T cells into suppressive-function Tregs (20).
Since the percentage of Tregs was not correlated with disease stage in our series of advanced pancreatic carcinoma patients, it is tempting to speculate that pancreatic tumor-induced Treg expansion occurs early in tumorigenesis and, once established, remains constant during the late phase of tumor progression. This has been reported to occur in ovarian, breast, head and neck cancers and gastrointestinal carcinomas (16,21–24). However, due to late presentation and delayed diagnosis, the timing of Treg expansion in pancreatic cancer is difficult to assess. Moreover, in contrast to findings in ovarian carcinoma and breast cancer (21,22), Treg frequency appears not to be correlated with poor survival in pancreatic carcinoma patients. However, since our study only investigated patients with locally advanced or metastatic cancer, the significance of Tregs as potential prognostic and predictive indicators must be evaluated prospectively, in a large cohort of patients with operable tumors.
There is mounting evidence that the composition of CD4+ cell functional subsets is dynamic and is controlled by several key cytokine networks, which play pivotal roles in promoting T regulatory or inflammatory responses upon microenvironment request. Naturally occurring CD4+CD25+FoxP3+ suppressor cells (nTregs), which govern self-tolerance, derive from the thymus, but CD4+FoxP3+ immunoregulatory cells (iTregs) related to IL-10 and/or to TGF-β can be induced in the periphery during immune response.
The conversion of CD4+FoxP3− to CD4+FoxP3+ Treg occurs at immune-privileged sites and has been shown in vivo to depend on TGF-β (25). Tumor cells themselves, including pancreatic cancer cells (26), and tumor-educated immune cells, can locally produce abundant TGF-β. It is therefore likely that the Treg pool in tumor bearers includes newly generated Tregs, alongside proliferating nTregs. However, it has recently been shown that TGF-β, under inflammatory conditions, is important in inducing Treg and IL-17-producing cells (Th17) from T CD4+ naïve cells, firmly linking these two subsets, which possess opposing functions (27). A T-cell activation state, which produces IL-17 as principle effector cytokine, can be promoted by the combined action of TGF-β and IL-6 (7); IL-23, a cytokine produced by myeloid DC and activated macrophages, is required for their expansion and maintenance (8).
We addressed the relationship between Tregs and the third arm of the CD4+ T-cell effectors, Th17, in pancreatic cancer patients, simultaneously investigating plasma levels of IL-23, IL-17A, IL-6 and TGF-β1, the crucial soluble factors orchestrating the Treg/Th17 balance (28). We found a significant elevation in the levels TGF-β1 and IL-6 and slightly elevated IL-17A levels in advanced pancreatic carcinoma patients compared to normal donors; levels of IL-23 did not differ significantly. Circulating cytokine levels were not correlated with the percentage of Tregs. However, the finding that IL-6 levels reflect disease status suggests that its overexpression may be crucial, not only in determining tumor growth, invasion, angiogenesis and cachexia, but also in controlling the Th17/Treg balance in this tumor type. IL-6 participates in differentiation of both T-cell subsets, whereas it induces Th17 differentiation, acting jointly with TGF-β, and inhibits TGF-β-induced Treg differentiation (28).
Notably, among the cytokines studied, TGF-β1 and IL-17A were significantly associated with poor prognosis. In the case of IL-17A, this is the first report of such an association in cancer. Whereas, during early tumor formation, TGF-β can function as a tumor suppressor and tends to prevent tumorigenesis, in established tumors overproduction of TGF-β is often associated with poor prognosis (29). TGF-β can suppress or alter the activation, maturation, and differentiation of innate and adaptive immune cells, including natural killer (NK) cells, DC, macrophages, neutrophils, and CD4+ and CD8+ T effector cells (29), thus promoting an overall tolerogenic state. However, depending on the cytokine milieu, TGF-β can also control the ratio between Tregs and Th17 cells in the tumor microenvironment (30). Since IL-17A was an independent factor that negatively affected prognosis in our patients, and since Th17 cells are the sole cellular source of IL-17 in the human tumor microenvironment, we may speculate that, at a certain point during tumor development, Tregs are converted to Th17, stimulated by high levels of IL-6 and TGF-β1; this contributes to tumor progression in advanced pancreatic carcinoma. A similar event has been reported in hepatocellular carcinoma and non-small cell lung carcinoma patients (31,32), whereas in ovarian carcinoma, high levels of IL-17 predict improved patient survival (33). This contradiction may be accounted for by the intensity and nature of IL-17-producing cells and the other inflammatory and immune cells infiltrated in the tumor microenvironment. The precise situations in which IL-17 has pro-tumor or antitumor activity remain to be fully explored. However, increasing evidence points to the ability of IL-17 to induce IL-6 and promote chemotaxis of inflammatory cells, angiogenesis and invasion in cancer (34). Importantly, we found a significant association between IL-23 and IL-17A plasma concentrations in support of the hypothesis that, in advanced pancreatic carcinoma, Th17-polarizing (TGF-β, IL-6) and/or stabilizing (IL-23) cytokines are important molecular links between tumor-promoting pro-inflammatory processes and the failure of adaptive immune surveillance to infiltrate tumors. In chemical carcinogenesis and transplantable tumor models, and through genetic deletion in mice, IL-23 has been shown to directly participate in triggering a pro-inflammatory cytokine cascade and sustaining proliferation and/or survival of IL-17-producing Th17 cells (35), but reducing CD8+ T-cell infiltration, activation and function (36). Significant upregulation of IL-23 mRNA has been found in the overwhelming majority of carcinoma types, as has expression of IL-17 transcript (37) as the result of the tumor-induced pro-inflammatory environment.
Like IL-12, IL-23 is efficiently produced by activated macrophages and DC; secretion of these cytokines is reciprocally regulated and affects the nature and evolution of the initial innate and immune responses. It was recently reported that Stat3 signaling, operative in the tumor microenvironment, shifts inflammation from an antitumor IL-12 program to a tumor-enhancing IL-23 program, suggesting that preponderant production of IL-23 over IL-12 by antigen-presenting cells in tumor-draining lymph nodes ultimately skews the differentiation of tumor-specific T cells, possibly leading to dominance of Treg and Th17 responses over antitumor Th1 responses (38).
Further investigation is needed, as information concerning the relevance of the IL-23/IL-17A axis in cancer biology is scant and contradictory. However, since it has been shown that IL-18 participates in the differentiation of Th17 cells in synergy with IL-23 (39), and since elevated levels of free active IL-18 can play a paradoxically detrimental role in pancreatic carcinoma (40), it is tempting to hypothesize a functional link with the present findings, envisaging a new important mechanism whereby the Th17 effector function is augmented in cancer.
As curative surgery is only practicable in a small group of pancreatic carcinoma patients, systemic palliative chemotherapy remains the standard-of-care for patients with locally advanced or metastatic cancer. This study employed standard single-therapy (GEM), two-drug combination therapy (GEMOX), or a newer therapeutic approach consisting of BEV+CAPE+RT. At the time of first restaging, GEM administered alone significantly reduced the percentage of Treg cells, as well as IL-6 levels. By contrast, GEMOX significantly reduced IL-17A plasma levels and induced downregulation of IL-23. Combined administration of BEV+CAPE+RT produced no change in the frequency of Treg, nor IL-23 or IL-17A levels, but caused a statistically significant decrease in IL-6.
The apparently contradictory effect of GEM as a single agent and GEM combined with OX is unsurprising, since metabolic and biological interactions between drugs are possible and may result in synergistic, additive, or antagonistic actions. Elsewhere we report an antagonistic interaction between GEM and 5-FU in some pancreatic cancer cases (41).
Very few data exist regarding the effects of the chemotherapeutics used here on the levels of circulating Tregs, or on plasma levels of IL-23, IL-17A, IL-6 and TGF-β1. In colon carcinoma patients, GEM in association with other chemotherapeutics, including OX, has been shown to reduce Tregs in the peripheral blood (42). However, these analyses were performed at early time points, whereas ours were after 2–3 months, depending on the chemotherapy regime; this difference may account for the discrepancy in findings. GEM has, moreover, been found to cause a significant decrease in the number of myeloid suppressor cells, one of the major sources of TGF-β1 in the cancer microenvironment, present in the spleens of tumor-bearing animals (43). CAPE and/or RT were probably responsible for the effects observed here, since these two treatments have been shown to decrease circulating IL-6 and TGF-β1 levels in human and in animal models (44,45), whereas BEV is a humanized neutralizing monoclonal antibody that acts against vascular endothelial growth factor A.
Considering the relation of immunological parameters to clinical response, irrespective of the chemotherapeutic regimen, responders (PR/SD) had lower IL-23, IL-17A, and TGF-β1 levels than non-responders, indirectly suggesting that inflammatory status may play a role in inducing chemotherapy resistance. Potential limitations of these findings could be the different types of chemotherapy employed and the limited cohort. However, some preliminary conclusions may be drawn. GEM as a single agent and the combined treatment BEV+CAPE+RT reduced plasma levels of IL-6 (a cytokine that, in association with TGF-β1, induces pro-Th17 development) and produced more favorable effects in terms of overall survival (BEV+CAPE+RT: 100% of patients with SD, median overall survival 406 days; GEM: 69% with SD, 31% with DP, median overall survival 360 days) than did GEMOX (42% with SD, 31% with DP, median overall survival 186.5 days).
Current theories indicate a role of chronic inflammation in generating conditions that contribute to malignant transformation, e.g., the progressive accumulation of genetic mutations consequent on repeated DNA damage and cell regeneration in an environment favoring proliferation and neovascularization. The present data provide novel insight into the significance of the pro-inflammatory response in human pancreatic carcinoma progression. We may speculate that, under pathophysiological conditions, the persistence of a still-unidentified inflammatory trigger in a particular context, may transform potentially protective cytokines, such as IL-17A, IL-18 and IL-23 into procancer cytokines.
These observations support our hypothesis that, in pancreatic carcinoma, during chronic inflammatory processes linked to tumor development, Tregs may augment rather than suppress Th17-mediated immune responses. This pro-inflammatory switch may play a crucial role in determining tumor progression, and paradoxically appears to be associated with a less favorable outcome. We may assume that the tumor microenvironment converts inflammation, from an IL-12-type program with strong antitumor effects, due to activation of NK cells and cytotoxic CD8+ T lymphocytes, to a tumor-promoting IL-23-type program. It is known that the prevailing production of IL-23 (rather than IL-12) by DC and tumor antigen-stimulated macrophages in tumor-draining lymph nodes and microenvironment, respectively, may result in subversion of tumor-specific T-cell differentiation, with a prevalence of Treg and Th17-type responses over the antitumor Th1 response (35). Moreover, IL-23, acting in an autocrine/paracrine manner on DC and activated macrophages, induces production of IL-1, IL-6 and TNF-α, responsible for the marked cachexia in patients with advanced pancreatic carcinoma (36).
Aberrant Tregs have been reported in polyp-ridden mice. By promoting inflammation and suppressing Th functions, these provide a dual advantage for tumor growth (46). The discovery of the two opposing cellular and signal networks (Treg/Th17, IL-23/IL-17), critically involved in modulating inflammation, induced not only by autoimmunity or bacterial infection but also by the tumor microenvironment, has dramatically changed the view of cell-mediated immune responses. Importantly, and for the first time, our results show the impact of the Treg/Th17 balance in pancreatic carcinoma in a new perspective, highlighting the significance of IL-17A as a potential prognostic and predictive indicator and showing that chemotherapy, if appropriately associated with immunotherapy, may amplify antitumor effects.
Due to its control of tumor-promoting inflammatory and angiogenic pathways and its immunoregulatory effects on tumor immune surveillance, IL-17A may be an attractive target for tumor treatment. As the underlying mechanism regulating the Th17/Treg balance is better understood, it may lead to the development of effective pancreatic cancer vaccine strategies that circumvent these tumor-related mechanisms.
Acknowledgments
We thank Dr Manuela Ceccarelli and Dr Anna Castiglione [Unit of Cancer Epidemiology (CPO-Piemonte), Azienda Ospedaliera Universitaria San Giovanni Battista, Turin, Italy] for their assistance with statistical analysis and for critical reading of the manuscript. This study was supported by research funds from the Piedmont Regional Government (Regione Piemonte, Italy) to G.B.
References
- 1.Laheru D, Jaffee EM. Immunotherapy for pancreatic cancer - science driving clinical progress. Nat Rev Cancer. 2005;5:459–467. doi: 10.1038/nrc1630. [DOI] [PubMed] [Google Scholar]
- 2.Greer JB, Whitcomb DC. Inflammation and pancreatic cancer: an evidence-based review. Curr Opin Pharmacol. 2009;9:411–418. doi: 10.1016/j.coph.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 3.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 4.Sakaguchi S, Powrie F. Emerging challenges in regulatory T cell function and biology. Science. 2007;317:627–629. doi: 10.1126/science.1142331. [DOI] [PubMed] [Google Scholar]
- 5.Erdman SE, Rao VP, Olipitz W, et al. Unifying roles for regulatory T cells and inflammation in cancer. Int J Cancer. 126:1651–1665. doi: 10.1002/ijc.24923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Z, O’Shea JJ. Th17 cells: a new fate for differentiating helper T cells. Immunol Res. 2008;41:87–102. doi: 10.1007/s12026-007-8014-9. [DOI] [PubMed] [Google Scholar]
- 7.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 8.Boniface K, Blom B, Liu YJ, de Waal Malefyt R. From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunol Rev. 2008;226:132–146. doi: 10.1111/j.1600-065X.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrone S, Whiteside TL. Tumor microenvironment and immune escape. Surg Oncol Clin N Am. 2007;16:755–774. viii. doi: 10.1016/j.soc.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 10.Bellone G, Turletti A, Artusio E, et al. Tumor-associated transforming growth factor-beta and interleukin-10 contribute to a systemic Th2 immune phenotype in pancreatic carcinoma patients. Am J Pathol. 1999;155:537–547. doi: 10.1016/s0002-9440(10)65149-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bellone G, Carbone A, Smirne C, et al. Cooperative induction of a tolerogenic dendritic cell phenotype by cytokines secreted by pancreatic carcinoma cells. J Immunol. 2006;177:3448–3460. doi: 10.4049/jimmunol.177.5.3448. [DOI] [PubMed] [Google Scholar]
- 12.Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res. 2003;9:606–612. [PubMed] [Google Scholar]
- 13.Ichihara F, Kono K, Takahashi A, Kawaida H, Sugai H, Fujii H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin Cancer Res. 2003;9:4404–4408. [PubMed] [Google Scholar]
- 14.Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005;65:2457–2464. doi: 10.1158/0008-5472.CAN-04-3232. [DOI] [PubMed] [Google Scholar]
- 15.Viguier M, Lemaitre F, Verola O, et al. Foxp3 expressing CD4+CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J Immunol. 2004;173:1444–1453. doi: 10.4049/jimmunol.173.2.1444. [DOI] [PubMed] [Google Scholar]
- 16.Liyanage UK, Moore TT, Joo HG, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–2761. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 17.North RJ, Bursuker I. Generation and decay of the immune response to a progressive fibrosarcoma. I. Ly-1+2- suppressor T cells down-regulate the generation of Ly-1-2+ effector T cells. J Exp Med. 1984;159:1295–1311. doi: 10.1084/jem.159.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiura T, Kagamu H, Miura S, et al. Both regulatory T cells and antitumor effector T cells are primed in the same draining lymph nodes during tumor progression. J Immunol. 2005;175:5058–5066. doi: 10.4049/jimmunol.175.8.5058. [DOI] [PubMed] [Google Scholar]
- 19.Walker LS, Chodos A, Eggena M, Dooms H, Abbas AK. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J Exp Med. 2003;198:249–258. doi: 10.1084/jem.20030315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamagiwa S, Gray JD, Hashimoto S, Horwitz DA. A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J Immunol. 2001;166:7282–7289. doi: 10.4049/jimmunol.166.12.7282. [DOI] [PubMed] [Google Scholar]
- 21.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 22.Merlo A, Casalini P, Carcangiu ML, et al. FOXP3 expression and overall survival in breast cancer. J Clin Oncol. 2009;27:1746–1752. doi: 10.1200/JCO.2008.17.9036. [DOI] [PubMed] [Google Scholar]
- 23.Schaefer C, Kim GG, Albers A, Hoermann K, Myers EN, Whiteside TL. Characteristics of CD4+CD25+ regulatory T cells in the peripheral circulation of patients with head and neck cancer. Br J Cancer. 2005;92:913–920. doi: 10.1038/sj.bjc.6602407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kono K, Kawaida H, Takahashi A, et al. CD4(+)CD25high regulatory T cells increase with tumor stage in patients with gastric and esophageal cancers. Cancer Immunol Immunother. 2006;55:1064–1071. doi: 10.1007/s00262-005-0092-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stein-Streilein J, Taylor AW. An eye’s view of T regulatory cells. J Leukoc Biol. 2007;81:593–598. doi: 10.1189/jlb.0606383. [DOI] [PubMed] [Google Scholar]
- 26.Bellone G, Smirne C, Mauri FA, et al. Cytokine expression profile in human pancreatic carcinoma cells and in surgical specimens: implications for survival. Cancer Immunol Immunother. 2006;55:684–698. doi: 10.1007/s00262-005-0047-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee YK, Mukasa R, Hatton RD, Weaver CT. Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol. 2009;21:274–280. doi: 10.1016/j.coi.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 28.Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. 2010;40:1830–1835. doi: 10.1002/eji.201040391. [DOI] [PubMed] [Google Scholar]
- 29.Wrzesinski SH, Wan YY, Flavell RA. Transforming growth factor-beta and the immune response: implications for anticancer therapy. Clin Cancer Res. 2007;13:5262–5270. doi: 10.1158/1078-0432.CCR-07-1157. [DOI] [PubMed] [Google Scholar]
- 30.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 31.Zhang JP, Yan J, Xu J, et al. Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol. 2009;50:980–989. doi: 10.1016/j.jhep.2008.12.033. [DOI] [PubMed] [Google Scholar]
- 32.Chen X, Wan J, Liu J, et al. Increased IL-17-producing cells correlate with poor survival and lymphangiogenesis in NSCLC patients. Lung Cancer. 69:348–354. doi: 10.1016/j.lungcan.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 33.Kryczek I, Banerjee M, Cheng P, et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood. 2009;114:1141–1149. doi: 10.1182/blood-2009-03-208249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaffen SL. An overview of IL-17 function and signaling. Cytokine. 2008;43:402–407. doi: 10.1016/j.cyto.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langowski JL, Zhang X, Wu L, et al. IL-23 promotes tumour incidence and growth. Nature. 2006;442:461–465. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- 36.Langowski JL, Kastelein RA, Oft M. Swords into plowshares: IL-23 repurposes tumor immune surveillance. Trends Immunol. 2007;28:207–212. doi: 10.1016/j.it.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 37.Numasaki M, Fukushi J, Ono M, et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood. 2003;101:2620–2627. doi: 10.1182/blood-2002-05-1461. [DOI] [PubMed] [Google Scholar]
- 38.Kortylewski M, Xin H, Kujawski M, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor micro-environment. Cancer Cell. 2009;15:114–123. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 40.Carbone A, Vizio B, Novarino A, et al. IL-18 paradox in pancreatic carcinoma: elevated serum levels of free IL-18 are correlated with poor survival. J Immunother. 2009;32:920–931. doi: 10.1097/CJI.0b013e3181b29168. [DOI] [PubMed] [Google Scholar]
- 41.Bellone G, Carbone A, Busso V, et al. Antagonistic interactions between gemcitabine and 5-fluorouracil in the human pancreatic carcinoma cell line Capan-2. Cancer Biol Ther. 2006;5:1294–1303. doi: 10.4161/cbt.5.10.3152. [DOI] [PubMed] [Google Scholar]
- 42.Correale P, Rotundo MS, Del Vecchio MT, et al. Regulatory (FoxP3+) T-cell tumor infiltration is a favorable prognostic factor in advanced colon cancer patients undergoing chemo or chemoimmunotherapy. J Immunother. 33:435–441. doi: 10.1097/CJI.0b013e3181d32f01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–6721. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 44.Fengming K, Anscher MS, Zhiping X, Jirtle RL. Elevated circulating transforming growth factor beta1 (TGFβ1) levels decreased after radiotherapy in patients with lung cancer, cervical cancer and Hodgkin’s disease: a possible tumor marker. Int J Radiat Oncol Biol Phys. 1995;32:239. [Google Scholar]
- 45.Fujimoto-Ouchi K, Onuma E, Shirane M, Mori K, Tanaka Y. Capecitabine improves cancer cachexia and normalizes IL-6 and PTHrP levels in mouse cancer cachexia models. Cancer Chemother Pharmacol. 2007;59:807–815. doi: 10.1007/s00280-006-0338-y. [DOI] [PubMed] [Google Scholar]
- 46.Gounaris E, Blatner NR, Dennis K, et al. T-regulatory cells shift from a protective anti-inflammatory to a cancer-promoting proinflammatory phenotype in polyposis. Cancer Res. 2009;69:5490–5497. doi: 10.1158/0008-5472.CAN-09-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]