

Abstract
AIM: To determine the effect of tumor necrosis factor alpha (TNF-α) on intestinal permeability (IP) in mice with fulminant hepatic failure (FHF), and the expression of tight junction proteins.
METHODS: We selected D-lactate as an index of IP, induced FHF using D-galactosamine/lipopolysaccharide and D-galactosamine/TNF-α, assessed the results using an enzymatic-spectrophotometric method, transmission electron microscopy, immunohistochemistry, Western blotting and real-time quantitative polymerase chain reaction. The effect of the administration of anti-TNF-α immunoglobulin G (IgG) antibody, before the administration of D-galactosamine/lipopolysaccharide, on TNF-α was also assessed.
RESULTS: IP was significantly increased in the mouse model of FHF 6 h after injection (13.57 ± 1.70 mg/L, 13.02 ± 1.97 mg/L vs 3.76 ± 0.67 mg/L, P = 0.001). Electron microscopic analysis revealed tight junction (TJ) disruptions, epithelial cell swelling, and atrophy of intestinal villi. Expression of occludin and claudin-1 mRNA was significantly decreased in both FHF models (occludin: 0.57 ± 0.159 fold vs baseline, P = 0.000; claudin-1: 0.3067 ± 0.1291 fold vs baseline, P = 0.003), as were the distribution density of proteins in the intestinal mucosa and the levels of occludin and claudin-1 protein (occludin: 0.61 ± 0.0473 fold vs baseline, P = 0.000; claudin-1: 0.6633 ± 0.0328 fold vs baseline, P = 0.000). Prophylactic treatment with anti-TNF-α IgG antibody prevented changes in IP (4.50 ± 0.97 mg/L vs 3.76 ± 0.67 mg/L, P = 0.791), intestinal tissue ultrastructure, and the mRNA levels of occludin and claudin-1 expression (occludin: 0.8865 ± 0.0274 fold vs baseline, P = 0.505; claudin-1: 0.85 ± 0.1437 fold vs baseline, P = 0.1), and in the protein levels (occludin: 0.9467 ± 0.0285 fold vs baseline, P > 0.05; claudin-1: 0.9533 ± 0.0186 fold vs baseline, P = 0.148).
CONCLUSION: Increased in IP stemmed from the downregulation of the TJ proteins occludin and claudin-1, and destruction of the TJ in the colon, which were induced by TNF-α in FHF mice.
Keywords: Tumor necrosis factor alpha, Fulminant hepatic failure, Intestinal permeability, Occludin, Claudin-1
INTRODUCTION
Fulminant hepatic failure (FHF) is a devastating disease with high mortality. The intestinal mucosal barrier plays an important role in the body’s protection against luminal pathogens and antigenic molecules. Increased intestinal permeability (IP) and disruption of the intestinal mucosal barrier have been observed in many hepatic diseases[1-4] that cause intestinal endotoxemia (IETM), also called gut-derived endotoxin. High levels and incidence of IETM have been reported in many hepatic diseases[5,6]. The IETM results in excessive inflammatory responses, with serious hepatic necrosis, further severe hepatitis, and even accelerated liver failure.
There are two potential pathways for passive permeation across the intestinal epithelia: the transcellular pathway (through the cells) and the paracellular pathway (between cells). The tight junction (TJ) is a semipermeable barrier that allows certain solutes (depending upon their size and charge) to pass through the paracellular pathway between the cells from one fluid component to the other, and even regulates the permeability of the paracellular spaces. It is thought that TJ may be functionally altered in model disease states, even when the intestinal epithelium remains confluent[7]. Structurally, TJs are composed of cytoplasmic proteins, including the zonula occludens (ZO) proteins, ZO-1, ZO-2, and ZO-3[8,9], as well as two distinct transmembrane proteins, occludin and claudins[10,11], especially claudin-1, the most widely distributed protein in the intestinal epithelium. Occludin and claudins are the major transmembrane proteins that interact with intracellular plaque proteins such as ZO-1, ZO-2, and ZO-3, which in turn interact with the actin cytoskeleton to anchor occludin and other transmembrane proteins at the apical end of the lateral membrane[1]. It has been shown that TJ proteins and IP could be modulated by many factors, including shock[12], hyperthermia[13], alcoholic or nonalcoholic liver disease[2,3], and referred to the cytokines, tumor necrosis factor alpha (TNF-α), and interleukin (IL)-1, and nitric oxide (NO)[14-17]. There are few studies on IP and its mechanism of modulation in FHF.
TNF-α is a key pro-inflammatory cytokine with a broad-spectrum of effects, and is primarily secreted by monocytes and macrophages. It can induce a hepatocyte necrosis in vivo or in vitro, and is elevated in FHF[18,19]. TNF-α can increase IP in vitro[14,17]; thus its role in the possible alteration of IP in FHF should be considered. D-lactate is produced by indigenous bacteria in the gastrointestinal tract[20]. Mammals do not have the enzyme systems to rapidly metabolize D-lactate. Evaluation of D-lactate levels indicates readily the change of IP[21]. We have tested TNF-α concentrations and intestinal ZO-1 expression in the FHF model in mice[22]. In this study, we utilized the same model and controls to investigate the changes in the permeability of the intestinal mucosa and attempted to elucidate the underlying molecular mechanisms by examining TJ ultrastructure and by measuring the expression of the TJ proteins, occludin, and claudin-1.
MATERIALS AND METHODS
Animals and treatment
Male, six-to eight-week-old BALB/c mice (China Medical University) were obtained from the China Medical University (Shenyang, China). They were housed and cared for in rooms maintained at a constant temperature and humidity. Food and water were allowed ad libitum. The mice were fasted overnight for the experiment. All animal experimental procedures were approved by the Ethics Committee of China Medical University before the commencement of the study. All mice were randomly divided into eight groups (n = 7 per group). One group of mice was given intraperitoneal injections of D-galactosamine (GalN; 800 mg/kg body weight; Sigma, Saint Louis, United States) and lipopolysaccharide (LPS; 10 μg/kg body weight; Sigma) to induce acute liver failure (ALF). A second ALF-induction group was also given intraperitoneal injections of GalN (800 mg/kg body weight) and TNF-α (10 μg/kg body weight; Sigma). First group was given anti-TNF-α immunoglobulin G (IgG) (100 μg per mouse; US Biological, United States) antibody treatment prior to ALF induction. The anti-TNF-α IgG antibody was injected via the vena caudalis 30 min before GalN/LPS administration. There were four control groups, which were injected intraperitoneally with GalN, LPS, TNF-α, or normal saline (NS). In summary, the eight groups were: (1) GalN/LPS; (2) GalN/TNF-α; (3) GalN control; (4) LPS control; (5) TNF-α control; (6) NS control; and (7) anti-TNF-α antibody and GalN/LPS (named anti-TNF-α IgG antibody pretreated group). Mice in first group were euthanized 2, 6, 9, 12 and 24 h after treatment. Other aforementioned groups were euthanized 6 h after administration of GalN/LPS.
D-lactate determination
The plasma from systemic blood samples was obtained and subjected to a deproteination and neutralization process by acid/base precipitation using perchloric acid and potassium hydroxide. The protein-free plasma was then assayed for D-lactate concentration by enzymatic-spectrophotometric method[23].
Detection and observation of colonic mucosal ultrastructure
Ultrathin (70 nm) colon sections were examined using a transmission electron microscope (Hitachi H-600, Japan).
Immunohistochemistry of occludin, claudin-1
Frozen colon tissue sections (5 μm thick) were fixed on glass slides by incubating them in acetone for 10 min at 4 °C. The slides were incubated with 3% H2O2 for 20 min at room temperature and indirectly immunolabeled using an ABC kit (Takara, Japan) according to the manufacturer’s instructions. Slides were then blocked in goat serum for 30 min at 37 °C and incubated with a rabbit anti-mouse polyclonal occludin, claudin-1 antibody (dilution, 1:50; Santa Cruz Biotechnology, United States) at 4 °C overnight. For the negative controls, the primary antibody was replaced with phosphate buffered saline (PBS). This incubation was followed by incubation with biotinylated goat anti-rabbit IgG (Histostain-Plus kit, ZYMED) diluted 1:300 in PBS for 2 h at room temperature. Sections were rinsed in PBS and then in distilled water. The slides were stained with 3,3’-diaminobenzidine and counterstained with hematoxylin.
Western blotting analysis of occludin, claudin-1
Intestinal tissue samples were homogenized in lysis buffer [20 mmol Tris-HCl (pH 7.5), 1% Triton X 100, 0.2 mol NaCl, 2 mmol ethylene diamine tetraacetic acid, 2 mmol ethylene glycol tetraacetic acid, 1 mol dithiothreitol and 2 mol aprotinin]. Proteins (50 μg) were electrophoresed using sodium dodecyl sulfate polyacrylamide gel electrophoresis (8%) and transferred to a nitrocellulose membrane. Membranes were blocked with non-fat dried milk in tris buffered saline containing 0.05% Tween-20 (TTBS) for 1 h at room temperature and incubated with a rabbit anti-mouse polyclonal occludin, claudin-1 antibody (diluted 1:400; Santa Cruz Bio-technology) at 4 °C overnight. After three washes in TTBS, the membranes were reacted with a 1:2000 dilution of alkaline phosphatase-labeled goat anti-rabbit IgG (Santa Cruz Biotechnology) for 2 h at room temperature. The immunoreaction was visualized using α-dianisidine and β-naphthyl acid phosphate (Sigma, United States).
RNA isolation and real-time quantitative polymerase chain reaction
Total RNA was isolated from intestinal tissues using TRI-zol Reagent (Invitrogen, United States). RNA was purified using DNase I and depurified using PI-PCI-EHCO. SYBR-green-based real-time polymerase chain reaction (PCR) [TaKaRa SYBR reverse transcription-PCR kit, Japan] was used to measure relative gene expression in each sample. PCR was performed using Taq DNA polymerase (Qiagen, Valencia, United States) and oligonucleotide primers for mouse were occludin (forward 5’-GCTTATCTTGGGAGCCTGGACA-3’, reverse 5’-GTCATTGCTTGGTGCATAATGATTG-3’), claudin-1 (forward 5’-AGACCTGGATTTGCATCTTGGTG-3’, reverse 5’-TGCAACATAGGCAGGACAAGAGTTA-3’) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH: forward 5’-TGTGTCCGTCGTGGATCTGA-3’, reverse 5’-TTGCTGTTGAAGTCGCAGGAG -3’). PCR conditions were as follows: one cycle at 95 °C for 30 min followed by 45 cycles of PCR amplification, each consisting of 95 °C for 5 s and 57 °C for 20 s. The concentration of mRNA was calculated according to the standard curve and then normalized to that of GAPDH.
Statistical analysis
SPSS version 13.0 Software was used to perform the statistical analyses. All datas were analyzed using analysis of variance followed by a least-squares difference test. A P < 0.05 was used to indicate statistical significance.
RESULTS
D-lactate
Compared to the normal control, plasma D-lactate levels increased after 2 h and reached significant levels 6 h after injection in GalN/LPS-treated mice (13.57 ± 1.70 mg/L vs 3.76 ± 0.67 mg/L, P < 0.01) (Figure 1A). The levels also increased significantly 6 h after injection in GalN/TNF-α- and TNF-α-treated mice (13.02 ± 1.97 mg/L, 12.48 ± 2.49 mg/L vs 3.76 ± 0.67 mg/L, P < 0.01), but did not show an increase in LPS-, GalN- and prophylactically anti-TNF-α IgG- treated groups (6.60 ± 1.73 mg/L, 4.69 ± 1.46 mg/L, 4.50 ± 0.97 mg/L vs 3.76 ± 0.67 mg/L, P > 0.05) (Figure 1B).
Figure 1.
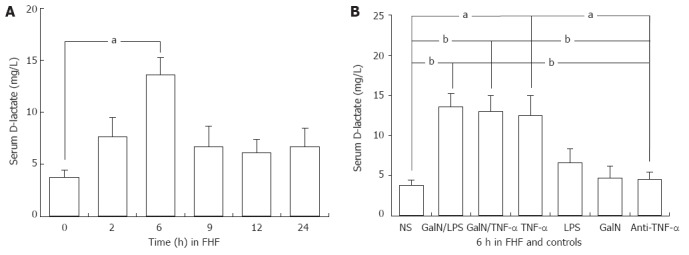
Systemic blood D(-)-lactate levels in the mouse model of fulminant hepatic failure and controls. Systemic blood D (-)-lactate levels 2, 6, 9, 12 and 24 h after injection in D-galactosamine (GalN)/lipopolysaccharide (LPS)-treated mice, and 6 h after injection in the saline, GalN, LPS, tumor necrosis factor alpha (TNF-α), anti-TNF-α immunoglobulin G antibody-pretreated groups. n = 5 rats for each time point. NS: Normal saline; FHF: Fulminant hepatic failure. The level of significance was set at aP < 0.05, bP < 0.01 vs baseline (NS group). Data are reported as the mean ± SD.
Ultrastructural characteristics of the colonic mucosa
We observed obvious ultrastructural changes in the intestinal mucosa 6 h after GalN/LPS administration. Some epithelial cell microvilli were disordered and distorted, and they were sparsely distributed. The epithelial cells were swollen or shrunken. Mitochondrial swelling was observed; cristae breakdown and disrupted tight junctions were also noted. The changes in the intestinal mucosa of mice treated with GalN/TNF-α were less severe than those of mice treated with GalN/LPS. There was no disruption of TJs in the control groups, including GalN, LPS and TNF-α groups; only less swelling of epithelial cells and distorted microvilli were observed. Pathological changes in the group prophylactically treated with antibody were less severe than those in the ALF groups, and even similar to the NS control group (Figure 2).
Figure 2.

Transmission electron microscopy of mouse intestine (8000×). Transmission electron microscopy of mice intestine from the D-galactosamine (GalN)/lipopolysaccharide (LPS) and GalN/tumor necrosis factor alpha (TNF-α) groups (A and B), the control groups (C, D, E and F), and the group that received antibody prior to fulminant hepatic failure induction (G) (8000×). A: At 6 h after injection in the GalN/LPS group. The mitochondria of the endothelial cells were loose. Tight junctions (TJs) (arrow) were disrupted. Organelles were swollen and had reduced electron density; some microvilli were loose. A TJ (arrow) was disrupted; B: At 6 h after injection in the GalN/TNF-α group, mitochondria, organelles and microvilli were similar to A. A TJ (arrow) was disrupted; C: The GalN control group; D: The LPS control group. Epithelial cells were slightly shrunken and TJs (arrow) were intact; E: TNF-α-treated group. TJ (arrow) was intact; F: The normal saline control group. Epithelial cells and TJ (arrow) were intact; G: Anti-TNF-α antibody and the GalN/LPS group. Epithelial cells were slightly shrunken and TJs (arrows) between endothelial cells were intact.
Expression of occludin and claudin-1 proteins in the colon
Immunohistochemical analysis revealed a high density of occludin and claudin-1 expression in the lateral membrane of the intestinal epithelium in the anti-TNF-α IgG antibody-pretreated group and the saline group. The traces of occludin and claudin-1 proteins weakened in the intestinal tissue 2 h after GalN/LPS treatment, and were most pale 9 h after the injections (Figure 3A and B). The traces slightly weakened even in the TNF-α control group.
Figure 3.

The tight junction proteins, occludin and claudin-1 determined by immunohistochemistry (400×). A: Occludin staining in the saline group; B-D: Occludin staining 2 h, 6 h, and 9 h after injection in the D-galactosamine (GalN)/lipopolysaccharide (LPS)-treated mice; E-H: Occludin staining 6 h after injection in the anti-tumor necrosis factor alpha (TNF-α) immunoglobulin G (IgG) antibody pretreated group within the TNF-α-treated group; the LPS-treated group and the D-GalN-treated group, respectively; I: Claudin-1 staining in the saline group; J-L: Claudin-1 staining 2, 6, and 9 h after injection in the GalN/LPS-treated mice; M-P: Claudin-1 staining 6 h after injection in the anti-TNF-α IgG antibody pretreated group, the TNF-α-treated group, the LPS-treated group and the D-GalN-treated group, respectively. The mucosal tissue sections were double-labeled for proteins (brown color). Labeled sections were analyzed immunohisto-chemically. Decreased protein staining in the epithelial cells were observed at 2-9 h after injection in the GalN/LPS-treated mice and the TNF-α-treated group, and was absent in the other controls and the antibody pretreated group.
The protein level of the NS group was set to 1 as the baseline. Western blotting analysis showed that occludin and claudin-1 expressions decreased significantly in the GalN/LPS-treated mice, particularly 6 h and 9 h after the GalN/LPS injections (occludin: 0.6733 ± 0.0433, 0.61 ± 0.0473 fold vs baseline, P < 0.01; claudin-1: 0.8600 ± 0.0208, 0.6633 ± 0.0328 fold vs baseline, P < 0.01) (Figure 4A and C). Expressions of these proteins were decreased significantly in the GalN/TNF-α and TNF-α groups 6 h after injection (occludin: 0.6133 ± 0.0145 fold vs baseline, P < 0.01, 0.8933 ± 0.01 fold vs baseline, P < 0.05; claudin-1: 0.6267 ± 0.0145 fold vs baseline, P < 0.01, 0.92 ± 0.0173 fold vs baseline, P < 0.05), which were less in the later group (Figure 4B and D). The protein expression in the antibody-treated group was close to the normal range (occludin: 0.9467 ± 0.0285 fold vs baseline, P > 0.05; claudin-1: 0.9533 ± 0.0186 fold vs baseline, P > 0.05) (Figure 4B and D).
Figure 4.

Mucosal colonic tissues from fulminant hepatic failure and control mice were analyzed for the protein expression of occludin and claudin-1 by Western blotting analysis. A: Occludin expression decreased significantly 6, 9 and 12 h after D-galactosamine (GalN)/lipopolysaccharide (LPS) injection; B: Occludin expression decreased significantly 6 h after injection in the GalN/ tumor necrosis factor alpha (TNF-α)- and TNF-α-treated mice; C: Claudin-1 expression decreased significantly 6 h and 9 h after GalN/LPS injection; D: Claudin-1 expression decreased significantly 6 h after injection in the GalN/TNF-α- and TNF-α-treated mice. NS: Normal saline; FHF: Fulminant hepatic failure. aP < 0.05, bP < 0.01 vs baseline. Data are reported as the mean ± SD.
Expression of occludin and claudin-1 mRNA in the colon
The mRNA level of the NS group was set to 1 as baseline. We obtained a reasonable amplification curve and a standard curve of occludin, claudin-1 and GAPDH RNAs, respectively. The correlation coefficients of all standard curves were 0.999. Real-time PCR quantitative analysis showed that there were marked decreases in occludin and claudin-1 expression in ALF mice 6 h and 9 h after GalN/LPS treatment (occludin: 0.57 ± 0.159 fold vs baseline, 0.345 ± 0.0247 fold vs baseline, P < 0.01; claudin-1: 0.3067 ± 0.1291 fold vs baseline, 0.2233 ± 0.1155 fold vs baseline, P < 0.01) (Figure 5A and C), as well as 6 h after GalN/TNF-α treatment and TNF-α treatment (occludin: 0.2562 ± 0.0945 fold vs baseline, P < 0.01; 0.6421 ± 0.164 fold vs baseline, P < 0.05; claudin-1: 0.1366 ± 0.0661 fold vs baseline, P < 0.01; 0.5356 ± 0.1874 fold vs baseline, P < 0.05) (Figure 5B and D). The mRNA expression in the antibody-treated group was close to the normal range (occludin: 0.8865 ± 0.0274 fold vs baseline, P > 0.05; claudin-1: 0.85 ± 0.1437 fold vs baseline, P > 0.05) (Figure 5B and D).
Figure 5.

Real-time quantitative reverse transcription-polymerase chain reaction for occludin and claudin-1 mRNA in the fulminant hepatic failure groups and the control groups. A: Occludin significantly decreased 6 h, 9 h and 12 h after D-galactosamine (GalN)/lipopolysaccharide (LPS) injections in GalN/LPS-treated mice; B: Occludin markedly decreased 6 h after injection in the GalN/tumor necrosis factor alpha (TNF-α) or TNF-α treated mice; C: Claudin-1 significantly decreased 6 h, 9 h and 12 h after GalN/LPS injections in the GalN/LPS treated mice; D: Claudin-1 markedly decreased 6 h after injection in the GalN/TNF-α- or TNF-α-treated mice. NS: Normal saline; FHF: Fulminant hepatic failure; qPCR: Quantitative polymerase chain reaction. aP < 0.05, bP < 0.01 vs baseline. Data are reported as the mean ± SD.
DISCUSSION
The intestinal mucosa is the physical and metabolic barrier that separates cytotoxic components from gut lumen. Disruption of the gastrointestinal barrier function and the diffusion of luminal toxins and pathogens into the systemic circulation are central to the pathogenesis of a number of diseases. Increased IP has been reported in many hepatic diseases[2-4,6]. IETM shows an inclination correlation with the D-lactate levels in liver cirrhosis and severe injuries[24,25], and appears to be related to IP. The incidence of IETM reaches 80%-100% in severe hepatitis, and plays an important role in the secondary liver injury observed in FHF[9]. How is IETM generated in FHF? When derived from overproduced bacteria in the lumen of the colon and terminal ileum[26,27], endotoxin (LPS) is translocated through the intestinal mucosa and into blood, causing IETM. Binding to LPS-binding protein, high density lipoprotein and low density lipoprotein[28,29], IETM is transported into the portal vein and achieves a higher level in the portal circulation than in peripheral veins[30]. When beyond the detoxification capability of debilitated monocytes and Kupffer cells, IETM damages hepatocytes and accelerates liver failure, even causing systemic injury[9,31]. To detect IP, we utilized D-lactate as an index. IP reached a peak 6 h post-injection of GalN/LPS. In GalN/LPS-treated mice, TNF-α levels reached its first peak 2 h post-injection[22]. When TNF-α was blocked with anti-TNF-α IgG antibody, there was a significant decrease in the IP of the mice. Considering that TNF-α causes an increase in the epithelial barrier permeability in vivo and in vitro, it is concluded that TNF-α is a crucial factor in the increased IP in FHF mice.
Borgstrom et al[32] have showed that at least 85% of the passive flow across the mammalian small intestine takes the paracellular route. Since the cotransporters of the apical membrane are saturated at a low nutrient concentration (below 25 mmol for glucose), but after a meal, nutrient concentrations in the proximal intestine often exceed 200 mmol, it follows that the paracellular pathway may be the major route of nutrient uptake. Drewe et al[33] has indicated that LPS is taken up by the intestinal mucosa predominantly by a transcellular pathway through enterocytes. Under ischemic conditions, the permeability of LPS is increased mainly by an enhanced paracellular translocation across the gut wall, whereas little apoptosis was observed in the intestinal epithelium. In several studies TNF-α induced an increase in the TJ permeability of Caco-2 cells, and did not induce apoptosis in the cells[14,34], indicating that apoptosis was not responsible for the increase in epithelial TJ permeability. In our previous study, terminal dUTP nick end labeling-positive enterocytes were not seen in the intestine of GalN/LPS and GalN/TNF-α-treated mice until 12 h after injection[35]. The increased IP observed 6 h after GalN/LPS and GalN/TNF-α treatment was not related significantly to the transcellular pathway. Because TJ regulates the paracellular route in the leakage of macromolecules, we examined TJ with TEM and tested the protein and mRNA levels of TJ proteins. Disrupted TJ structures, down-regulated occludin and claudin-1 protein and RNA levels, and a lower density of proteins distributed in the intestinal mucosa were observed in GalN/LPS- and GalN/TNF-α-treated mice 6 h after injection. These were not observed in normal controls and in the group treated prophylactically with anti-TNF-α IgG antibody. Similar results were obtained with another TJ protein, ZO-1, in our previous study[22]. These observations indicate that TNF-α plays a central role in the regulation of TJ proteins, resulting in the disruption of TJ structure. Considering that TNF-α disrupts TJ structure and increases transepithelial permeability in vitro and in vivo, we conclude that the altered IP is induced by TNF-α via the destruction of TJ. The destruction of TJ by TNF-α was the result of down-regulation of TJ proteins. In the TNF-α-treated group, because of the lower extent of down-regulation of the TJ proteins, ZO-1[22], occludin and claudin-1 in the TNF-α-treated group, TJs were not visibly disrupted.
When IP was increased in GalN/LPS-treated mice 6 h after injection, the TJ structure was disrupted. In our previous study, ZO-1 showed an insignificant downregulated expression[22], whereas occludin and claudin-1 were significantly down-regulated in GalN/LPS-treated mice 6 h after injection. This indicated a crucial role of occludin and claudin-1 in the composition of TJ. In vitro, TNF-α had a dose-dependent increasing effect on the permeability of the epithelium[15], and IETM had a severe effect on IP[36]. TNF-α could stimulate the expression of IL-1 and NO, which could disrupt TJ structure and increase epithelium permeability[14,18,37,38], and even synergize with interferon-α to induce intestinal epithelial barrier dysfunction[38]. In vitro on Caco-2 cell monolayers, TNF-α decreased the expression of phosphorylated occludin in detergent-insoluble fractions, but did not affect the expression of non-phosphorylated occludin protein. It also caused a decrease in Caco-2 transepithelial resistance and an increase in transepithelial permeability to a paracellular marker, Lucifer yellow[15]. It seemed that the combined decreased effect on IP was maximized 6 h after injection of GalN/LPS in the mouse model of FHF. Few studies on the mechanism of TNF-α-mediated decrease in the induction of TJ proteins were found. This effect might be related to nuclear factor kappa B (NF-κB). NF-κB activation and nuclear translocation of NF-κB p65 has been observed when TNF-α induces an increase in the TJ permeability of Coca-2 cell monolayers[34]. In our previous experiments on Caco-2 cell monolayers, TNF-α induced an increase of TJ permeability to Lucifer yellow, which could be partially inhibited by preincubation with 2 μg/mL anti-TNF receptor I monoclonal antibody and anti-TNF receptor II monoclonal antibody (data not shown). This observation suggests that TNF receptor I and TNF receptor II mediate the TNF-α induced increase in tight junction permeability. Further studies are necessary to clarify the underlying signal transduction system of the TNF-α-induced decrease in the induction of TJ proteins and the increase in TJ permeability in vitro and in vivo.
In conclusion, we found that the increase in IP stems from the downregulation of TJ proteins, especially occludin and claudin-1, and the subsequent destruction of the TJ structure that were induced by TNF-α in the mouse model of FHF.
COMMENTS
Background
Fulminant hepatic failure (FHF) is a devastating disease with many complications. Intestinal endotoxemia can lead to the pathogenesis of hepatic failure. It was therefore essential to detect the intestinal permeability (IP) in hepatic failure as the source of intestinal endotoxemia.
Research frontiers
It has been shown that altered IP occurs in many hepatic diseases. It seems that these alterations are induced by a variety of cytokines. Moreover, specified dates and underlying mechanisms deserved to illustrate about pathogenesis of IP alteration.
Innovations and breakthroughs
This is the first study to report an increased IP in FHF. Their observations support the hypothesis that tumor necrosis factor alpha down-regulates intestinal tight junction proteins and constructed to the progress of IP alteration.
Applications
The study provides insights into the mechanisms of increased IP in FHF, which probably contributes to the progress of the disease.
Peer review
This study has shown the increased IP in FHF and illustrated a probable mechanism. It therefore contributes to the understanding of intestinal complications in FHF.
Footnotes
Supported by National Ministry of Health of China, No. 97100252
Peer reviewers: Tamara Vorobjova, Senior Researcher, Department of Immunology, University of Tartu, Ravila 19, 50411 Tartu, Estonia; Zhao-Xiang Bian, Professor, School of Chinese Medicine, Hong Kong Baptist University, 1/F Jockey Club School of Chinese Medicine Building, 7 Baptist University Road, Kowloon Tong, Hong Kong, China
S- Editor Gou SX L- Editor A E- Editor Li JY
References
- 1.Al-Sadi R, Ye D, Said HM, Ma TY. IL-1beta-induced increase in intestinal epithelial tight junction permeability is mediated by MEKK-1 activation of canonical NF-kappaB pathway. Am J Pathol. 2010;177:2310–2322. doi: 10.2353/ajpath.2010.100371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Mascianà R, Forgione A, Gabrieli ML, Perotti G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- 3.Rao RK, Seth A, Sheth P. Recent Advances in Alcoholic Liver Disease I. Role of intestinal permeability and endotoxemia in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;286:G881–G884. doi: 10.1152/ajpgi.00006.2004. [DOI] [PubMed] [Google Scholar]
- 4.Such J, Guardiola JV, de Juan J, Casellas JA, Pascual S, Aparicio JR, Solá-Vera J, Pérez-Mateo M. Ultrastructural characteristics of distal duodenum mucosa in patients with cirrhosis. Eur J Gastroenterol Hepatol. 2002;14:371–376. doi: 10.1097/00042737-200204000-00006. [DOI] [PubMed] [Google Scholar]
- 5.Han DW. Intestinal endotoxemia as a pathogenetic mechanism in liver failure. World J Gastroenterol. 2002;8:961–965. doi: 10.3748/wjg.v8.i6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin RS, Lee FY, Lee SD, Tsai YT, Lin HC, Lu RH, Hsu WC, Huang CC, Wang SS, Lo KJ. Endotoxemia in patients with chronic liver diseases: relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J Hepatol. 1995;22:165–172. doi: 10.1016/0168-8278(95)80424-2. [DOI] [PubMed] [Google Scholar]
- 7.Madara JL. Loosening tight junctions. Lessons from the intestine. J Clin Invest. 1989;83:1089–1094. doi: 10.1172/JCI113987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haskins J, Gu L, Wittchen ES, Hibbard J, Stevenson BR. ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J Cell Biol. 1998;141:199–208. doi: 10.1083/jcb.141.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morita K, Itoh M, Saitou M, Ando-Akatsuka Y, Furuse M, Yoneda K, Imamura S, Fujimoto K, Tsukita S. Subcellular distribution of tight junction-associated proteins (occludin, ZO-1, ZO-2) in rodent skin. J Invest Dermatol. 1998;110:862–866. doi: 10.1046/j.1523-1747.1998.00209.x. [DOI] [PubMed] [Google Scholar]
- 10.Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141:1539–1550. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szalay L, Umar F, Khadem A, Jafarmadar M, Fürst W, Ohlinger W, Redl H, Bahrami S. Increased plasma D-lactate is associated with the severity of hemorrhagic/traumatic shock in rats. Shock. 2003;20:245–250. doi: 10.1097/00024382-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Shapiro Y, Alkan M, Epstein Y, Newman F, Magazanik A. Increase in rat intestinal permeability to endotoxin during hyperthermia. Eur J Appl Physiol Occup Physiol. 1986;55:410–412. doi: 10.1007/BF00422742. [DOI] [PubMed] [Google Scholar]
- 14.Al-Sadi RM, Ma TY. IL-1beta causes an increase in intestinal epithelial tight junction permeability. J Immunol. 2007;178:4641–4649. doi: 10.4049/jimmunol.178.7.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui W, Li LX, Sun CM, Wen Y, Zhou Y, Dong YL, Liu P. Tumor necrosis factor alpha increases epithelial barrier permeability by disrupting tight junctions in Caco-2 cells. Braz J Med Biol Res. 2010;43:330–337. doi: 10.1590/S0100-879X2010007500020. [DOI] [PubMed] [Google Scholar]
- 16.Mullin JM, Snock KV. Effect of tumor necrosis factor on epithelial tight junctions and transepithelial permeability. Cancer Res. 1990;50:2172–2176. [PubMed] [Google Scholar]
- 17.Xu DZ, Lu Q, Deitch EA. Nitric oxide directly impairs intestinal barrier function. Shock. 2002;17:139–145. doi: 10.1097/00024382-200202000-00010. [DOI] [PubMed] [Google Scholar]
- 18.Muto Y, Nouri-Aria KT, Meager A, Alexander GJ, Eddleston AL, Williams R. Enhanced tumour necrosis factor and interleukin-1 in fulminant hepatic failure. Lancet. 1988;2:72–74. doi: 10.1016/s0140-6736(88)90006-2. [DOI] [PubMed] [Google Scholar]
- 19.Wang JH, Redmond HP, Watson RW, Bouchier-Hayes D. Role of lipopolysaccharide and tumor necrosis factor-alpha in induction of hepatocyte necrosis. Am J Physiol. 1995;269:G297–G304. doi: 10.1152/ajpgi.1995.269.2.G297. [DOI] [PubMed] [Google Scholar]
- 20.Perlmutter DH, Boyle JT, Campos JM, Egler JM, Watkins JB. D-Lactic acidosis in children: an unusual metabolic complication of small bowel resection. J Pediatr. 1983;102:234–238. doi: 10.1016/s0022-3476(83)80527-7. [DOI] [PubMed] [Google Scholar]
- 21.Murray MJ, Barbose JJ, Cobb CF. Serum D(-)-lactate levels as a predictor of acute intestinal ischemia in a rat model. J Surg Res. 1993;54:507–509. doi: 10.1006/jsre.1993.1078. [DOI] [PubMed] [Google Scholar]
- 22.Song HL, Lv S, Liu P. The roles of tumor necrosis factor-alpha in colon tight junction protein expression and intestinal mucosa structure in a mouse model of acute liver failure. BMC Gastroenterol. 2009;9:70. doi: 10.1186/1471-230X-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brandt RB, Siegel SA, Waters MG, Bloch MH. Spectrophotometric assay for D-(-)-lactate in plasma. Anal Biochem. 1980;102:39–46. doi: 10.1016/0003-2697(80)90314-0. [DOI] [PubMed] [Google Scholar]
- 24.Ruan P, Gong ZJ, Zhang QR. Changes of plasma D(-)-lactate, diamine oxidase and endotoxin in patients with liver cirrhosis. Hepatobiliary Pancreat Dis Int. 2004;3:58–61. [PubMed] [Google Scholar]
- 25.Sun XQ, Fu XB, Zhang R, Lu Y, Deng Q, Jiang XG, Sheng ZY. Relationship between plasma D(-)-lactate and intestinal damage after severe injuries in rats. World J Gastroenterol. 2001;7:555–558. doi: 10.3748/wjg.v7.i4.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kasravi FB, Wang L, Wang XD, Molin G, Bengmark S, Jeppsson B. Bacterial translocation in acute liver injury induced by D-galactosamine. Hepatology. 1996;23:97–103. doi: 10.1002/hep.510230114. [DOI] [PubMed] [Google Scholar]
- 27.Wells CL, Erlandsen SL. Localization of translocating Escherichia coli, Proteus mirabilis, and Enterococcus faecalis within cecal and colonic tissues of monoassociated mice. Infect Immun. 1991;59:4693–4697. doi: 10.1128/iai.59.12.4693-4697.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Viktorov AV, Yurkiv VA. Binding of LPS and LPS--LDL complexes to rat hepatocytes. Bull Exp Biol Med. 2005;139:441–443. doi: 10.1007/s10517-005-0317-z. [DOI] [PubMed] [Google Scholar]
- 29.Vosbeck K, Tobias P, Mueller H, Allen RA, Arfors KE, Ulevitch RJ, Sklar LA. Priming of polymorphonuclear granulocytes by lipopolysaccharides and its complexes with lipopolysaccharide binding protein and high density lipoprotein. J Leukoc Biol. 1990;47:97–104. doi: 10.1002/jlb.47.2.97. [DOI] [PubMed] [Google Scholar]
- 30.Lumsden AB, Henderson JM, Kutner MH. Endotoxin levels measured by a chromogenic assay in portal, hepatic and peripheral venous blood in patients with cirrhosis. Hepatology. 1988;8:232–236. doi: 10.1002/hep.1840080207. [DOI] [PubMed] [Google Scholar]
- 31.Wheeler MD. Endotoxin and Kupffer cell activation in alcoholic liver disease. Alcohol Res Health. 2003;27:300–306. [PMC free article] [PubMed] [Google Scholar]
- 32.Borgstrom B, Dahlqvist A, Lundh G, Sjovall J. Studies of intestinal digestion and absorption in the human. J Clin Invest. 1957;36:1521–1536. doi: 10.1172/JCI103549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drewe J, Beglinger C, Fricker G. Effect of ischemia on intestinal permeability of lipopolysaccharides. Eur J Clin Invest. 2001;31:138–144. doi: 10.1046/j.1365-2362.2001.00792.x. [DOI] [PubMed] [Google Scholar]
- 34.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 35.Song HL, Lu S, Liu P. Tumor necrosis factor-alpha induces apoptosis of enterocytes in mice with fulminant hepatic failure. World J Gastroenterol. 2005;11:3701–3709. doi: 10.3748/wjg.v11.i24.3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osman NE, Weström B, Karlsson B. Serosal but not mucosal endotoxin exposure increases intestinal permeability in vitro in the rat. Scand J Gastroenterol. 1998;33:1170–1174. doi: 10.1080/00365529850172520. [DOI] [PubMed] [Google Scholar]
- 37.Lau KS, Nakashima O, Aalund GR, Hogarth L, Ujiie K, Yuen J, Star RA. TNF-alpha and IFN-gamma induce expression of nitric oxide synthase in cultured rat medullary interstitial cells. Am J Physiol. 1995;269:F212–F217. doi: 10.1152/ajprenal.1995.269.2.F212. [DOI] [PubMed] [Google Scholar]
- 38.Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409–419. doi: 10.1016/s0002-9440(10)62264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]