

Background: ZnR signaling is found in tissues where frequent pH changes are encountered.
Results: ZnR signaling in colonocytes is pH-dependent. Asp313 is critical for ZnR pH sensing.
Conclusion: pH regulation of ZnR signaling is tuned to the pH range that is relevant in colonic lumen.
Significance: pH regulation of ZnR signaling affects colonocytes survival and ion transport.
Keywords: Calcium Imaging, Calcium Intracellular Release, Cell pH, Epithelial Cell, pH Regulation, Zinc, Zinc Signaling, Zinc Transport
Abstract
Zinc activates a specific Zn2+-sensing receptor, ZnR/GPR39, and thereby triggers cellular signaling leading to epithelial cell proliferation and survival. Epithelial cells that express ZnR, particularly colonocytes, face frequent changes in extracellular pH that are of physiological and pathological implication. Here we show that the ZnR/GPR39-dependent Ca2+ responses in HT29 colonocytes were maximal at pH 7.4 but were reduced by about 50% at pH 7.7 and by about 62% at pH 7.1 and were completely abolished at pH 6.5. Intracellular acidification did not attenuate ZnR/GPR39 activity, indicating that the pH sensor of this protein is located on an extracellular domain. ZnR/GPR39-dependent activation of extracellular-regulated kinase (ERK)1/2 or AKT pathways was abolished at acidic extracellular pH of 6.5. A similar inhibitory effect was monitored for the ZnR/GPR39-dependent up-regulation of Na+/H+ exchange activity at pH 6.5. Focusing on residues putatively facing the extracellular domain, we sought to identify the pH sensor of ZnR/GPR39. Replacing the histidine residues forming the Zn2+ binding site, His17 or His19, or other extracellular-facing histidines to alanine residues did not abolish the pH dependence of ZnR/GPR39. In contrast, replacing Asp313 with alanine resulted in similar Ca2+ responses triggered by ZnR/GPR39 at pH 7.4 or 6.5. This mutant also showed similar activation of ERK1/2 and AKT pathways, and ZnR-dependent up-regulation of Na+/H+ exchange at pH 7.4 and pH 6.5. Substitution of Asp313 to His or Glu residues restored pH sensitivity of the receptor. This indicates that Asp313, which was shown to modulate Zn2+ binding, is an essential residue of the pH sensor of GPR39. In conclusion, ZnR/GPR39 is tuned to sense physiologically relevant changes in extracellular pH that thus regulate ZnR-dependent signaling and ion transport activity.
Introduction
Extracellular zinc activates a selective zinc sensing G-protein-coupled receptor (ZnR)2 that serves as a possible link between Zn2+ and its well established roles in epithelial function (1, 2). Upon interaction with extracellular Zn2+ at physiological concentrations, ZnR triggers the IP3 pathway to release intracellular Ca2+ (3, 4). In colonocytes, keratinocytes, and prostate cancer cells, ZnR mediates extracellular Zn2+-dependent activation of MAPK and PI3K pathways and thereby enhances cell proliferation and survival (5–7) as well as regulates ion transport (8, 9). We further demonstrated that GPR39 is the endogenous ZnR that activates Zn2+-dependent signaling in keratinocytes, colonocytes, and neurons (9–11). Analysis of GPR39 KO mice indicated that these mice have accelerated gastric emptying and increased secretions (12), thus linking this receptor to digestive system function. In addition, activation of the Na+/H+ exchanger by ZnR was absent in colon epithelium obtained from GPR39 KO mice (11), also linking GPR39 to the activity of the ZnR in this tissue (5).
Changes in pH during physiological or pathological conditions are common in tissues exhibiting ZnR activity, among them brain, skin, colon, and salivary gland. These pH changes are of fundamental importance in regulating a vast array of physiological processes ranging from hormonal secretion to cell survival (13–16). Probably most prominent is the regulation of pH in the gut, which varies significantly under normal physiological activity after mucosal secretion of buffers such as bicarbonate or luminal production and absorption of short chain fatty acids (17–19). Abnormal acidic colonic pH is associated with intestinal bowel disease, and alkalization of fecal pH has been monitored in colorectal cancer (17–20). On the cellular level, changes in extracellular pH were shown to interfere with CaCo-2 colonocytes migration, proliferation, and differentiation (14). The cellular mechanisms linking pH changes to these processes in the digestive system are not well understood. Most notable, it is unclear if colonocytes have a pH sensor that is tuned to sense perturbations of colonic pH at the physiologically relevant range.
Sensors of pH changes on receptors and channels are composed of acidic and histidine residues (21). These residues also coordinate Zn2+ in high affinity binding sites of numerous proteins (22, 23). Thus, pH changes may affect the interaction of Zn2+ with its binding sites. Previous studies indicate that histidine and aspartate residues also compose the Zn2+ binding site on ZnR/GPR39 (24). We find that ZnR-dependent Ca2+ release and subsequent activation of kinase pathways and ion transport are highly regulated by extracellular pH in HT29 colonocytes. Our results further show that an aspartate residue, putatively facing the extracellular domain in ZnR/GPR39, is critical for pH sensing by this receptor.
EXPERIMENTAL PROCEDURES
Cell Culture
HT29-Cl and HEK293 cells were grown in standard DMEM medium containing, 100 units/ml penicillin, 0.1 mg/ml streptomycin, 2 mm glutamine, and 10% (v/v) fetal calf serum (Biological Industries) in a 5% CO2 humidified atmosphere at 37 °C. Cells were routinely checked for mycoplasma detection.
Molecular Techniques
pCMV6-entry (C-terminal Myc- and DDK-tagged) human GPR39 cDNA coding plasmid was from OriGene (OriGene Technologies Inc.). All site-directed mutagenesis were performed by the QuikChange II site-directed mutagenesis kit (Stratagene, Agilent Technologies). Primers used for the generation of the mutations were as follows: His17 to Ala (H17A; 5′-CAAATCATTGATGCCAGTCATGTC-3′, 5′-GACATGACTGGCATCAATGATTTG-3′); His19 to Ala (H19A; 5′-ATTGATCACAGTGCTGTCCCCGAG-3′ and 5′-CTCGGGGACAGCACTGTGATCAAT-3′); His17 and His19 to Ala (H17A/H19A; 5′-ATCATTGATGCCAGTGCTGTCCCCGAG-3′ and 5′-CTCGGGGACAGCACTGGCATCAATGAT-3′); His198 and His199 to Ala (H198A/H199A; 5′-TCCAGCACCCGCGCCGCCGAGCAGCCC-3′ and 5′-GGGCTGCTCGGCGGCGCGGGTGCTGGA-3′); Asp313 to Ala (D313A; 5′-CCAAGCACGCCTGGACGAGGTCCTACTTCC-3′ and 5′-GGACCTCGTCCAGGCGTGCTTGGGTTTGG-3′); Asp313 to His (D313H; 5′-CCAAGCACCACTGGACGAGGTCCTACTTCCGGG-3′ and 5′-CCCGGAAGTAGGACCTCGTCCAGTGGTGCTTGG-3′), Asp313 to Glu (D313E; 5′- CCAAGCACGAGTGGACGAGGTCCTACTTCCGGG-3′ and 5′-CCCGGAAGTAGGACCTCGTCCACTCGTGCTTGG-3′). Using the same primer templates we also generated a 3MUT (H17A/H19A/D313A) and 4MUT (H17A/H19A/H198A/H199A). All mutations were verified using sequence analysis.
Cell Transfection
For GPR39 overexpression, HEK293 cells were seeded on glass cover slides 24 h before transfection in standard DMEM culture media as described above. Cells were transfected using Jet-PEI (Polyplus-transfection, France) according to the manufacturer's protocol, with 0.3 μg of plasmid DNA coding for WT or mutant GPR39 as described (25), and used 48 h post transfection.
For GPR39 silencing, HT29 cells were seeded in 60-mm plates 24 h before transfection in standard DMEM media as described above. Cells were transfected with siRNA (Sigma) using Lipofectamine 2000 according to the manufacturer's protocol. siRNA used for GPR39 silencing was 5′-CCAUGGAGUUCUACAGCAU-3′ and for siRNA control (scrambled) sequence was 5′-GCCCAGAUCCCUGUACGU-3′.
Immunoblot Analysis for Expression of GPR39 and Mutants
Protein expression was monitored using Western blot analysis. HT29 cells or HEK293 cells were seeded on 60-mm plates and transfected as described above. At 48 h after transfection, cells were harvested into lysis buffer (50 mm HEPES (pH 7.5), 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 10% (v/v) glycerol, 1% (v/v) Triton X-100, 10 μm MgCl2, 20 mm p-nitrophenyl phosphate, 1 mm Na3VO4, 25 mm NaF) in the presence of Protease Inhibitor Mixture (1:25 Complete, Roche). Lysates were placed on ice for 10 min and then centrifuged for 30 min (14,000 rpm) at 4 °C. Supernatants (cytosolic fraction) were collected, protein concentrations were determined using Bio-Rad protein assay, SDS sample buffer was added, and samples were boiled for 5 min and then frozen at −80 °C until used. Whole cell lysates (50 μg) were separated on 10% SDS-PAGE and blotted onto nitrocellulose membranes. Proteins were detected using antibodies raised against human GPR39 (for HT29 cells, Abcam), FLAG (for the tagged GPR39 plasmid, GenScript U.S.A Inc.) or β-actin (Cell Signaling Technology, Inc.). Densitometric analysis of expression level was performed using EZQuant-Gel software (EZQuant). Protein levels were normalized to actin levels and are presented as a percentage of the expression level of WT GPR39. Each graph represents an average of at least three independent experiments.
Analysis of Kinase Activation
HT29 cells were seeded on 60-mm plates and serum-starved for 24 h as described above. Cells were Zn2+- or control-treated (100 μm for 10 min) in Ca2+-free Ringer's solution and incubated for an additional 10 min. For analysis of kinase activation in HEK293 cells, the cells were transfected with WT or mutant GPR39 constructs, as described above, and serum starved for 24 h. Cells were Zn2+- or control-treated (100 μm for 2 min) in Ca2+-free Ringer's solution without further incubation. Cells were harvested, and samples were prepared as described above. Kinase phosphorylation was assayed with 10% SDS-PAGE (5) and analyzed using antibodies raised against the double-phosphorylated ERK1/2 and total ERK1/2 (Sigma) or phosphorylated AKT (Santa-Cruz Biotechnology) and total AKT (Sigma). Densitometry analysis of expression level was performed as described above. Levels of pERK1/2 and pAKT were normalized against the total ERK1/2 or AKT protein, respectively. Phosphorylation of ERK1/2 or AKT is presented as a percentage of the phosphorylation level triggered by application of 100 μm Zn2+ at pH 7.4. Each bar graph represents an average of at least three independent experiments.
Fluorescent Imaging
The imaging system consisted of an Axiovert 100 inverted microscope (Zeiss), Polychrome V monochromator (TILL Photonics), and a SensiCam cooled charge-coupled device (PCO) (3). Fluorescent imaging measurements were acquired with Imaging Workbench 5 (Indec). All results shown are the means of at least three independent experiments, with averaged responses of 30 cells in each experiment.
Calcium imaging was conducted as previously described (3). Briefly, cells were loaded with Fura-2 acetoxymethyl ester (30 min 2.5 μm; TEFLabs) in Ringer's solution with 0.1% BSA and subsequently washed for at least 15 min. Coverslides were then mounted in a microscope chamber that allowed rapid solution exchange. Fura-2 was excited at 340 and 380 nm and imaged with a 510-nm long-pass filter. Results are presented as the ratio (R) of the fluorescence signal obtained upon excitation at 340 nm/380 nm. In the bar graphs, Δ/R/s is the averaged initial rate of fluorescence change as calculated from the traces.
pH imaging was conducted as previously described (3). Cells were loaded for 12 min with 1.25 μm 2,7-bis(carboxyethyl)-5-(and -6)-carboxyfluorescein (BCECF)- acetoxymethyl ester (TEFLabs) in 0.1% (w/v) BSA in Ringer's solution and then washed for at least 15 min. The excitation was at 440 and 480 nm, and emission was monitored through a 510-nm long-pass filter. Results are presented as the ratio (R) of the fluorescence signal obtained upon excitation at 480 nm/440 nm. In the bar graphs, ΔR/s is the averaged initial rate of fluorescence change as calculated from the traces. For pHi calibration, nigericin was added to KCl Ringer's (120 mm KCl replacing NaCl) solution at pH 6.8, 7, and 7.2, the relative fluorescence was monitored, and a linear calibration curve was produced (26).
The NH4Cl prepulse paradigm was applied to monitor Na+/H+ exchanger activity or to induce intracellular acidification (5). Briefly, cells were perfused with Ringer's solution containing NH4Cl (30 mm), and an intracellular equilibrium of NH4+ and NH3 was reached leading to intracellular alkalization. Replacing the extracellular buffer with Na+-free Ringer's solution (iso-osmotically replaced by N-methyl d glucamine) caused rapid acidification of the cells. The addition of Na+ to the extracellular solution allowed the recovery of pHi after activation of the Na+/H+ exchange (NHE), and its activity was estimated as the rate of pHi recovery (dpHi/dt) from the acid load.
Statistical Analysis
Data are expressed as the means ± S.E. Each treatment was compared with the control or Zn2+ treatment, and statistical significance between the groups was evaluated using Student's t test or analysis of variance. *, p < 0.05.
RESULTS
ZnR/GPR39 Signaling Is Regulated by Extracellular pH
We first determined the role of GPR39 in mediating the Zn2+-dependent intracellular Ca2+ (Cai2+ ) release in HT29 cells loaded with Fura-2. The Cai2+ release triggered by Zn2+ at pH 7.4 was largely abolished after GPR39 silencing using siRNA constructs (Fig. 1A), indicating that the response is mediated by ZnR/GPR39. The Cai2+ response triggered via the purinergic Gq-dependent receptor, activated by ATP (25 μm), served as the control. The ATP-dependent Cai2+ release persisted after ZnR/GPR39 silencing (Fig. 1A), indicating that the metabotropic pathway is intact. We then asked if ZnR/GPR39-dependent Ca2+ release in HT29 cells is regulated by pH at values found in the lumen of the colon (19). HT29 cells were loaded with Fura-2, and the initial rate of Cai2+ rise was determined after application of Zn2+ (200 μm) at extracellular pH levels of 6.5–8. The ZnR response decreased by about 50% at pH of 7.7 or 8 and by about 62% at pH of 7.1 (Fig. 1, B and C). This response was completely abolished at pH 6.5 (Fig. 1, B and C). To determine if such pH changes affect other metabotropic responses, we analyzed the pH dependence of the Ca2+ response triggered by ATP (25 μm). In agreement with a previous study (27), the Ca2+ responses triggered by ATP in HT29 cells were insensitive to extracellular pH changes (Fig. 1D). Thus, our findings indicate that changes in pH selectively modulate the ZnR-dependent Ca2+ responses.
FIGURE 1.
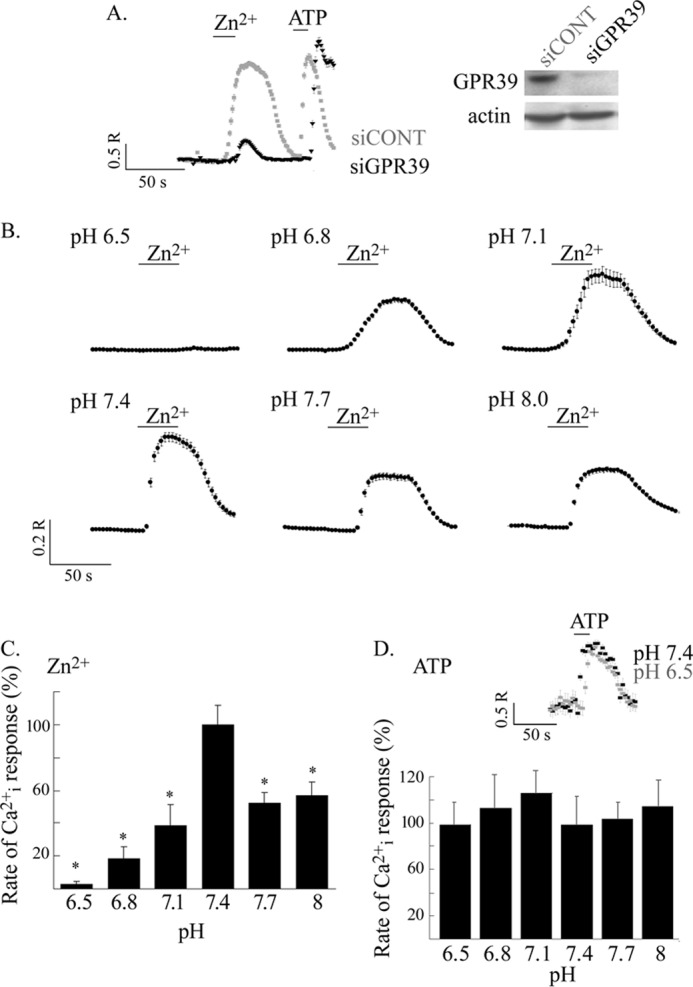
ZnR/GPR39-dependent Ca2+ response in HT29 colonocytes is regulated by extracellular pH. A, Zn2+-dependent Ca2+ responses were monitored in HT29 cells transfected with an siRNA construct targeted to silence GPR39 (siGPR39) or a scrambled siRNA. Zn2+ (200 μm) and subsequently ATP (25 μm) were applied in Ringer's solution to cells loaded with Fura-2 at the indicated times (left panel). Representative immunoblots of GPR39 expression and actin used for loading control (right panel) are shown. As shown, the Zn2+-dependent Ca2+ response is largely attenuated when the expression of ZnR/GPR39 is knocked down. Shown are representative results from three independent experiments. B, pH dependence of ZnR response was monitored in HT29 colonocytes loaded with Fura-2 as in A. Representative Ca2+ responses at the indicated pH values are shown. C, the initial rates of the Zn2+-dependent Ca2+ responses were determined (from B) and are presented as the percentage of the response obtained at pH 7.4 (n = 6; *, p < 0.05). D, averaged initial rates of the Ca2+ responses triggered by ATP (25 μm) at the indicated pH values are presented as the percentage of the response at pH 7.4. The inset shows representative responses to ATP at pH 7.4 versus 6.5 (n = 4; *, p < 0.05).
Exposure of HT29 cells to extracellular acidic pH (pH 6.5) may induce translocation of the ZnR from the plasma membrane or irreversibly inactivate the ZnR. To determine if the pH-dependent loss of ZnR activity was related to these events, we treated the cells with Ringer's solution at pH 6.5 for 2 min and then applied Zn2+ in Ca2+ -free Ringer's solution at pH 7.4 while monitoring the Ca2+ response. Similar rates of Ca2+ release were monitored in cells pretreated at pH 6.5 or kept at pH 7.4 throughout the experiment (Fig. 2A). This result indicates that the effect of pH on ZnR is transient and not related to irreversible changes of ZnR/GPR39.
FIGURE 2.
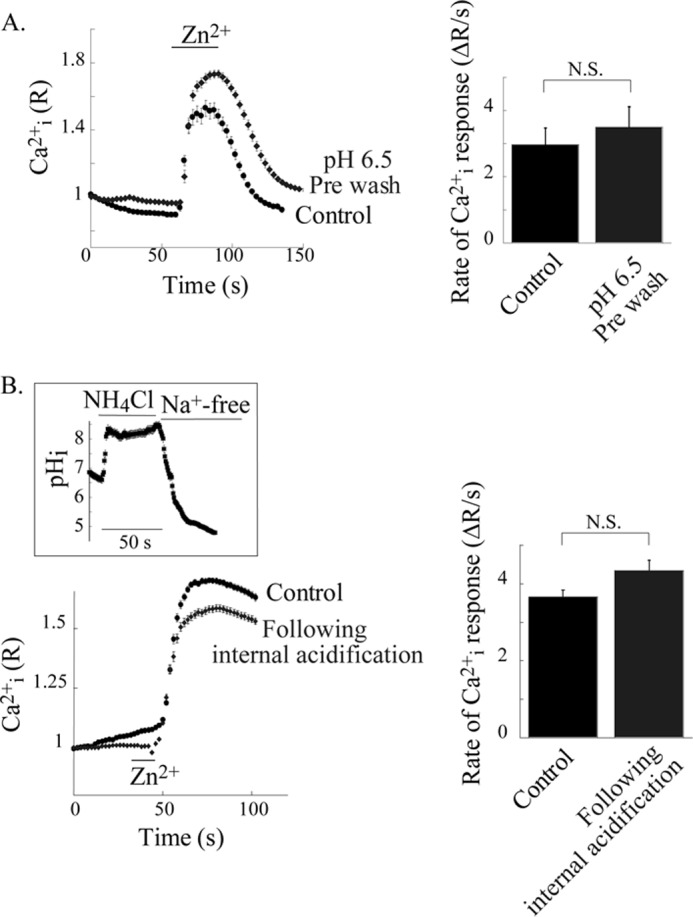
pH-dependent regulation of ZnR response is reversible and not mediated by intracellular pH changes. A, reversibility of pH dependence of ZnR response was determined in HT29 cells that were incubated in Ringer's solution at pH 6.5 or 7.4 (control) for 2 min. Subsequently Zn2+ (200 μm) was applied in Ringer's solution at pH 7.4. Representative traces of the Zn2+-dependent Ca2+ response in control cells versus the pretreated cells (pH 6.5) are shown (left panel). Averaged initial rates of fluorescence change triggered by Zn2+ are shown (right panel, n = 4). N.S., not significant. B, ZnR response was determined in HT29 cells after induction of internal acidification using the NH4Cl prepulse paradigm (see the inset showing an example of intracellular pH in BCECF-loaded cells that were treated with this paradigm). After intracellular acidification, Zn2+ (200 μm) was applied in Na+-free Ringer's solution (pH 7.4) to cells that were loaded with Fura-2. Control cells were maintained in Na+-free Ringer's solution (pH 7.4) for the same time and then treated with Zn2+ (200 μm). Representative traces of Fura-2 fluorescence changes in cells subjected to the NH4Cl prepulse paradigm versus control cells (without NH4Cl) are shown (left panel). In the right panel, averaged initial rates of Fura-2 fluorescence change after the addition of Zn2+ are shown (n = 4).
Changes in extracellular pH may modulate the intracellular pH and thereby regulate ZnR responses. Hence, we determined the effect of pHi acidification on ZnR activity using the NH4Cl prepulse paradigm, which does not affect extracellular pH but induces steep intracellular acidification (see “Experimental Procedures” and inset of Fig. 2B). Application of Na+-free Ringer's solution (iso-osmotically replaced by NMG, pH 7.4) after cellular acidification prevents NHE activity and, hence, pHi recovery. Using this paradigm, pHi is maintained at values of less than 5 (Fig. 2B, inset). The addition of Zn2+ (at extracellular pH of 7.4) triggered a ZnR-dependent Ca2+ response that was monitored with Fura-2 fluorescence (Fig. 2B). Despite the strong intracellular acidification, ZnR-dependent Ca2+ response was unaltered. The Ca2+ response triggered by ATP, which served as the control, was also insensitive to intracellular acidification (rate of Ca2+ rise in control cells was 13.2 ± 1.4 ΔR/s versus 11.1 ± 4.3 ΔR/s after intracellular acidification). Altogether our results indicate that extracellular pH modulates ZnR/GPR39 activity, but this receptor was insensitive to intracellular pH changes.
Activation of ZnR triggers Zn2+-dependent phosphorylation of AKT and ERK1/2 (5) leading to colonocytes proliferation and survival (11). We, therefore, asked if the Zn2+-dependent activation of these signaling pathways will be modulated by pH-dependent regulation of ZnR. Western blot analysis was used to compare the Zn2+-dependent phosphorylation of AKT and ERK1/2 after exposure to Zn2+ (100 μm, 10 min) in Ringer's solution at pH 7.4 or 6.5. Application of Zn2+ at pH 7.4 to HT29 cells was followed by an ∼20-fold increase in phosphorylation of AKT (pAKT) and a 10-fold increase in ERK1/2 phosphorylation (pERK1/2) compared with non-treated cells (Fig. 3, A and B). In contrast, application of Zn2+ at pH 6.5 was followed by only a 3-fold increase of pAKT level and 2-fold increase of pERK1/2 compared with control cells (maintained at pH of 6.5 for the same time but not treated with Zn2+; Fig. 3, A and B). Thus, application of Zn2+ at pH 6.5 resulted in reduced pAKT (30 ± 4.5%) and pERK1/2 (25 ± 2.9%) levels compared with the phosphorylation level monitored after application of Zn2+ at pH 7.4 (100%).
FIGURE 3.
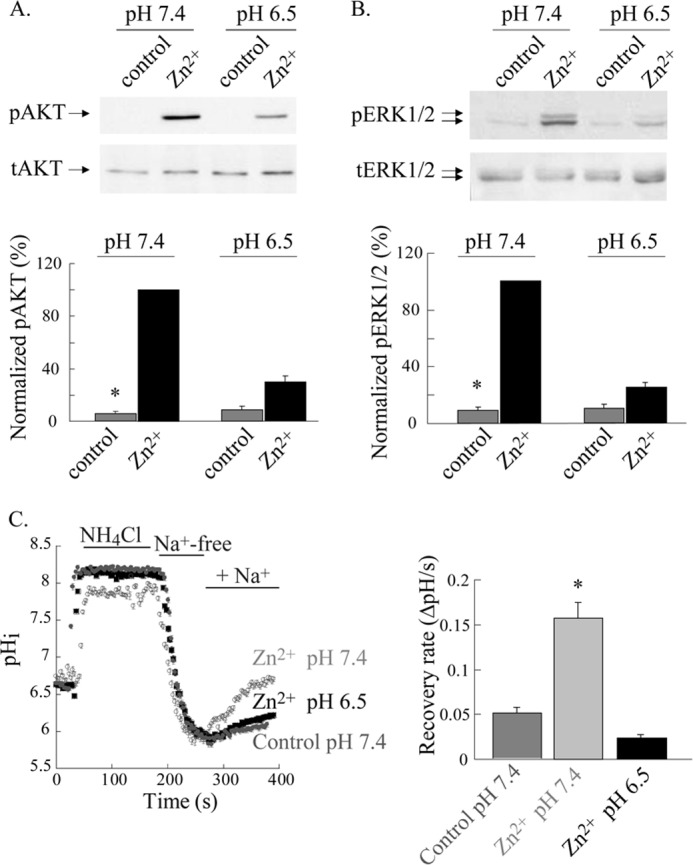
ZnR-dependent activation of AKT and ERK1/2 and up-regulation of NHE transport are eliminated at extracellular acidic pH. A and B, AKT and ERK1/2 phosphorylation in HT29 cells was monitored after application of Zn2+ (100 μm, 10 min) in Ringer's solution at pH 7.4 or 6.5. Immunoblots using antibodies against phospho- or total-AKT (A) and ERK1/2 (B) are shown (upper panels). Phosphorylated protein levels were normalized to the total protein and are presented as the percentage of the phosphorylation induced by Zn2+ at pH 7.4 (lower panels; n = 4; *, p < 0.05). C, ZnR-dependent up-regulation of NHE activity was monitored in HT29 cells using the NH4Cl prepulse paradigm. Intracellular pH was monitored in BCECF-loaded cells, and the recovery rates from internal acidification upon the addition of Na+-containing Ringer's solution were monitored. Rates were compared between cells pretreated with Zn2+ (100 μm, 2 min) applied in Ringer's solution at the indicated pH or in control cells that were incubated in Zn2+-free Ringer's solution at pH 7.4. Representative traces (left panel) and the averaged recovery rates (right panel) are shown. n = 3; *, p < 0.05.
Zn2+, via ZnR, up-regulates NHE and thereby regulates pHi (5). We, therefore, asked whether changes in extracellular pH regulate the ZnR-dependent activity of NHE in HT29 cells. Cells were preincubated with Zn2+ (100 μm, 2 min) at either pH 7.4 or 6.5, and the activity of NHE was subsequently monitored using the NH4Cl prepulse paradigm (5). Activity of NHE is established from the rate of pHi recovery after acidification that is monitored in BCECF-loaded cells upon the addition of Na+-containing Ringer's solution (see “Experimental Procedures”). Consistent with our previous results, pretreatment of the cells with Zn2+ at pH 7.4 resulted in ZnR-dependent up-regulation of NHE activity (Fig. 3C and Ref. 5). Whereas Zn2+ pretreatment at pH 7.4 resulted in a recovery rate of 0.16 ± 0.02 pHi/s, the recovery rate after Zn2+ pretreatment at pH 6.5 was reduced to 0.023 ± 0.004 pHi/s. Moreover, pHi recovery rates in cells that were pretreated with Zn2+ at pH 6.5 were similar to those of control cells (0.05 ± 0.006 pHi/s), suggesting that up-regulation of NHE activity by Zn2+ is eliminated when Zn2+ is applied at pH 6.5.
Altogether our results indicate that ZnR activity and signaling in HT29 colonocytes is largely attenuated at pH 6.5 or 8 and is maximal at pH 7.4. Thus, it is regulated at a narrow pH range that is physiologically relevant in the lumen of the colon (19).
Histidine Residues That Participate in Zn2+ Binding Do Not Act as the pH Sensor of GPR39
We then set out to identify the pH sensor site on ZnR/GPR39. Based on the role of His residues in numerous pH sensors (21), we initially focused on the His residues suggested to form the Zn2+ binding site on GPR39 (24). Using site-directed mutagenesis, we created GPR39 mutants in which these residues were replaced. We then expressed the wild type (WT) and mutant GPR39 in HEK293 cells, which allow effective overexpression, and asked if extracellular pH also regulates the Zn2+-dependent response in the overexpressing cells. We, therefore, first performed analysis of pH dependence of the initial rate of the Zn2+-dependent Ca2+ rise in HEK293 cells transfected with WT GPR39. As shown in Fig. 4A, application of 200 μm Zn2+ at extracellular pH 6.5 was followed by an ∼2-fold reduction in the Zn2+-dependent Ca2+ response compared with the response at pH 7.4 (average rates were: 2.03 ± 0.36 ΔR/s at pH 7.4 and 1.11 ± 0.26 ΔR/s at pH 6.5). Hence, a smaller pH dependence of the Zn2+-dependent Ca2+ response was monitored in overexpressing HEK293 cells compared with the effect monitored for the endogenous ZnR/GPR39 in HT29 cells. This may result from altered pathway regulation of the downstream signaling leading to Ca2+ release or from different levels of surface expression of GPR39 between these cell lines (28, 29). We, therefore, asked if the pH dependence is maintained at various Zn2+ concentrations. We monitored a pH-dependent 2-fold reduction in the ZnR/GPR39 response in all Zn2+ concentrations that induced a ZnR/GPR39 response (Fig. 4B). These results indicate that ZnR/GPR39 activity in HEK293 cells is also strongly regulated by extracellular pH.
FIGURE 4.
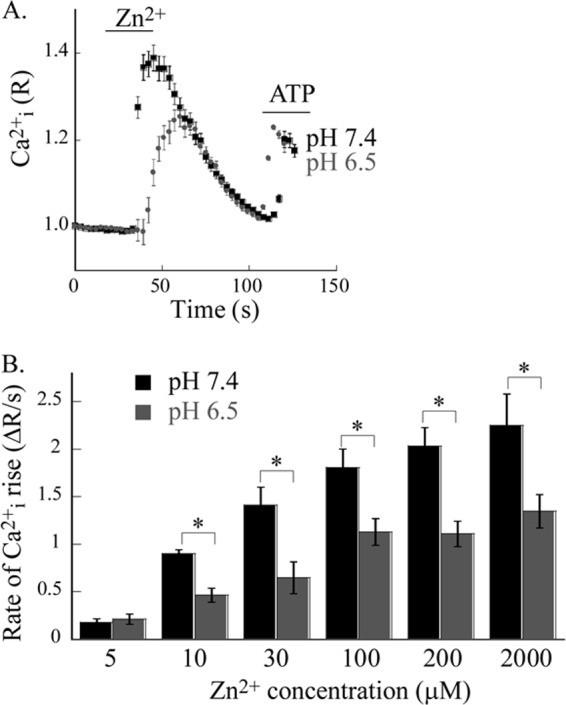
Ectopically expressed ZnR/GPR39 in HEK293 cells is regulated by extracellular acidic pH. A, pH dependence of ZnR responses was determined in HEK293 cells transfected with hGPR39 that were loaded with Fura-2. Representative traces after application of Zn2+ (200 μm) and ATP (25 μm) at pH 6.5 and 7.4 are shown. B, averaged initial rates of Zn2+-dependent Ca2+ rise at the indicated Zn2+ concentrations applied in Ringer's solution at pH 6.5 (gray bars) versus pH 7.4 (black bars; n = 3, *, p < 0.05). Note that the response of ZnR/GPR39 is attenuated to about 50% at pH 6.5 that over the whole range of Zn2+ concentrations.
The extracellular Zn2+ binding site of GPR39 is composed of two histidine residues, His17 and His19 (Fig. 5A and Ref. 24). We first replaced these residues to alanine (H17A, H19A, and a double mutant H17A/H19A). In addition, we studied the role of two His residues (His198 and His199) found on the putative extracellular loop 2, which were also suggested to regulate the Zn2+-dependent signaling (24). We generated a double GPR39 mutant in which His198 and His199 were substituted with Ala (H198A/H199A) and an additional mutant replacing all four His residues involved in the Zn2+ binding site, H17A/H19A/H198A/H199A, to Ala (4MUT). Immunoblot analysis of these constructs showed that expression levels of GPR39 H17A, H19A, H17A/H19A, H198A/H199A, or H17A/H19A/H198A/H199A (termed 4MUT) constructs in HEK293 cells were similar to that of WT GPR39 (Fig. 5B). The initial rate of the Zn2+-dependent Cai2+ rise was monitored using Fura-2-loaded cells. In H17A-, H19A-, and H17A/H19A-transfected cells, the initial rate of the Ca2+ rise was reduced to 66 ± 6, 44 ± 2, or 34 ± 11%, respectively, of the response of WT GPR39 (Fig. 5C). Such a decrease in the Zn2+-dependent activity of these mutants is in agreement with the role of these residues in the Zn2+ binding site. Similarly, the 4MUT construct shows only 30 ± 6% response compared with WT GPR39, consistent with the low activity of the H17A/H19A mutant. Note that the Zn2+-dependent Ca2+ rise triggered in cells expressing the GPR39 H17A/H19A mutant was small but always apparent, as observed using Fura-2 measurements, in contrast to loss of activity that was described when IP3 release was monitored in a previous study (24). The H198A/H199A mutant showed only slightly reduced Zn2+-dependent Ca2+ response, 82 ± 8%, suggesting that these residues are not directly involved with the binding site. We then studied the effect of extracellular pH on the Zn2+-dependent Ca2+ response in cells expressing the His to Ala mutants. Cells overexpressing the mutants were loaded with Fura-2, and the initial rate of Ca2+ release was determined after application of Zn2+ at extracellular pH 6.5. The initial rate of Ca2+ release that was monitored for all the His mutants showed at least 50% reduction when Zn2+ was applied at pH 6.5 compared with pH 7.4 (Fig. 6, A–F). This pH-dependent effect of the His mutants was similar to that of WT GPR39. Importantly, despite the reduced activity monitored at pH 7.4, all 5 mutants manifested a pH-dependent Zn2+-induced Ca2+ response. Thus, our results indicate that replacing the putative Zn2+ binding His residues to the pH insensitive Ala residue reduces the Zn2+-dependent activity but does not change the pH sensitivity of the ZnR/GPR39 response. This indicates that extracellular histidine residues that form the Zn2+ binding site or are involved in its regulation do not participate in pH sensing by GPR39.
FIGURE 5.
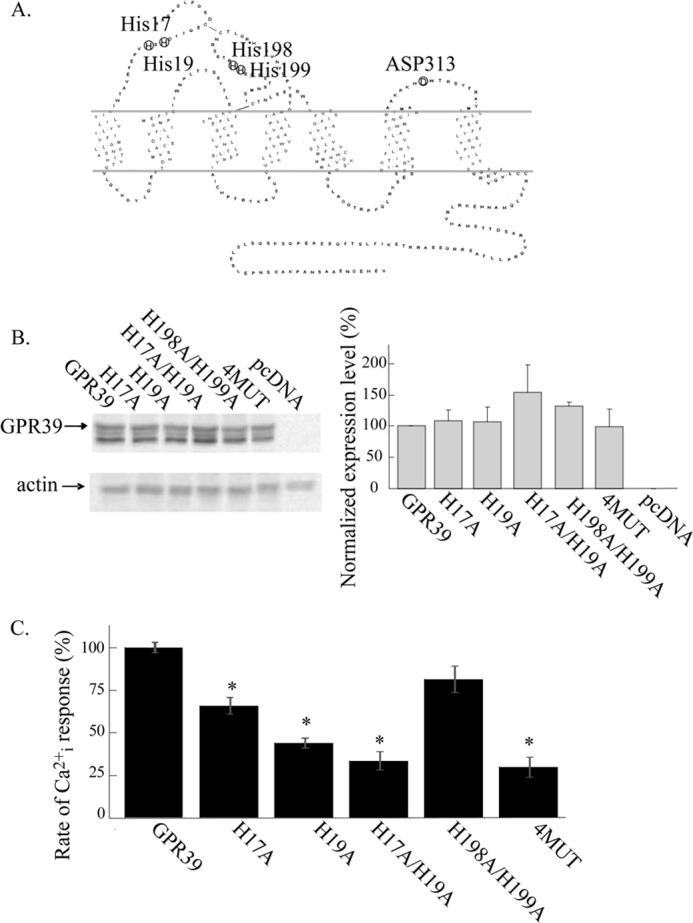
GPR39 mutant expression and their Zn2+-dependent Ca2+ responses. A, shown is a serpentine diagram of GPR39 highlighting the residues forming the Zn2+ binding site (His17, His19), a regulator of this site, Asp313, and other extracellular His residues (His198, His199) that were suggested to interact with the binding site. B, expression of ZnR/GPR39 was studied in HEK293 cells that were transfected with WT GPR39 or the indicated mutants (see A, 4MUT is the H17A/H19A/H198A/H199A) or control pcDNA vector. Immunoblot analysis of the indicated FLAG-tagged GPR39 mutants (see “Experimental Procedures” and left panel) is shown with actin as the loading control. Expression levels of the mutants normalized to actin are presented as a percentage of the expression of WT GPR39 (right panel, n = 4). C, initial rates of Zn2+ (200 μm)-dependent Ca2+ responses monitored in Fura-2-loaded cells expressing the indicated mutants (as in Fig. 4A) are presented as the percentage of the WT GPR39 response (n = 4; *, p < 0.05). Clear Zn2+-dependent Ca2+ rises were observed in all mutants but were smaller in mutants of the Zn2+ binding site.
FIGURE 6.
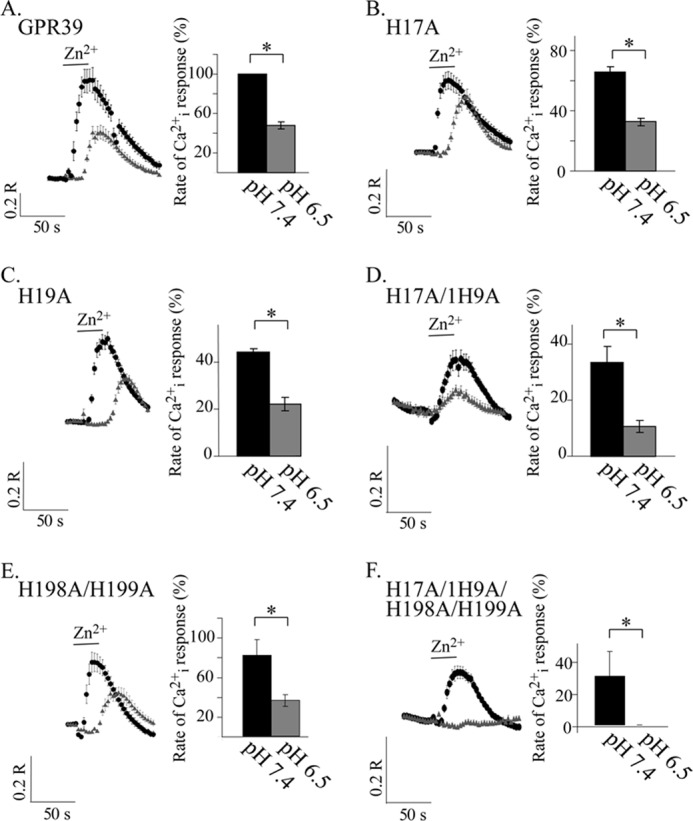
GPR39 His mutants do not lose pH sensitivity, similar to the WT GPR39. A–F, Zn2+ (200 μm)-dependent Ca2+ responses were measured in Fura-2-loaded cells at pH 7.4 (black circles) or 6.5 (gray triangles). The fluorescent signals were measured in HEK293 cells expressing WT GPR39 (A) or the indicated mutants (B–F). Shown are representative fluorescence traces monitored after application of Zn2+ in each of the pH values (left panels) and the average initial rate of the responses (right panels) presented as a percentage of the response triggered by 200 μm Zn2+ applied at pH 7.4 in cells expressing WT GPR39 (n = 5; *, p < 0.05).
Asp313 Is Involved in the pH Regulation of GPR39
Asp313, putatively located in extracellular loop 3 of GPR39, was also suggested to regulate the interaction of Zn2+ with the metal-ion binding site (24). We next sought to determine if Asp313 may contribute to pH sensing by GPR39. We, therefore, substituted Asp313 to Ala (D313A) and also generated a triple mutant by replacing Asp313 along with His17 and His19 to Ala (3MUT). Expression levels of these mutants in HEK293 cells were similar to WT GPR39 (Fig. 7A). We then loaded cells with Fura-2 and monitored the Zn2+-dependent Cai2+ release as in Fig. 6. The initial rate of Cai2+ rise at pH 7.4 was reduced in cells expressing the D313A construct to 41 ± 6% compared with WT GPR39, an effect that was similar to the response of other mutants of the binding site (see Fig. 5C). However, the Zn2+-dependent Ca2+ response in cells expressing the D313A mutant was strikingly similar at pH 7.4 and 6.5 (Fig. 7B). Thus, expression of the D313A mutant results in loss of pH sensitivity and retains the full Zn2+-dependent Ca2+ response at pH 6.5. Importantly, although the activity of the D313A mutant at pH 7.4 is similar to that of H19A or H17A mutants, only this mutant is pH-insensitive. Finally, the initial rate of the Zn2+-dependent Ca2+ response of cells expressing the 3MUT (H17A/H19A/D313A) construct was 21 ± 4% that of the WT GPR39 response at pH 7.4. This greatly reduced Zn2+-dependent activity is expected when all three residues suggested to compose the main Zn2+ binding site are replaced (24). As expected, the Zn2+-dependent response of cells expressing the 3MUT (H17A/H19A/D313A) construct was similar at pH 6.5 and 7.4 (Fig. 7C). Despite the smaller Ca2+ response triggered in cells expressing this mutant, the 3MUT (H17A/H19A/D313A) construct was pH-insensitive. This finding is in contrast to the effect monitored in cells expressing the H17A/H19A mutant or the 4MUT (H17A/H19A/H198A/H199A), which also showed similarly small Zn2+-dependent Ca2+ response but maintained pH sensitivity. Thus, our results indicate that Asp313 is an essential part of the pH sensor of GPR39.
FIGURE 7.
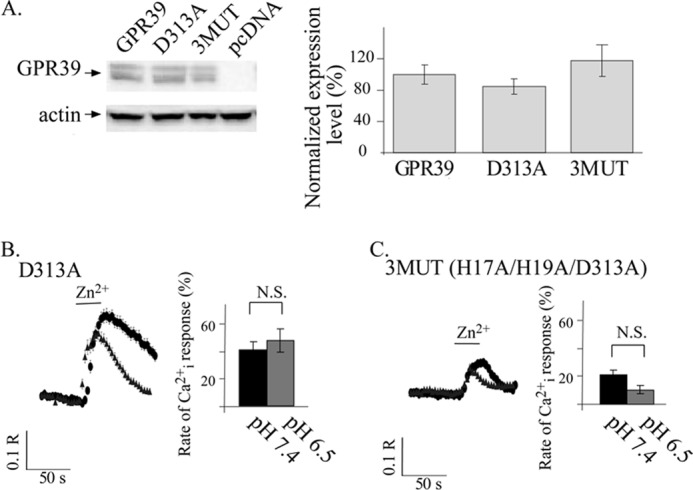
Asp313 is essential for pH sensing by GPR39. A, immunoblot analysis of HEK293 cell lysates was performed in cells expressing FLAG-tagged WT GPR39, D313A GPR39 mutant, a triple mutant (H17A/H19A/D313A, termed 3MUT), or control pcDNA vector. Expression levels were normalized to actin and are presented as the percentage of WT GPR39 expression level (right panel, n = 4). B and C, shown are representative Fura-2 traces of Zn2+-dependent Cai2+ release at pH 7.4 (black circles) or 6.5 (gray triangles) as monitored in HEK293 cells expressing the D313A (B) or 3MUT (C, H17A/H19A/D313A) constructs (left panels). The average initial rates of Ca2+ release (right panels) are presented as a percentage of the response triggered by 200 μm Zn2+ applied at pH 7.4 in cells expressing WT GPR39 that are shown in Fig. 6A (n = 5; *, p < 0.05). Replacing Asp313 with Ala (in D313A or H17A/H19A/D313A mutants) was followed by loss of the pH dependence of the ZnR response. N.S., not significant.
We then asked if the loss of pH regulation of the D313A mutant would also effect the pH dependence of the downstream signaling pathways or transport activity regulated by ZnR/GPR39. We first determined if AKT and ERK1/2 phosphorylation triggered in cells expressing the D313A mutant are regulated by extracellular pH. HEK293 cells expressing either the WT GPR39 or D313A mutant were treated with Zn2+ (100 μm, 2 min) as described in Fig. 3 and under “Experimental Procedures.” Immunoblot analysis of the Zn2+-dependent phosphorylation of AKT (Fig. 8A) and ERK1/2 (Fig. 8B) in HEK293 cells transfected with WT GPR39 showed robust activation of these kinases when Zn2+ was applied at pH 7.4 but not at pH 6.5. This is consistent with the pH regulation of WT ZnR/GPR39 activity in HT29 cells. In contrast, Zn2+-dependent phosphorylation of AKT (Fig. 8A) and ERK1/2 (Fig. 8B) in the D313A-transfected HEK293 cells was similar at both values. Thus, although Zn2+-dependent AKT and ERK1/2 activation is regulated by acidic extracellular pH in WT GPR39-expressing cells, it is insensitive to pH when cells express the D313A mutant. These results are in agreement with the loss of pH sensitivity of the Zn2+-dependent Ca2+ response in cells expressing the D313A mutant.
FIGURE 8.

Regulation by acidic pH of the Zn2+-dependent signaling is eliminated in HEK293 cells expressing the D313A mutant. A and B, AKT and ERK1/2 phosphorylation in HEK293 cells expressing either WT GPR39 or D313A mutant were monitored after application of Zn2+ (100 μm, 2 min) in Ringer's solution at pH 7.4 or 6.5. Immunoblots using antibodies against phospho- or total-AKT (A) and ERK1/2 (B) are shown (upper panels). Phosphorylated protein levels were normalized to the total protein and are presented as percentage of the phosphorylation induced by Zn2+ at pH 7.4 (lower panels; n = 4; *, p < 0.05 compared with WT control; #, p < 0.05 compared with D313A control). C and D, ZnR-dependent up-regulation of NHE activity was monitored in HEK293 cells expressing WT GPR39 (C) or D313A (D) using the NH4Cl prepulse paradigm. Intracellular pH was monitored in BCECF-loaded cells, and the recovery rates from internal acidification upon the addition of Na+-containing Ringer's solution were monitored. Rates were compared between cells pretreated with Zn2+ (100 μm) applied in Ringer's solution at the indicated pH or in control cells that were incubated in Zn2+-free Ringer's solution at pH 7.4. Representative traces of the pHi recovery after the NH4+ prepulse paradigm (top panel, for simplicity only the recovery phase after the addition of Na+ is shown) and the averaged recovery rates (bottom panel) are shown (n = 3; *, p < 0.05 compared with WT control; #, p < 0.05 compared with D313A control). N.S., not significant.
Next, we asked if expression of the D313A mutant will also alter the pH sensitivity of the ZnR-dependent up-regulation of NHE activity. The rate of Na+/H+ exchange activity was compared in HEK293 cells expressing WT GPR39 or the D313A mutant at pH 6.5 and 7.4 using the same experimental setting used for HT29 cells (Fig. 3). Cells were loaded with BCECF and pretreated with Zn2+ (100 μm, 2 min) at pH 7.4 or 6.5. Next, the cells were subjected to the NH4Cl prepulse paradigm, and the recovery rate from acidification was monitored. In HEK293 cells transfected with WT GPR39, the rate of NHE activity after pretreatment with Zn2+ at pH 7.4 was increased compared with control (recovery rates of 0.11 ± 0.04 pHi/s with Zn2+ versus 0.010 ± 0.004 in controls, Fig. 8C). In contrast, cells pretreated with Zn2+ at pH 6.5 showed a lower rate of recovery (0.009 ± 0.006 pHi/s) that was similar to the recovery rate of control cells. We then tested the pH recovery rates in HEK293 cells transfected with the D313A mutant. Consistent with the lower ZnR activity of the D313A mutant (Fig. 5), cells expressing this mutant showed a more moderate up-regulation of the pHi recovery rate after pretreatment with Zn2+ at pH 7.4 (0.04 ± 0.01 pHi/s after Zn2+ versus 0.01 ± 0.002 pHi/s in the control cells; Fig. 8D). Remarkably, in cells expressing the D313A mutant, up-regulation of NHE-dependent pHi recovery rate was maintained when Zn2+ was applied at pH 6.5 (0.03 ± 0.01 pHi/s; Fig. 8D). Altogether, expression of D313A mutation eliminated the pH dependence of ZnR signaling, including the effect of acidic pH on Zn2+-dependent Ca2+ response, AKT, and ERK1/2 phosphorylation and regulation of NHE activity.
Replacing Asp313 to His or Glu Residues Restores the pH Sensitivity of GPR39
Finally, we asked if other residues that participate in pH sensing and Zn2+ binding (23, 30, 31), such as His or Glu, can rescue the pH sensitivity of the Zn2+-dependent response. Hence, we replaced Asp313 to His (D313H) or Glu (D313E) residues and studied the Zn2+-dependent Ca2+ response at pH 7.4 and 6.5. The expression levels of the D313H and D313E mutants were similar to those of WT GPR39 (Fig. 9A). The Zn2+-dependent Cai2+ release triggered by the D313H mutant at pH 7.4 was similar to that of WT GPR39 (90 ± 12% of the response obtained with WT GPR39). Interestingly, the Ca2+ response of cells expressing the D313H mutant at pH 6.5 was 42 ± 8% that of the response obtained with WT GPR39 at pH 7.4 (Fig. 9B). Hence, replacing Asp313 to a His residue restored the pH sensitivity of the Zn2+-dependent Ca2+ response. We then measured the initial rate of the Zn2+-dependent Ca2+ rise in cells expressing the D313E mutant. When Zn2+ was applied at pH 7.4, the response was 55 ± 9% (Fig. 9C) of the response obtained in WT GPR39 (pH 7.4). However, when Zn2+ was applied at pH 6.5 to cells expressing the D313E mutant, the Ca2+ response was only 21 ± 3% that of the response obtained in WT GPR39 (Fig. 9C). Although the D313E mutant showed a partial decrease of the Zn2+-dependent Ca2+ response compared with WT GPR39, the pH sensitivity of this mutant was also fully restored and was similar to that of the WT ZnR/GPR39. Both the D313E and D313H mutants exhibited a pH-sensitive ZnR response and thus rescued the GPR39 pH sensor.
FIGURE 9.
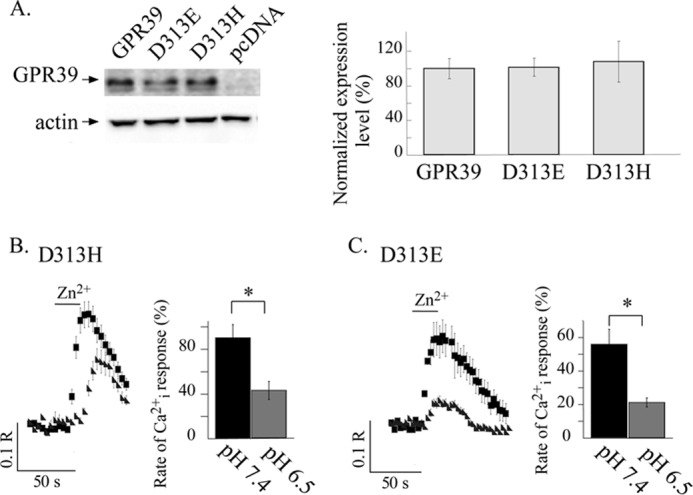
Replacing Asp313 with Glu or His residues rescues pH sensing by GPR39. A, shown is immunoblot analysis of HEK293 cell lysates was performed in cells expressing FLAG-tagged WT GPR39, D313E, or D313H GPR39 mutants or control pcDNA vector. Expression levels were normalized to actin and are presented as the percentage of WT GPR39 expression level (right panel, n = 4). B and C, shown are representative traces of Zn2+-dependent Ca2+ responses at pH 6.5 and 7.4 as monitored in HEK293 cells expressing the D313H or D313E constructs as indicated (left panels). The average initial response rates (right panels) are presented as a percentage of the response triggered by 200 μm Zn2+ applied at pH 7.4 in cells expressing WT GPR39 (n = 4; *, p < 0.05). Cells expressing the mutants in which Asp313 was replaced with Glu or His residues exhibit a pH-sensitive ZnR response.
DISCUSSION
Activity of a Zn2+ sensing receptor, which mediates between changes in extracellular Zn2+ and cellular signaling, was introduced more than a decade ago (3). Yet, only recently was the endogenous ZnR molecularly linked to GPR39 using siRNA and a genetic knock-out mouse model (9, 10). Here we show that ZnR/GPR39 signaling activity is highly regulated by extracellular pH. ZnR/GPR39 activity is maximal at pH 7.4 but is attenuated already at pH 7.7 or 7.1. The endogenous ZnR signaling is totally abolished at pH 6.5 in HT29 cells. Thus, ZnR/GPR39 is adjusted as a physiologically relevant pH sensor, regulated within a narrow range. Such small pH changes are found in the lumen of the colon (19, 20).
Under normal conditions, changes in luminal colonic pH do not easily affect the periapical side of the colonocytes, which are buffered by secretion of bicarbonate-rich mucus. This creates a relatively constant microenvironment for the cells with changes in the narrow pH range of 7–7.5 (32, 33). Hence, under physiological conditions the pH changes that occur at the apical surface of colonocytes regulate Zn2+-dependent intracellular signaling via ZnR/GPR39. Regulation of the periapical pH is mediated by bicarbonate secretion, and thus a smaller effect of alkaline pH on ZnR/GPR39 activity allows the Zn2+ receptor to be less sensitive to the secretion process. Most pH sensors in the colon are expressed in afferent neurons and sensitized to much lower pH values, i.e. the transient receptor potential channels and the acid sensing ion channels that are activated at pH below 6 (15, 34, 35). Indeed these receptors are involved in pain sensation and inflammatory hyperalgesia (36). In contrast, our data indicate that ZnR/GPR39 tightly regulates the activity of the epithelial cells at the physiological pH range. We currently do not know the exact localization of ZnR/GPR39 in colonocytes; however, our functional analysis using native colon tissue indicate that it is activated by luminal application of Zn2+, suggesting that ZnR/GPR39 is found on the apical surface (5, 11). Interestingly, a Ca2+-sensing receptor, CaSR, is a G-protein-coupled receptor that is found on the basolateral side of colonocytes and is tuned to sense alkaline pH changes monitored on this side of the cells (27, 37, 38). It will be of interest to determine if the concerted activity of the CaSR and ZnR shape the response of colonocytes to pH and regulate short chain fatty acid transport and bicarbonate secretion (39). In the gastric mucosa Zn2+ is indeed involved in regulation of acid secretion via a Ca2+-dependent mechanism (40).
Although physiological pH is kept within a narrow range, large changes in colon pH, in particular chronic acidification, are often encountered during inflammatory bowel diseases (19). Our results indicate that such drop in pH will result in profound attenuation of ZnR/GPR39 Ca2+ response and in turn suppress Zn2+-dependent ERK1/2 and AKT kinase activation. These signaling pathways are associated with enhanced proliferation and survival, processes that were induced by ZnR/GPR39 in epithelial tissues (5, 6, 11, 41). Zinc was suggested to ameliorate symptoms of colitis (42, 43) via an unknown mechanism, yet impairment of ZnR-dependent signaling may contribute to the epithelial erosion encountered during inflammatory bowel disease.
The Na+/H+ exchanger in the colon plays a key role in solute transport and pH homeostasis (41, 44, 45). The latter function is of particular importance considering the high concentration of short chain fatty acids generated by bacterial fermentation in the gut lumen. Our data indicate that Zn2+-dependent activation of NHE is completely diminished when Zn2+ is applied at pH 6.5. This was evident in HT29 cells endogenously expressing GPR39 (Fig. 3) and was reproduced in HEK293 cells overexpressing WT GPR39 (Fig. 7). This finding is also consistent with our previous studies demonstrating that the activation NHE by Zn2+ is mediated through ERK1/2 signaling (5, 10), which is also inhibited at this pH. Activation of an apical NHE, however, acidifies the periapical domain. Thus, down-regulation of ZnR/GPR39 activity at low pH may provide a feedback mechanism to regulate the periapical pH level.
Acidic residues are essential components of pH sensors in numerous proteins (46). Similar acidic residues also compose Zn2+ binding sites (47). A link between Zn2+ and pH sensors has been previously described in regulating the purinergic P2X Ca2+ channels (21, 48). Our results indicate that ZnR/GPR39 interaction with Zn2+ ions via His and Asp residues is also regulated by pH in HT29 colonocytes and in HEK293 cells ectopically expressing the receptor. After internal acidification, no change in ZnR/GPR39 response was monitored, indicating that only extracellular pH regulates the response. Such regulation by extracellular, but not intracellular, pH was also observed for the CaSR where protonation of residues on the ligand binding site was suggested to mediate pH sensing (27). A similar mechanism may be induced by oxidation and regulate Zn2+ and Ca2+ signaling in gastrointestinal cells (49, 50).
The effect of pH on receptor activity may result from conformational changes after different protonation states. Indeed, such pH-sensitive conformational changes have been shown to regulate activity of ion and water channels, affinity of proteins for binding actin filaments, and activity of viral and bacterial proteins controlling host cell entry (46). A family of lipid sensing G-protein-coupled receptors has been suggested to sense extracellular protons via His residues that affect the conformation of the receptors and directly trigger their activation even in the absence of the lipid ligand (51). Our results do not show ZnR/GPR39 signaling in the absence of Zn2+, the endogenous ligand of this receptor, arguing against such mechanism. The attenuation of Zn2+-dependent signaling in pH 6.5 was short-termed and reversible, as subsequent Zn2+-dependent (applied at pH 7.4) Ca2+ response was observed. This reversal of the pH-dependent inhibition of ZnR/GPR39 response does not support a mechanism involving changes in the surface expression of the receptor. Another possible mechanism for pH regulation of receptor function was suggested for the CaSR, where changes in Glu residues at the ligand binding site affect the ability of the receptor to interact with its ligand (27). Similar allosteric regulation of the binding site was suggested for the mGluR4 receptor by pH (52). The Zn2+ binding site on GPR39 is composed of His17 and His19 residues located at the extracellular N-terminal segment (24). The location of the His residues in the extracellular N-terminal region of the receptor corresponds to our extracellular pH dependence. However, despite the reduced Ca2+ response of the ZnR/GPR39 H17A, H19A, and H17A/H19A mutants, they all retained their pH sensitivity. These results indicate that these His residues on the Zn2+ binding site are not essential for ZnR/GPR39 pH sensing.
Previous functional analysis of Asp313 in GPR39 suggested that replacing this residue with Ala resulted in high constitutive IP3 release (24). Likely due to this constitutive activity, further Zn2+-dependent response was not observed. We find that replacing Asp313 with Ala resulted in a mutant that retained significant Zn2+-dependent activity, albeit lower than that of WT GPR39. Furthermore, replacement of all three residues: Asp313 along with His17 and His19 results in a mutant that shows very low residual activity that is maintained at a similar rate at pH 6.5; thus, this triple mutant loses the ZnR/GPR39 pH dependence. Importantly, mutation of Asp313 to alanine is sufficient to completely eliminate the ZnR/GPR39 pH sensitivity. Moreover, replacement of Asp313 with Ala also reversed the inhibitory effect of extracellular acidic pH on the ZnR-dependent AKT and MAPK activation and up-regulation of Na+/H+ exchange. In contrast, the H17A/H19A double mutant as well as the 4MUT (H17A/H19A/H198A/H199A) also showed low residual activity compared with WT GPR39, but acidic pH further lowered this activity. Thus, a major conclusion of this study is that the Asp313 residue is essential for pH sensing.
Replacing Asp313 with His or Glu residues, which are often found in pH sensors of proteins, restored the pH sensitivity of the receptor. Hence, the presence of a residues capable of protonation-deprotonation fully rescued the pH site on GPR39. Our findings are also consistent with previous findings indicating that Asp/Glu residues are critical for pH sensors of numerous channels and receptors among them, most notable Asp94 on human hemoglobin (53) and Asp269 on Tsr chemoreceptor in Escherichia coli (54). Thus, Asp313 in GPR39 may serve as a regulatory residue of the Zn2+ binding site that undergoes protonation at low extracellular pH. This alters the ability of the Zn2+ binding site to interact with Zn2+, and thus the downstream signaling is reduced.
Although previous and our studies show that His17 and His19 are major constituents of the Zn2+ binding site on GPR39, several aspects regarding the activity of GPR39 mutants in this work differ from those shown in the previous study (24). In the previous study GPR39 and specifically the D313A mutant showed constitutive IP3 release in the overexpressing cells. We do not see constitutive Ca2+ response of GPR39 or of its D313A mutant as measured using Fura-2. Another major difference is that the activity of the H17A/H19A mutant showed no Zn2+-dependent IP3 release in the previous study (24). However. we found that this mutant exhibits a small but clearly apparent Zn2+-dependent Ca2+ response that is ∼30% of the response of the WT GPR39. Moreover, we saw clear Zn2+-dependent activity of the D313A mutant and measured significant Zn2+-dependent up-regulation of NHE activity as well as AKT and MAPK phosphorylation. Finally, our results show that the triple mutant of the suggested Zn2+ binding site (H17A/H19A/D313A) maintains Zn2+-dependent activity. This residual response after the deletion of key Zn2+ binding residues suggests that additional sites may participate in Zn2+ binding of GPR39, as is also suggested for the Ca2+-sensing receptor, CaSR (27). The basis for the differences in the Zn2+-dependent activity between the present and previous (24) studies is not fully clear but may result from the different assays used. For example, whereas IP3 release was monitored in the previous study, we measured the subsequent Ca2+ release and kinase activity. Therefore, a possible explanation is that the constitutive release of IP3 may not be sufficiently strong to trigger Ca2+ release. Alternatively, the constitutive activity may also be attributed to varying residual Zn2+ often found in physiological solutions (10, 55–57). Finally, the apparent constitutive activity of the H17A/H19A or D313A mutants in the previous study (24) may have masked their Zn2+-dependent activity. In contrast, the low base-line activity in the Ca2+ measurements allowed us to monitor the Zn2+-dependent Ca2+ activity and downstream signaling of the mutants.
Altogether our results identify a strong pH dependence of ZnR/GPR39 signaling. In colonocytes this modulates Zn2+-dependent signaling and thereby activation of cell survival pathways and regulation of ion transport within a physiologically relevant pH range. We further find that Asp313 is essential for pH sensing by ZnR/GPR39.
This work was supported by Israel Science Foundation Grant 513/09 (to M. H.).
- ZnR
- zinc-sensing receptor
- IP3
- inositol 1,4,5-triphosphate
- NHE
- Na+/H+ exchanger
- pHi
- intracellular pH
- BCECF
- 2,7-bis(carboxyethyl)-5-(and -6)-carboxyfluorescein
- CaSR
- Ca2+-sensing receptor.
REFERENCES
- 1. Hershfinkel M., Silverman W. F., Sekler I. (2007) The zinc sensing receptor, a link between zinc and cell signaling. Mol. Med. 13, 331–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maret W. (2001) Cross-talk of the group IIa and IIb metals calcium and zinc in cellular signaling. Proc. Natl. Acad. Sci. U.S.A. 98, 12325–12327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hershfinkel M., Moran A., Grossman N., Sekler I. (2001) A zinc-sensing receptor triggers the release of intracellular Ca2+ and regulates ion transport. Proc. Natl. Acad. Sci. U.S.A. 98, 11749–11754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Popovics P., Stewart A. J. (2011) GPR39. A Zn2+-activated G protein-coupled receptor that regulates pancreatic, gastrointestinal, and neuronal functions. Cell. Mol. Life Sci. 68, 85–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Azriel-Tamir H., Sharir H., Schwartz B., Hershfinkel M. (2004) Extracellular zinc triggers ERK-dependent activation of Na+/H+ exchange in colonocytes mediated by the zinc-sensing receptor. J. Biol. Chem. 279, 51804–51816 [DOI] [PubMed] [Google Scholar]
- 6. Dubi N., Gheber L., Fishman D., Sekler I., Hershfinkel M. (2008) Extracellular zinc and zinc citrate, acting through a putative zinc sensing receptor, regulate growth, and survival of prostate cancer cells. Carcinogenesis 29, 1692–1700 [DOI] [PubMed] [Google Scholar]
- 7. Sharir H., Hershfinkel M. (2005) The extracellular zinc-sensing receptor mediates intercellular communication by inducing ATP release. Biochem. Biophys. Res. Commun. 332, 845–852 [DOI] [PubMed] [Google Scholar]
- 8. Segal D., Ohana E., Besser L., Hershfinkel M., Moran A., Sekler I. (2004) A role for ZnT-1 in regulating cellular cation influx. Biochem. Biophys. Res. Commun. 323, 1145–1150 [DOI] [PubMed] [Google Scholar]
- 9. Chorin E., Vinograd O., Fleidervish I., Gilad D., Herrmann S., Sekler I., Aizenman E., Hershfinkel M. (2011) Up-regulation of KCC2 activity by zinc-mediated neurotransmission via the mZnR/GPR39 receptor. J. Neurosci. 31, 12916–12926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sharir H., Zinger A., Nevo A., Sekler I., Hershfinkel M. (2010) Zinc released from injured cells is acting via the Zn2+-sensing receptor, ZnR, to trigger signaling leading to epithelial repair. J. Biol. Chem. 285, 26097–26106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cohen L., Azriel-Tamir H., Arotsker N., Sekler I., Hershfinkel M. (2012) Zinc sensing receptor signaling, mediated by GPR39, reduces butyrate-induced cell death in HT29 colonocytes via up-regulation of clusterin. PLoS One 7, e35482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moechars D., Depoortere I., Moreaux B., de Smet B., Goris I., Hoskens L., Daneels G., Kass S., Ver Donck L., Peeters T., Coulie B. (2006) Altered gastrointestinal and metabolic function in the GPR39-obestatin receptor-knockout mouse. Gastroenterology 131, 1131–1141 [DOI] [PubMed] [Google Scholar]
- 13. Sharma M., Sahu K., Dube A., Gupta P. K. (2005) Extracellular pH influences the mode of cell death in human colon adenocarcinoma cells subjected to photodynamic treatment with chlorin p6. J. Photochem. Photobiol. B 81, 107–113 [DOI] [PubMed] [Google Scholar]
- 14. Perdikis D. A., Davies R., Zhuravkov A., Brenner B., Etter L., Basson M. D. (1998) Differential effects of mucosal pH on human (Caco-2) intestinal epithelial cell motility, proliferation, and differentiation. Dig. Dis. Sci. 43, 1537–1546 [DOI] [PubMed] [Google Scholar]
- 15. Holzer P. (2009) Acid-sensitive ion channels and receptors. Handb. Exp. Pharmacol. 194, 283–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sandoval M., Burgos J., Sepúlveda F. V., Cid L. P. (2011) Extracellular pH in restricted domains as a gating signal for ion channels involved in transepithelial transport. Biol. Pharm. Bull 34, 803–809 [DOI] [PubMed] [Google Scholar]
- 17. Walker A. R., Walker B. F. (1992) Fecal pH and colon cancer. Gut 33, 572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thornton J. R. (1981) High colonic pH promotes colorectal cancer. Lancet 1, 1081–1083 [DOI] [PubMed] [Google Scholar]
- 19. Nugent S. G., Kumar D., Rampton D. S., Evans D. F. (2001) Intestinal luminal pH in inflammatory bowel disease. Possible determinants and implications for therapy with aminosalicylates and other drugs. Gut 48, 571–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McDougall C. J., Wong R., Scudera P., Lesser M., DeCosse J. J. (1993) Colonic mucosal pH in humans. Dig. Dis. Sci. 38, 542–545 [DOI] [PubMed] [Google Scholar]
- 21. Acuña-Castillo C., Coddou C., Bull P., Brito J., Huidobro-Toro J. P. (2007) Differential role of extracellular histidines in copper, zinc, magnesium, and proton modulation of the P2X7 purinergic receptor. J. Neurochem. 101, 17–26 [DOI] [PubMed] [Google Scholar]
- 22. Gore A., Moran A., Hershfinkel M., Sekler I. (2004) Inhibitory mechanism of store-operated Ca2+ channels by zinc. J. Biol. Chem. 279, 11106–11111 [DOI] [PubMed] [Google Scholar]
- 23. Maret W. (2012) New perspectives of zinc coordination environments in proteins. J. Inorg. Biochem. 111, 110–116 [DOI] [PubMed] [Google Scholar]
- 24. Storjohann L., Holst B., Schwartz T. W. (2008) Molecular mechanism of Zn2+ agonism in the extracellular domain of GPR39. FEBS Lett. 582, 2583–2588 [DOI] [PubMed] [Google Scholar]
- 25. Holst B., Egerod K. L., Schild E., Vickers S. P., Cheetham S., Gerlach L. O., Storjohann L., Stidsen C. E., Jones R., Beck-Sickinger A. G., Schwartz T. W. (2007) GPR39 signaling is stimulated by zinc ions but not by obestatin. Endocrinology 148, 13–20 [DOI] [PubMed] [Google Scholar]
- 26. Boyarsky G., Ganz M. B., Sterzel R. B., Boron W. F. (1988) pH regulation in single glomerular mesangial cells. II. Na+-dependent and -independent Cl−-HCO3 exchangers. Am. J. Physiol. 255, C857–C869 [DOI] [PubMed] [Google Scholar]
- 27. Quinn S. J., Bai M., Brown E. M. (2004) pH sensing by the calcium-sensing receptor. J. Biol. Chem. 279, 37241–37249 [DOI] [PubMed] [Google Scholar]
- 28. Savdie C., Ferguson S. S., Vincent J., Beaudet A., Stroh T. (2006) Cell-type-specific pathways of neurotensin endocytosis. Cell Tissue Res. 324, 69–85 [DOI] [PubMed] [Google Scholar]
- 29. Aramori I., Ferguson S. S., Bieniasz P. D., Zhang J., Cullen B., Cullen M. G. (1997) Molecular mechanism of desensitization of the chemokine receptor CCR-5. Receptor signaling and internalization are dissociable from its role as an HIV-1 co-receptor. EMBO J. 16, 4606–4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vallee B. L., Auld D. S. (1990) Zinc coordination, function, and structure of zinc enzymes and other proteins. Biochemistry 29, 5647–5659 [DOI] [PubMed] [Google Scholar]
- 31. Maret W. (2006) Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid Redox Signal. 8, 1419–1441 [DOI] [PubMed] [Google Scholar]
- 32. Genz A. K., v Engelhardt W., Busche R. (1999) Maintenance and regulation of the pH microclimate at the luminal surface of the distal colon of guinea-pig. J. Physiol. 517, 507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chu S., Montrose M. H. (1995) Extracellular pH regulation in microdomains of colonic crypts. Effects of short-chain fatty acids. Proc. Natl. Acad. Sci. U.S.A. 92, 3303–3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dong X., Ko K. H., Chow J., Tuo B., Barrett K. E., Dong H. (2011) Expression of acid-sensing ion channels in intestinal epithelial cells and their role in the regulation of duodenal mucosal bicarbonate secretion. Acta Physiol. 201, 97–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holzer P. (2011) Acid sensing by visceral afferent neurones. Acta Physiol. 201, 63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Malin S. A., Christianson J. A., Bielefeldt K., Davis B. M. (2009) TPRV1 expression defines functionally distinct pelvic colon afferents. J. Neurosci. 29, 743–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Goo T., Akiba Y., Kaunitz J. D. (2010) Mechanisms of intragastric pH sensing. Curr. Gastroenterol. Rep. 12, 465–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hofer A. M., Curci S., Doble M. A., Brown E. M., Soybel D. I. (2000) Intercellular communication mediated by the extracellular calcium-sensing receptor. Nat. Cell Biol. 2, 392–398 [DOI] [PubMed] [Google Scholar]
- 39. Bachmann O., Seidler U. (2011) News from the end of the gut. How the highly segmental pattern of colonic HCO transport relates to absorptive function and mucosal integrity. Biol. Pharm. Bull. 34, 794–802 [DOI] [PubMed] [Google Scholar]
- 40. Liu J., Kohler J. E., Blass A. L., Moncaster J. A., Mocofanescu A., Marcus M. A., Blakely E. A., Bjornstad K. A., Amarasiriwardena C., Casey N., Goldstein L. E., Soybel D. I. (2011) Demand for Zn2+ in acid-secreting gastric mucosa and its requirement for intracellular Ca2+. PLoS One 6, e19638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chu J., Chu S., Montrose M. H. (2002) Apical Na+/H+ exchange near the base of mouse colonic crypts. Am. J. Physiol. Cell Physiol. 283, C358–C372 [DOI] [PubMed] [Google Scholar]
- 42. Luk H. H., Ko J. K., Fung H. S., Cho C. H. (2002) Delineation of the protective action of zinc sulfate on ulcerative colitis in rats. Eur. J. Pharmacol. 443, 197–204 [DOI] [PubMed] [Google Scholar]
- 43. Finamore A., Massimi M., Conti Devirgiliis L., Mengheri E. (2008) Zinc deficiency induces membrane barrier damage and increases neutrophil transmigration in Caco-2 cells. J. Nutr. 138, 1664–1670 [DOI] [PubMed] [Google Scholar]
- 44. Guan Y., Dong J., Tackett L., Meyer J. W., Shull G. E., Montrose M. H. (2006) NHE2 is the main apical NHE in mouse colonic crypts but an alternative Na+-dependent acid extrusion mechanism is up-regulated in NHE2-null mice. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G689–G699 [DOI] [PubMed] [Google Scholar]
- 45. Arena E. A., Longo W. E., Roberts K. E., Geibel P., Nateqi J., Brandstetter M., Geibel J. P. (2012) Functional role of NHE4 as a pH regulator in rat and human colonic crypts. Am. J. Physiol. Cell Physiol. 302, C412–C418 [DOI] [PubMed] [Google Scholar]
- 46. Srivastava J., Barber D. L., Jacobson M. P. (2007) Intracellular pH sensors. Design principles and functional significance. Physiology 22, 30–39 [DOI] [PubMed] [Google Scholar]
- 47. Elling C. E., Frimurer T. M., Gerlach L. O., Jorgensen R., Holst B., Schwartz T. W. (2006) Metal ion site engineering indicates a global toggle switch model for seven-transmembrane receptor activation. J. Biol. Chem. 281, 17337–17346 [DOI] [PubMed] [Google Scholar]
- 48. Hargitai D., Pataki A., Raffai G., Füzi M., Dankó T., Csernoch L., Várnai P., Szigeti G. P., Zsembery A. (2010) Calcium entry is regulated by Zn2+ in relation to extracellular ionic environment in human airway epithelial cells. Respir. Physiol. Neurobiol. 170, 67–75 [DOI] [PubMed] [Google Scholar]
- 49. Walsh B. M., Naik H. B., Dubach J. M., Beshire M., Wieland A. M., Soybel D. I. (2007) Thiol-oxidant monochloramine mobilizes intracellular Ca2+ in parietal cells of rabbit gastric glands. Am. J. Physiol. Cell Physiol. 293, C1687–C1697 [DOI] [PubMed] [Google Scholar]
- 50. Cima R. R., Dubach J. M., Wieland A. M., Walsh B. M., Soybel D. I. (2006) Intracellular Ca2+ and Zn2+ signals during monochloramine-induced oxidative stress in isolated rat colon crypts. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G250–G261 [DOI] [PubMed] [Google Scholar]
- 51. Tomura H., Mogi C., Sato K., Okajima F. (2005) Proton-sensing and lysolipid-sensitive G-protein-coupled receptors. A novel type of multifunctional receptors. Cell. Signal. 17, 1466–1476 [DOI] [PubMed] [Google Scholar]
- 52. Levinthal C., Barkdull L., Jacobson P., Storjohann L., Van Wagenen B. C., Stormann T. M., Hammerland L. G. (2009) Modulation of group III metabotropic glutamate receptors by hydrogen ions. Pharmacology 83, 88–94 [DOI] [PubMed] [Google Scholar]
- 53. Lukin J. A., Ho C. (2004) The structure-function relationship of hemoglobin in solution at atomic resolution. Chem. Rev. 104, 1219–1230 [DOI] [PubMed] [Google Scholar]
- 54. Umemura T., Matsumoto Y., Ohnishi K., Homma M., Kawagishi I. (2002) Sensing of cytoplasmic pH by bacterial chemoreceptors involves the linker region that connects the membrane-spanning and the signal-modulating helices. J. Biol. Chem. 277, 1593–1598 [DOI] [PubMed] [Google Scholar]
- 55. Bozym R. A., Thompson R. B., Stoddard A. K., Fierke C. A. (2006) Measuring picomolar intracellular exchangeable zinc in PC-12 cells using a ratiometric fluorescence biosensor. ACS Chem Biol. 1, 103–111 [DOI] [PubMed] [Google Scholar]
- 56. Bozym R. A., Chimienti F., Giblin L. J., Gross G. W., Korichneva I., Li Y., Libert S., Maret W., Parviz M., Frederickson C. J., Thompson R. B. (2010) Free zinc ions outside a narrow concentration range are toxic to a variety of cells in vitro. Exp. Biol. Med. (Maywood) 235, 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wilson M., Hogstrand C., Maret W. (2012) Picomolar concentrations of free zinc(II) ions regulate receptor protein-tyrosine phosphatase β activity. J. Biol. Chem. 287, 9322–9326 [DOI] [PMC free article] [PubMed] [Google Scholar]