

Background: For TRIM/RBCC family members, the stomach-specific TRIM50 has no defined biological role.
Results: TRIM50 was associated with intracellular vesicles to contribute to their dynamic movement in cultured cells, and mutant gastric parietal cells from Trim50 knock-out mice exhibited abnormal rearrangement of intracellular vesicles.
Conclusion: TRIM50 is essential for vesicular dynamics in parietal cells.
Significance: Our finding unveiled the basic biological role of TRIM50.
Keywords: E3 Ubiquitin Ligase, Gene Knock-out, Membrane Trafficking, Phosphoinositides, PI 3-Kinase (PI3K), RBCC/TRIM Family, SPRY Domain, Tubulovesicle
Abstract
Of the TRIM/RBCC family proteins taking part in a variety of cellular processes, TRIM50 is a stomach-specific member with no defined biological function. Our biochemical data demonstrated that TRIM50 is specifically expressed in gastric parietal cells and is predominantly localized in the tubulovesicular and canalicular membranes. In cultured cells ectopically expressing GFP-TRIM50, confocal microscopic imaging revealed dynamic movement of TRIM50-associated vesicles in a phosphoinositide 3-kinase-dependent manner. A protein overlay assay detected preferential binding of the PRY-SPRY domain from the TRIM50 C-terminal region to phosphatidylinositol species, suggesting that TRIM50 is involved in vesicular dynamics by sensing the phosphorylated state of phosphoinositol lipids. Trim50 knock-out mice retained normal histology in the gastric mucosa but exhibited impaired secretion of gastric acid. In response to histamine, Trim50 knock-out parietal cells generated deranged canaliculi, swollen microvilli lacking actin filaments, and excess multilamellar membrane complexes. Therefore, TRIM50 seems to play an essential role in tubulovesicular dynamics, promoting the formation of sophisticated canaliculi and microvilli during acid secretion in parietal cells.
Introduction
TRIM/RBCC family proteins are characterized by the RBCC/tripartite motif, consisting of a Ring domain followed by one or two B-box domains and then a coiled-coil domain (1). The RBCC motif always appears at the N-terminal regions of the family members, whereas their C-terminal regions contain various domains and motifs, including the PRY-SPRY domain (2), NHL/WD repeat (3), and pleckstrin homology domain (4). More than 70 TRIM family members are predicted in the mammalian genomes and classified into ∼10 subfamilies based on their C-terminal regions. Recent studies indicate that the RBCC motif has the conserved biochemical ability to act as E3 ligases for ubiquitin, small ubiquitin-like modifier and interferon-stimulated protein of 15 kDa (ISG15). The Ring domain of the RBCC motif probably recognizes ubiquitin or its related molecules, whereas the coiled-coil domain, together with the B-box domain, is likely assigned as a substrate-specific binding pocket (5). The E3-ligase activities for several TRIM family members have been shown to link to various cellular functions, including proliferation, differentiation, and viral infection, whereas the roles of the divergent C-terminal domains remain largely unknown (1).
Our recent studies defined mitsugumin 53 (MG53),4 a muscle-specific TRIM family protein (TRIM72), as an essential component of the cell membrane repair machinery (6, 7). Knock-out mice lacking MG53 show progressive myopathy, and mutant muscle cells are defective in the ability to repair membrane injury (6). MG53 acts as a sensor of extracellular oxidation and recruits intracellular vesicles to the injury site for membrane patch formation in cooperation with other repair-related proteins, including dysferlin and caveolin-3 (8–10). Furthermore, MG53 facilitates steady-state intracellular vesicular trafficking for the quality control of cell-surface membrane proteins (11). In the primary structure of MG53, the RBCC motif is followed by the PRY-SPRY domain. Although the role of the PRY-SPRY domain remains largely unknown, it is present in ∼25 TRIM family members. Of these family members, MG53 exhibits the highest sequence identity with TRIM50 (12). In this study, we tested the hypothesis that TRIM50 may share similar biological functions with MG53 in vesicular trafficking. We employed biochemical, immunohistochemical, and live cell imaging tools to define the cellular function of TRIM50, and we also generated Trim50 knock-out mice to define its physiological role.
MATERIALS AND METHODS
DNA Cloning and Immunochemistry
Based on the reported mouse Trim50 cDNA sequence (GenBankTM NM 178240), PCR primers were designed to amplify the full-length cDNA using stomach RNA preparations as templates. Northern blot analysis was carried out using total RNA preparations from adult C57BL/6J mouse tissues (13). The full-length and partial Trim50 cDNAs were cloned into the pMalp2 vector (New England Biolabs) to produce maltose-binding protein (MBP) fusion proteins. The MBP-TRIM50 fusion proteins were purified from bacterial cultures according to the manufacturer's instructions for monoclonal antibody production (13), lipid dot-blot assay (6), and in vitro ubiquitination assay (14).
Wistar rats were immunized with MBP-TRIM50 fusion protein, and hybridoma cells were produced by fusion of rat lymphocytes with NS-1 cells (15). The specificity of a monoclonal antibody against TRIM50 (mAb84) was confirmed with the lack of immunoreactivity in the Trim50 knock-out stomach (supplemental Fig. S3). To detect authentic organelle marker proteins, we used primary antibodies against the α-subunit of H+/K+-ATPase (Medical & Biological Laboratories), Na+/K+-ATPase (Upstate), pepsinogen (Abcam), KCNQ1 (Santa Cruz Biotechnology), actin (Sigma), ezrin (Santa Cruz Biotechnology), calnexin (Santa Cruz Biotechnology), mannose 6-phosphate receptor (Abcam), Golgi matrix 130-kDa protein (Abcam), lysosomal associated membrane protein 1 (Abcam), early endosomal antigen 1 (BD Biosciences), Rab11 (Abcam), JNK (Cell Signaling), and GAPDH (Sigma). To survey the lipid binding activity, purified MBP-TRIM50 proteins were applied to PIP2-Strip membranes (Echelon) according to the manufacturer's instructions, and protein-lipid interaction was visualized using an anti-MBP antibody (New England Biolabs) as described previously (6, 13).
Gastric Cell and Membrane Preparations
Mucosal cells were prepared from the mouse stomach and separated by density gradient centrifugation as described previously (16). Briefly, the stomach was removed and turned inside out, and the inverted sac was treated with a Ca2+-free solution containing 0.025% Pronase E. After the cell debris was removed, the tissue was further digested in a solution containing 1 mm CaCl2 and 0.05% Pronase E. The separated cells were collected and resuspended in DMEM for loading onto a discontinuous Optiprep (Axis-Shield) gradient composed of solutions with densities of 1.139, 1.095, 1.073, and 1.049 g/ml. After centrifugation at 1,000 × g for 8 min, the cells on the top of each layer were collected for Western blot analysis (17).
Biochemical cell fractionation from the gastric mucosa was carried out as described previously (18). The fundic mucosae from mice fasted for 24 h were homogenized in a buffer containing 250 mm sucrose, 1 mm EGTA, and 5 mm Tris-HCl (pH 7.4). The homogenate was centrifuged at 1,000 × g for 10 min to remove cell debris and nuclei (P1), and the supernatant was further centrifuged at 13,500 × g for 30 min. The resulting pellet (P2) was resuspended in the homogenizing buffer, loaded onto a 7 and 18% Ficoll step gradient, and centrifuged at 100,000 × g for 2 h to recover the stimulation-associated vesicles from the interface between the Ficoll layers. The supernatant recovered in the 13,500 × g centrifugation was further centrifuged at 100,000 × g for 30 min to acquire the microsomal membranes (P3) and cytosolic fraction (S3).
Tracking of TRIM50-containing Vesicles
Human gastric adenocarcinoma AGS cells (American Type Culture Collection) were grown in DMEM supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 10 μg/ml streptomycin. A GFP-TRIM50 expression plasmid was constructed by inserting the mouse Trim50 cDNA into the 3′ end of the GFP-C1 vector (Clontech). AGS cells were plated in glass-bottom dishes and cultured for 24 h to reach ∼70% confluency. The cells were transfected with the GFP-TRIM50 plasmid using Lipofectamine 2000 (Invitrogen) and visualized by live-cell confocal imaging (Bio-Rad) at 24–48 h after transfection. The movements of GFP-TRIM50 containing vesicles in AGS cells were analyzed using a modified vesicle trafficking software originally developed by Jaqaman et al. (19, 20). To examine the pharmacological effects of drugs that influence the motor activity and the dynamic movement of GFP-TRIM50-labeled vesicles, the vesicular velocity was calculated from the absolute difference between 50 consecutive frames.
Knock-out Mouse Generation
All of the animal experiments in this study were conducted with the approval of the Animal Research Committee according to the regulations on animal experimentation at Kyoto University. The generation of Trim50 knock-out mice was carried out as described previously (21). The targeting vector (supplemental Fig. S3) was constructed with Trim50 genomic DNA fragments, the neomycin resistance gene from pMC1Neo poly(A) (Stratagene), the diphtheria toxin gene from pMC1-DT-A, and pBluescript SK(−) (Stratagene). The linearized vector was transfected into embryonic stem J1 cells, and several clones carrying the expected homologous mutation were selected by Southern blot analysis. Chimeric mice generated with the positive clone were crossed with C57BL/6J mice and transmitted the mutant gene to their pups. To determine the mouse genotypes, PCR analysis was conducted using the following primers: TRIM50-M2, CGGCAGGATAGAGGACTGACTGC; TRIM50-M5, GGCAGCCTCAGAGCATCTATCAC.
Gastric Acid Measurements
Gastric acid secretion was examined as described previously (22). Mice were fasted for 24 h and then injected with histamine (2 mg/kg, s.c.) or cimetidine (100 mg/kg, i.p.). After 15 min, the gastroesophageal and pyloric junctions were clamped, and the stomach was removed. The stomach was immersed in 2 ml of oxygen-saturated normal saline and opened along the lesser curvature. The solution containing the gastric contents was centrifuged at 500 × g for 5 min, and the supernatant was recovered for measuring pH and titrating to pH 6.5 with 0.01 n NaOH; the titration value was normalized to the stomach wet weight.
Total H+/K+-ATPase activity in microsomal fractions (20 μg of protein) from gastric mucosa was measured in a 1-ml reaction mixture containing 15 mm KCl, 3 mm MgSO4, 1 mm ATP, 5 mm NaN3, 2 mm ouabain, and 17.4 mm Tris-HCl (pH 6.8) in the presence or absence of 10 μm of the H+/K+-ATPase inhibitor SCH28080, as described previously (18). After incubation for 20 min at 37 °C, the reaction was terminated by an ice-cold stop solution containing 12% HClO4 and 3.6% ammonium molybdate to monitor the inorganic phosphate released.
Morphological Analyses
Morphological analyses were carried out as described previously (23, 24). Mice were fasted for 24 h and treated with cimetidine (100 mg/kg, i.p. injection 30 min before sacrifice) for the resting phase or histamine (2 mg/kg, s.c. injection 15 min before sacrifice) for the stimulated phase. For light- microscopic analyses, anesthetized mice were perfused with a fixative containing 4% paraformaldehyde, 0.1% glutaraldehyde (GTA), and 0.1 m phosphate buffer (pH 7.4) (PB). The isolated stomachs were further immersed in the fixative at 4 °C overnight and dehydrated for paraffin embedding. Paraffin sections were stained with H&E or 1% Alcian blue (pH 2.5), periodic acid-Schiff reagent. For the immunohistochemical analysis of TRIM50, the isolated stomachs were plunged into an isopentane/propane mixture cooled by liquid nitrogen. Freeze substitution was carried out in 0.1% GTA in acetone at −80 °C for 16 h, and then the tissues were gradually warmed to room temperature. After being washed with ethanol, the specimens were embedded in paraffin. Paraffin sections were treated in methanol containing 0.3% H2O2 for 30 min to block endogenous peroxidase activity and incubated in PBS containing 5% normal horse serum and 1% bovine serum albumin for 10 min to block nonspecific binding. After incubation with primary antibodies followed by biotinylated secondary antibodies, the immunoreactivities were visualized using an avidin-biotinylated horseradish peroxidase complex kit (Vector Laboratories) or catalyzed signal amplification system (Dako).
For electron microscopic analyses, anesthetized mice were perfused with a fixative containing 2% paraformaldehyde, 2.5% GTA, and PB, and small pieces of the gastric mucosa were further immersed in the fixative at 4 °C overnight. After being rinsed with PB, the small pieces of gastric mucosa were post-fixed with 1% OsO4 in PB on ice for 2 h and then embedded into epoxy resin. Ultrathin sections (60–80 nm in thickness) were stained with 2% uranyl acetate and Reynolds' lead citrate. For the immunolabeling of H+/K+-ATPase, the anesthetized mice were perfused with a fixative containing 4% paraformaldehyde, 0.1% GTA, and PB, and the small pieces of gastric mucosa were further immersed in the fixative at 4 °C overnight. After rinsing with PB, the small pieces of gastric mucosa were embedded into Lowicryl K4M resin (Structure Probe). Ultrathin sections were incubated with horse serum and bovine albumin to block nonspecific binding. After incubation with a monoclonal anti-H+/K+-ATPase antibody, the sections were reacted with a biotinylated secondary antibody and anti-biotin IgG (Vector Laboratories) conjugated with 8-nm colloidal gold (23). The ultrathin sections were analyzed using a transmission electron microscope (JEOL 1200EX) operating at 80 kV.
RESULTS
Expression of TRIM50 in Gastric Parietal Cells
The previous study has demonstrated that Trim50 mRNA is detected in the stomach (12). Based on the reported sequences in DNA databases, PCR primers were designed for amplifying the full-length mouse Trim50 cDNA using stomach RNA preparations as templates. Sequencing-amplified cDNAs identified an insertion of a Lys residue between Arg-242 and Phe-243 in the TRIM50 protein-coding sequence deposited in the databases (supplemental Fig. S1). A phylogenic tree generated from sequence comparison revealed the closest homology between TRIM50 and MG53 among the TRIM family members containing the PRY-SPRY domain (Fig. 1A). Northern blotting with various mouse tissues indicated that Trim50 mRNA is specifically expressed in the stomach (Fig. 1B).
FIGURE 1.

Specific expression of TRIM50 in gastric parietal cells. A, phylogenetic tree of TRIM/RBCC subfamily members containing the PRY and SPRY domains. Our BLAST search found ∼25 members categorized into the subfamily in mammalian protein databases. Using ClustalW software, the phylogenetic tree was generated based on their multiple sequence alignment. TRIM50 shows the highest sequence identity with MG53. The scale bar indicates substitutions per aligned position. B, Northern blot analysis of TRIM50 in mouse tissues. Total RNA preparations (25 μg per lane) from mouse tissues were analyzed using a probe derived from exon 2 of the Trim50 gene. C, phase-contrast images of fractionated gastric mucosal cells. Dispersed mucosal cells were prepared from mouse stomach tissue and separated on a Percoll density gradient; the fractions numbered 1, 2, and 3 correspond to low, intermediate, and high density fractions, respectively. Of the mucosal cells, parietal cells with a large size (∼25 μm in diameter) were enriched in fraction 1. D, detection of TRIM50 in gastric mucosal cell fractions. Total cell lysates prepared from each cell fraction (1 or 10 μg of protein) were analyzed using antibodies to TRIM50, H+/K+-ATPase (H/K-P, parietal cell marker), pepsinogen (Peps) (chief cell marker), and actin (loading control). E, immunohistochemical staining of TRIM50 in mouse gastric mucosa. Immunoreactivity of mAb84 was specifically observed in parietal cells. The scale bars indicate 50 μm (C and E).
To identify the cell type expressing TRIM50 in the stomach, we developed a specific monoclonal antibody from rats immunized with the recombinant TRIM50 protein. Single mucosal cells were prepared from mouse stomachs and separated by density gradient centrifugation for Western blotting (Fig. 1C). Of the cell fractions, the larger sized parietal cells containing H+/K+-ATPase, and the smaller sized chief cells expressing pepsinogen were abundantly enriched in the low and high density fractions, respectively (Fig. 1D). Immunoreactivities against TRIM50 and H+/K+-ATPase were associated during the cell preparation, indicating that TRIM50 is specifically expressed in parietal cells. This conclusion was confirmed by our immunohistochemical staining of TRIM50 in gastric tissue sections (Fig. 1E).
Association of TRIM50 with Tubulovesicles
TRIM50 immunoreactivity was not uniform in the cytoplasm of parietal cells (Fig. 1E), suggesting that TRIM50 does not behave like a typical cytosolic protein. To examine the subcellular localization of TRIM50 in parietal cells, we performed cell fractionation from gastric mucosal homogenates by differential centrifugation (Fig. 2A). Western blotting using the separated fractions demonstrated that TRIM50 was predominantly recovered in the high density membrane fraction P2 but not in the cytosolic fraction S3 (Fig. 2B). In density gradient centrifugation of the microsomal vesicles, TRIM50 was further enriched in the stimulation-associated vesicle fraction as demonstrated by its co-migration during the fractionation with the marker proteins, including H+/K+-ATPase and ezrin.
FIGURE 2.

Subcellular localization of TRIM50 in parietal cells. A, flow chart for mouse gastric mucosal cell fractionation by differential centrifugation. Ho, total homogenate of gastric mucosa; P1, precipitates (ppt) after low speed centrifugation (nuclei and cell debris); P2, precipitates after high speed centrifugation (high density microsomal membrane); SAV, stimulation-associated vesicles, fraction prepared from P2 by Ficoll density gradient centrifugation (apical canalicular membrane-rich fraction); P3, precipitates after ultracentrifugation (low density microsomal membrane); S3, supernatant (sup) after the ultracentrifugation (cytosolic fraction). B, preparations (1 or 10 μg opf protein) during the cell fractionation were subjected to Western blot analysis using antibodies specific to TRIM50, H+/K+-ATPase (H/K-P; canalicular and tubulovesicular membrane marker), ezrin (canalicular and tubulovesicular membrane marker), Na+/K+-ATPase (Na/K-P; basolateral membrane marker), KCNQ1 (tubulovesicular membrane marker), and JNK (cytoplasmic kinase). C, immunohistochemical detection of H+/K+-ATPase and TRIM50 in parietal cells during the resting and histamine-stimulated phases. The scale bar indicates 10 μm.
During histamine-induced structural transformation of tubulovesicles into canaliculi connected with the apical surface, the subcellular localization of H+/K+-ATPase is dramatically changed to facilitate gastric acid secretion in parietal cells (23, 25). Consistent with the previous observations, histamine induced a clear redistribution of H+/K+-ATPase from the cytoplasmic region to the apical side, leading to formation of dense immunoreactive spots near the cell periphery (Fig. 2C). Although TRIM50 does not contain a membrane-spanning segment or lipid-modification motif in the primary structure, similar staining features were observed between H+/K+-ATPase and TRIM50 under resting conditions. Therefore, the immunostaining, together with the cell fractionation, strongly indicates that TRIM50 is predominantly localized in tubulovesicles. However, TRIM50 immunolabeling became more condensed in cell interior regions during the stimulated phase, and this transitional profile was clearly different from the peripherally confined redistribution of H+/K+-ATPase. Notice that the TRIM50 and KCNQ1 channels were recovered in different fractions during cell fractionation (Fig. 2B). In contrast to H+/K+-ATPase, KCNQ1 channels localized in tubulovesicles do not undergo the histamine-induced subcellular translocation during acid secretion (26). Taken together, our histochemical observations likely suggest that TRIM50 resides in the cell interior canaliculi generated by tubulovesicular fusion during the stimulated phase.
Tracking of TRIM50-associated Vesicles
For further resolution of the subcellular distribution of TRIM50, cultured cells were transfected with an expression plasmid carrying the Trim50 cDNA. The immunofluorescence signals for the ectopically expressed TRIM50 protein were never distributed evenly in the cytoplasm; instead, they existed in patterns of cytosolic vesicles (supplemental Fig. S2A). The punctate and meshwork structures labeled by TRIM50 were partly overlapped with the distribution of endoplasmic reticulum and lysosome markers but did not merge with endosome and Golgi markers. Thus, TRIM50 may exist in a vesicular population that is different from these major intracellular organelles. We have previously demonstrated that MG53 ectopically expressed is also associated with intracellular vesicles distinct from the authentic organelles (8). When C2C12 cells expressing GFP-MG53 are stabbed with a microcapillary glass, MG53-associated vesicles immediately nucleate into the injury site for membrane repair (6). In contrast, no accumulation of TRIM50-associated vesicles was observed after the surface injury in C2C12 cells expressing GFP-TRIM50 (supplemental Fig. S2B). Therefore, TRIM50 could not contribute to the membrane repair mechanism in C2C12 cells.
To monitor the intracellular distribution and movement of the TRIM50-labeled vesicles, we introduced the GFP-TRIM50 expression construct into cultured AGS cells and examined them under a confocal microscope. Punctate patterns of GFP-TRIM50-associated vesicles were clearly identified in the cells, and live-cell imaging revealed dynamic movement of the TRIM50-associated vesicles under basal conditions (supplemental movie). In our attempt to characterize the vesicular movement using various chemical inhibitors, prolonged treatment (30 min to 1 h of incubation) of wortmannin, an inhibitor for phosphoinositide 3-kinase (PI3K), led to redistribution of the punctate GFP-TRIM50 labeling into a more cytosolic pattern (Fig. 3A). This observation may imply that the vesicular binding of TRIM50 and/or clustering of TRIM50-labeled vesicles occurs in a PI3K-dependent manner. Tracking of the TRIM50-containing vesicles was also performed using a computer software to calculate the vesicular mobility index “effective trafficking distance” (Fig. 3B). Even in short term treatment (10–20 min of incubation), wortmannin significantly decelerated the mobility of TRIM50-associated vesicles (Fig. 3C). Moreover, the movement of TRIM50-associated vesicles was inhibited by another PI3K inhibitor LY294002, whereas its inactive homologue LY303511 had no effect. The imaging data likely suggest that PI3K is involved in tethering TRIM50 to intracellular vesicles and thus enhances the mobility of the TRIM50-associated vesicles. However, no significant effects on the vesicular mobility were detected in the application of other chemical inhibitors that act on the myosin II motor (N-benzyl-p-toluene sulfonamide), actin assembly (latrunculin-B), or microtubule network (colchicine).
FIGURE 3.

Trafficking of TRIM50-containing vesicles in cultured cells. A, confocal microscopic imaging of AGS cells expressing GFP-TRIM50. The scale bar indicates 5 μm. The representative fluorescence images showed that prolonged wortmannin (WTN) treatment (10 μm for 1 h) reduces the cellular contents of TRIM50-associated vesicles. B, tracking of TRIM50-containing vesicles by confocal microscopic imaging. TRIM50-associated vesicles were tracked for 77 s (50 frames), and the moving traces are illustrated with red lines. The scale bars indicate 1 μm. Representative images show that the myosin II inhibitor N-benzyl-p-toluene sulfonamide (BTS, 40 μm, 20 min) does not inhibit the vesicular trafficking (left panels), whereas the PI 3-kinase inhibitor wortmannin (10 μm, 12 min) immobilizes the dynamic movements of TRIM50-containing vesicles (right panels). C, pharmacological characterization of the mobility of TRIM50-containing vesicles. The movements of TRIM50-containing vesicles were spatio-temporally examined to yield effective trafficking distance. The vesicular mobility was analyzed before and after inhibitor treatments: the myosin II inhibitor N-benzyl-p-toluene sulfonamide (BTS, 40 μm), actin assembly inhibitor latrunculin-B (LAT-B, 10 μm), microtubule inhibitor colchicines (COL, 20 μm), PI 3-kinase inhibitors wortmannin (WTN, 10 μm) and LY294002 (20 μm), and LY303511 (an inactive homologue of LY294002, 20 μm). The data represent the mean ± S.E. in three independent experiments (n >6 cells), and statistical differences are indicated by asterisks (**, p < 0.01 in Kolmogorov-Smirnov test).
Binding of TRIM50 to Phospholipids
To examine the mechanisms that underlie the vesicular association of TRIM50, we produced recombinant MBP-TRIM50 proteins (Fig. 4, A and B) and performed the established lipid dot-blot assay (Fig. 4C). In the protein-overlay experiments, the C-terminal PRY-SPRY domain preferentially reacted with phosphatidylinositol phosphates among the various lipids examined. In particular, strong binding activities were detected for phosphatidylinositol 3- and 5-monophosphates. Neither the PRY domain nor the SPRY domain alone exhibited such binding activity. To examine the role of the PRY-SPRY domain in the subcellular localization, GFP-TRIM50 proteins were expressed in cultured cells (Fig. 4D). As expected from results in the protein-overlay assay, GFP-TRIM50 was predominantly associated with cytoplasmic vesicles, whereas its PRY and SPRY deletion mutants no longer displayed clear punctate distribution.
FIGURE 4.

Phospholipid binding activity of TRIM50. A, scheme of MBP-TRIM50 fusion proteins. The numbers in parentheses indicate amino acid residues in the TRIM50 primary sequence. B, purification of recombinant fusion proteins from bacterial culture. Whole E. coli lysates and affinity-purified proteins were separated on an SDS-polyacrylamide gel and stained with Coomassie Blue. C, protein-overlay analysis using PIP Strips. The recombinant protein containing the PRY-SPRY domain (PR1) preferentially reacted with the phosphatidylinositol monophosphates PI(3)P and PI(5)P. Such binding activity was not detected in the fusion proteins carrying either the PRY (PR2) or SPRY domain (PR3) alone. D, fluorescence images of COS-7 cells expressing GFP-TRIM50 and its deletion mutants. The representative images indicate that both PRY and SPRY domains essentially contribute to the vesicular binding activity of TRIM50. The scale bar indicates 10 μm.
Our observations suggest that the PRY and SPRY domains cooperatively form a phospholipid-binding pocket structure to associate with cytoplasmic vesicles. The PI3K inhibitors disturb the metabolism of phosphatidylinositol phosphates and may disrupt the active movement of TRIM50-associated vesicles in cultured cells (Fig. 3). In a more physiological point of view, the lipid binding activities likely contribute to the attachment of TRIM50 to the tubulovesicular and canalicular membranes in parietal cells. Phosphoinositide species are enriched at distinct sites in intracellular membrane systems; for example, phosphatidylinositol 4-phosphate, 3-phosphate, and 4,5-bisphosphate are generally concentrated in the Golgi complex, endosome, and plasma membrane, respectively (27). Although the subcellular distribution of phospholipids has not been closely examined in parietal cells, our biochemical data may imply that the phosphatidylinositol monophosphates are enriched in the tubulovesicular membrane.
Impaired Gastric Acid Secretion in Trim50 Knock-out Mice
To examine the physiological role of TRIM50, we generated knock-out mice (supplemental Fig. S3). In the vector used for introducing a targeted mutation, the genomic segment composed of the partial sequences of the first intron and second exon in the Trim50 gene is replaced by the neomycin resistance gene. Embryonic stem cells carrying the targeted mutation were selected and used to produce chimeric mice. The Trim50 knock-out mice thus generated have a mixed genetic background of C57BL and 129 strains, and they exhibited no abnormalities in growth or reproduction under our conventional housing conditions. In gastric mucosal preparations from the mutant mice, light microscopic observations detected no abnormalities in the overall cellular population and morphology as shown in H&E staining profiles (supplemental Fig. S4). Moreover, the mucosal layer from the mutant mice retained normal features in both Alcian blue/periodic acid-Schiff reagent staining for mucous cells and immunohistochemical staining of H+/K+-ATPase for parietal cells. Therefore, the genetic ablation of TRIM50 does not impact the development and differentiation of the gastric mucosa.
Because TRIM50 is associated with the tubulovesicular and canalicular membranes in parietal cells, we focused on gastric acid secretion in Trim50 knock-out mice. To examine histamine-induced gastric acid secretion, the pH and amount of acid were measured in gastric secretions from the knock-out mice at ∼10 and 20 weeks of age (Fig. 5A). In both age groups, histamine highly stimulated gastric acid secretion, suggesting that the basic machinery for acid secretion remained intact in the Trim50 knock-out parietal cells. However, the pH of the gastric secretions was significantly more alkaline in the knock-out mice than in wild-type mice. Consistently, the amount of acid in gastric secretions of the knock-out mice was significantly reduced compared with that of control mice (Fig. 5B).
FIGURE 5.

Impaired gastric acid secretion in Trim50 knock-out mice. A, gastric acid pH in Trim50 knock-out mice. Mice (9–10 or 19–20 weeks old) were fasted for 24 h, and the stomach contents were examined 15 min after histamine injection (2 mg/kg s.c.). B, gastric acid secretion in Trim50 knock-out mice. Total acid and base equivalents were determined by titration with 0.01 n NaOH or 0.01 n HCl. The data were normalized to the blotted wet weight of the stomach. The horizontal bars represent the mean values of the data for each group. C, normal histological features of the Trim50 knock-out stomach. Gastric mucosa was examined by immunohistochemical staining of H+/K+-ATPase. The scale bar indicates 50 μm. D, normal H+/K+-ATPase activity in gastric mucosal membrane preparations from Trim50 knock-out mice. SCH28080-sensitive and K+-dependent ATPase activity was determined in the microsomal preparations. E, normal H+/K+-ATPase content in gastric mucosal membrane preparations from Trim50 knock-out mice. The total microsomal preparations were examined by Western blotting using antibody to H+/K+-ATPase, and the immunoreactive signals (inset images) were digitalized and normalized to the mean value of the data from control mice. The numbers of mice examined are shown in parentheses, and statistical differences between the genotypes are indicated by asterisks (**, p < 0.01 in t test).
Because H+/K+-ATPase is responsible for acid secretion in parietal cells, we examined whether TRIM50 deficiency would reduce the protein content or catalytic activity of H+/K+-ATPase. Immunohistochemical observation revealed similar profiles of H+/K+-ATPase staining between Trim50 knock-out and wild-type cells (Fig. 5C). We also detected normal enzymatic activity and protein content of H+/K+-ATPase in gastric mucosal preparations derived from the knock-out mice (Fig. 5, D and E). Therefore, the loss of TRIM50 leads to impairment in acid secretion without affecting H+/K+-ATPase in parietal cells.
Abnormal H+/K+-ATPase-containing Membranes in Trim50 Knock-out Parietal Cells
During histamine-induced acid secretion in parietal cells, the H+/K+-ATPase-rich tubulovesicles fuse together and connect with the apical plasma membrane to form intracellular canaliculi (28, 29). To examine the cellular mechanism underlying the impaired acid secretion, we next designed electron microscopic analyses in resting and stimulated Trim50 knock-out parietal cells, which were induced by the application of a histamine receptor antagonist and agonist, respectively. In the resting phase, we detected no obvious ultrastructural abnormality in Trim50 knock-out parietal cells. However, disorganized canaliculi were irregularly developed in Trim50 knock-out cells during the histamine-stimulated phase (Fig. 6A). The formation of canaliculi seemed to be random or pleomorphic in the mutant cells, and such deranged formation was not detected in wild-type cells. Moreover, Trim50 knock-out cells formed atypical microvilli upon histamine stimulation. The microvilli in wild-type cells exhibited firm cylindrical morphology (average diameter of 97.0 nm) and were always equipped with the central core of actin filaments. In contrast, the mutant cells frequently exhibited swollen microvilli (average diameter of 151.9 nm) lacking the central actin filaments. Such swollen microvilli were never detected in wild-type cells.
FIGURE 6.

Abnormal ultrastructural features in Trim50 knock-out parietal cells. A, parietal cells in the resting and stimulated phases were examined by electron microscopy. In the resting phase, no difference was observed between the genotypes (left column). Note the irregular formation of intracellular canaliculi (IC) and excessive multilamellar membrane complexes (arrowhead) in Trim50 knock-out parietal cells during the stimulated phase (middle column). High magnification views clarify the differences between the firm cylindrical microvilli with central actin filaments (arrows) in wild-type and the swollen microvilli leading to pleomorphic canaliculi in Trim50 knock-out parietal cells (right column). The scale bars indicate 1 μm (left and middle columns) or 0.2 μm (right column). B, excessive multilamellar membrane complexes in Trim50 knock-out parietal cells. Electron microscopic immunolabeling shows specific labeling for H+/K+-ATPase on the multilamellar membrane complexes in stimulated Trim50 knock-out parietal cells. The scale bar indicates 0.5 μm. C, quantitative evaluation of the multilamellar membrane complexes in wild-type and Trim50 knock-out cells. Parietal cells (n = 30 in each genotype) were randomly selected, and the multilamellar membrane complexes (minor axis >0.5 μm, with triple or more lamellar membranes) were evaluated in electron micrographs using the freeware program Image J (version 1.45p). The data represent the mean ± S.E., and statistical differences are indicated by asterisks (*, p < 0.05 and **, p < 0.01 in t test).
In Trim50 knock-out parietal cells, large numbers of multilamellar membrane complexes were generated during the stimulated phase (Fig. 6, A and B). Such membrane complexes have been previously detected in normal parietal cells during the reverting phase after histamine stimulation, and they seem to represent autophagosome or multivesicular body-like characteristics for recovering canalicular membranes (24, 30). Indeed, a colloidal gold-conjugated antibody targeting H+/K+-ATPase efficiently decorated the multilamellar membrane complexes in the mutant cells, demonstrating that the membrane complexes are originally derived from tubulovesicles (24). Our statistical analysis confirmed that the membrane complexes obviously accumulated in the Trim50 knock-out cells during the stimulation phase (Fig. 6C). Taken together, the deranged canaliculi, swollen microvilli, and accumulated multilamellar membrane complexes likely suggest the severe impairment of tubulovesicular dynamics upon histamine stimulation in Trim50 knock-out parietal cells (Fig. 7).
FIGURE 7.
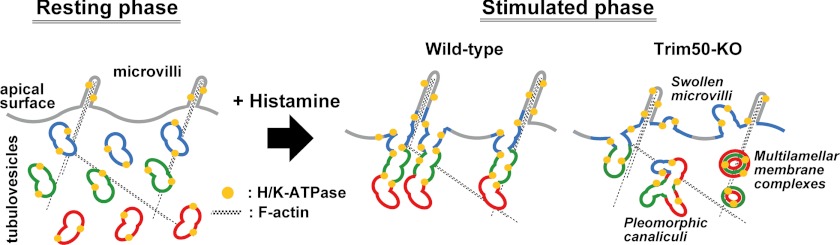
Proposed abnormal membrane dynamics in Trim50 knock-out parietal cells. Trim50 deficiency likely inhibits the elaborate transformation of tubulovesicles to canaliculi upon histamine stimulation and thus impairs efficient gastric acid secretion. In Trim50 knock-out cells, tubulovesicles probably undergo atypical membrane fusion and form pleomorphic canaliculi and swollen microvilli lacking the core of microfilaments. Such aberrant structures are likely recovered as the multilamellar membrane complexes for lysosomal degradation after the stimulated phase, and Trim50 knock-out cells regain normal membranous features in the resting phase.
DISCUSSION
The muscle-specific TRIM protein MG53 contains a C-terminal PRY-SPRY domain and is localized in small vesicles below the plasma membrane. The PRY-SPRY domain of MG53 exhibits a binding affinity for phosphatidylserine (6), whereas the corresponding domain in TRIM50 preferentially interacted with phosphatidylinositol species (Fig. 4). Although the biological role of the PRY-SPRY domain is not established, binding to phospholipid species may be a common feature among the PRY-SPRY domains shared by several TRIM family members. In fact, MG53 has an essential role in membrane repair of striated muscle cells, and muscle fiber atrophy is progressively developed in Mg53 knock-out mice (6, 7). The highest sequence homology between MG53 and TRIM50 among the family members might reasonably predict that TRIM50 also contributes to membrane repair in parietal cells. However, this prediction was not supported by our observations that TRIM50-associated vesicles did not nucleate into injury sites in GFP-TRIM50-expressing C2C12 cells (supplemental Fig. S2) and that Trim50 knock-out mice maintained normal parietal cell density in the gastric mucosa (Fig. 5). Moreover, MG53 also plays a role in steady-state trafficking of intracellular vesicles to contribute to the maintenance of certain cell-surface proteins (11). From a molecular point of view, MG53 seems to facilitate vesicular mobility to promote membrane remodeling during the cell-surface repair and quality control processes (31). As discussed below, TRIM50 and MG53 may exert similar biological roles in vesicular trafficking below the cell membrane in muscle and parietal cells, respectively.
TRIM50 is predominantly localized in tubulovesicles during the resting phase in parietal cells (Fig. 2B). Immunohistological observations detecting no dramatic subcellular relocation upon histamine stimulation (Fig. 2C) suggest that TRIM50 mainly functions at the cell interior regions. The loss of TRIM50 significantly impaired histamine-induced gastric acid secretion without affecting the expression and activity of H+/K+-ATPase (Fig. 5). The H+/K+-ATPase relocation upon stimulation was also retained in Trim50 knock-out parietal cells (supplemental Fig. S4), suggesting that TRIM50 deficiency does not abolish membrane fusion between the apical surface and tubulovesicles. Therefore, it is likely that TRIM50 deficiency compromises inter-tubulovesicular fusion that should efficiently recruit deep-seated H+/K+-ATPase for acid secretion during the stimulated phase. In Trim50 knock-out parietal cells, the organized canalicular formation was disturbed, and pleomorphic canaliculi were aberrantly formed at the cell interior regions (Fig. 6). Because TRIM50 presumably plays a role in tubulovesicular dynamics, such as vesicular nucleation and fusion prearrangement, TRIM50-deficient tubulovesicles may undergo aberrant fusing and may fail to form systematic canaliculi that efficiently contribute to histamine-induced acid secretion (Fig. 7).
In the cultured cells expressing GFP-TRIM50, PI3K inhibitors decelerated the mobility of TRIM50-containing vesicles, and also detached TRIM50 from the intracellular vesicles as a result of long term exposure (Fig. 3). Accumulated evidence recently indicated that PI3K actively takes part in vesicular trafficking and fusion of intracellular vesicles (32, 33). Considering the reported observations, our data raise the possibility that PI3K and TRIM50 may coordinately stimulate tubulovesicular dynamics for acid secretion in parietal cells. However, controversy exists regarding the specific function of PI3K in acid secretion; Lang et al. (34) reported that a PI3K inhibitor blunts acid secretion in parietal cells, and Mettler et al. (35) observed that the inhibitor adversely facilitates acid secretion. Based on the different effects between the short and long term treatments of PI3K inhibitors in our cDNA expression experiments, it may be important to carefully examine the temporally confined effects of the inhibitors on acid secretion in parietal cells.
The dynamic organization of the cytoskeleton network often plays an essential role in vesicular trafficking. In parietal cells treated with the actin inhibitor cytochalasin, the actin network is massively disrupted, and pleomorphic canaliculi are irregularly generated in response to histamine (36). The cytochalasin-induced pleomorphic canaliculi bear some morphological resemblance to the deranged canaliculi observed in the Trim50 knock-out cells (Fig. 6). Therefore, similar defects in vesicular trafficking may be proposed in cytochalasin-treated and Trim50 knock-out cells. In addition, huntingtin-interacting protein 1-related (Hip1r) is an actin- and clathrin-binding protein, abundantly expressed in parietal cells. In Hip1r knock-out parietal cells, tubulovesicles almost completely disappear, and swollen microvilli develop in response to histamine (37). The physiological role of Hip1r is largely unknown, but the swollen microvilli may arise secondarily from defective tubulovesicular dynamics in both Trim50- and Hip1r knock-out parietal cells. Therefore, the ultrastructural defects reported in cytochalasin-treated and Hip1r knock-out parietal cells reasonably predict that actin-mediated tubulovesicular trafficking is impaired in Trim50 knock-out parietal cells, although the actin inhibitor latrunculin had no effect on the mobility of TRIM50-containing vesicles in our artificial cDNA expression system (Fig. 3).
In addition to the irregular canaliculi and swollen microvilli, Trim50 knock-out parietal cells formed excess multilamellar membrane complexes (Fig. 6). Such membrane complexes are usually generated during the recovering phase after acid secretion in wild-type parietal cells and likely represent the pinocytotic processes of apical and canalicular membranes for physiological membrane retrieval (24). In Trim50 knock-out parietal cells, tubulovesicles form aberrant canaliculi, which may be promptly recovered as multilamellar membrane complexes immediately after their formation. However, these membrane complexes are probably subjected to effective lysosomal degradation, because Trim50 knock-out cells exhibited normal intracellular features during the resting phase (Fig. 6).
In addition to its role in vesicular trafficking, MG53 likely contributes to proteolysis of specific targets by catalyzing ubiquitination in the RBCC motif. A recent study proposed that molecules functioning in the insulin-like growth factor signaling may be candidates for MG53 substrates (38). Therefore, it may also be important to characterize the putative ubiquitin E3 ligase activity of TRIM50. We designed in vitro ubiquitination assay using recombinant MBP-TRIM50 proteins, in which Lys-rich MBP could be a ubiquitin acceptor as a pseudo-substrate when TRIM50 would exert E3 ligase activity (supplemental Fig. S5). In this assay, MBP-TRIM50 protein catalyzed the auto-ubiquitination in the presence of ATP, E1, E2, and ubiquitin, suggesting that TRIM50 functions as a novel E3 ligase localized in tubulovesicles. It has been recently established that ubiquitination plays an indispensable role not only for proteasome-mediated degradation but also for intracellular vesicular dynamics, including endosomal and lysosomal sorting. Epsin is a ubiquitin-binding endocytic adaptor that is concentrated at clathrin-coated pits and coordinates the acquisition of bilayer curvature with coat recruitment and cargo selection (39). Of the epsin subtypes, epsin 3 is highly enriched at endocytic sites of apical canaliculi, and it contributes to endocytotic and membrane recycling processes in parietal cells (40). The experimental data on TRIM50 and epsin 3 may suggest that parietal cell-specific ubiquitin systems modulate membrane remodeling processes during gastric acid secretion. However, proposed TRIM50 substrates and epsin-targeted proteins, both of which must be ubiquitinated near the tubulovesicular/canalicular membrane, are unknown at the present. It is important to unveil the ubiquitin systems to understand the molecular basis of parietal cell-specific functions, and Trim50 knock-out mice will provide a useful model animal in future studies.
Supplementary Material
Acknowledgments
We thank Yuki Mizobe and Masako Nishida for technical assistance.
This work was supported, in whole or in part, by the National Institutes of Health. This work was also supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology, the Japan Society for the Promotion of Science, the Ministry of Health and Welfare, the Takeda Science Foundation, the UBE Foundation, and the Japan Foundation for Applied Enzymology.

This article contains supplemental Figs. S1–S5 and Movie 1.
- MG53
- mitsugumin 53
- MBP
- maltose-binding protein
- GTA
- glutaraldehyde.
REFERENCES
- 1. McNab F. W., Rajsbaum R., Stoye J. P., O'Garra A. (2011) Tripartite motif proteins and innate immune regulation. Curr. Opin. Immunol. 23, 46–56 [DOI] [PubMed] [Google Scholar]
- 2. Park E. Y., Kwon O. B., Jeong B. C., Yi J. S., Lee C. S., Ko Y. G., Song H. K. (2010) Crystal structure of PRY-SPRY domain of human TRIM72. Proteins 78, 790–795 [DOI] [PubMed] [Google Scholar]
- 3. Li D., Roberts R. (2001) WD-repeat proteins. Structure characteristics, biological function, and their involvement in human diseases. Cell. Mol. Life Sci. 58, 2085–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DiNitto J. P., Lambright D. G. (2006) Membrane and juxtamembrane targeting by PH and PTB domains. Biochim. Biophys. Acta 1761, 850–867 [DOI] [PubMed] [Google Scholar]
- 5. Meroni G., Diez-Roux G. (2005) TRIM/RBCC, a novel class of “single protein RING finger” E3 ubiquitin ligases. BioEssays 27, 1147–1157 [DOI] [PubMed] [Google Scholar]
- 6. Cai C., Masumiya H., Weisleder N., Matsuda N., Nishi M., Hwang M., Ko J. K., Lin P., Thornton A., Zhao X., Pan Z., Komazaki S., Brotto M., Takeshima H., Ma J. (2009) MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 11, 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang X., Xie W., Zhang Y., Lin P., Han L., Han P., Wang Y., Chen Z., Ji G., Zheng M., Weisleder N., Xiao R. P., Takeshima H., Ma J., Cheng H. (2010) Cardioprotection of ischemia/reperfusion injury by cholesterol-dependent MG53-mediated membrane repair. Circ. Res. 107, 76–83 [DOI] [PubMed] [Google Scholar]
- 8. Cai C., Weisleder N., Ko J. K., Komazaki S., Sunada Y., Nishi M., Takeshima H., Ma J. (2009) Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 284, 15894–15902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu H., Lin P., De G., Choi K. H., Takeshima H., Weisleder N., Ma J. (2011) Polymerase transcriptase release factor (PTRF) anchors MG53 protein to cell injury site for initiation of membrane repair. J. Biol. Chem. 286, 12820–12824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lin P., Zhu H., Cai C., Wang X., Cao C., Xiao R., Pan Z., Weisleder N., Takeshima H., Ma J. (2012) Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J. 26, 1875–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Masumiya H., Asaumi Y., Nishi M., Minamisawa S., Adachi-Akahane S., Yoshida M., Kangawa K., Ito K., Kagaya Y., Yanagisawa T., Yamazaki T., Ma J., Takeshima H. (2009) Mitsugumin 53-mediated maintenance of K+ currents in cardiac myocytes. Channels 3, 6–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Micale L., Fusco C., Augello B., Napolitano L. M., Dermitzakis E. T., Meroni G., Merla G., Reymond A. (2008) Williams-Beuren syndrome TRIM50 encodes an E3 ubiquitin ligase. Eur. J. Hum. Genet. 16, 1038–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takeshima H., Komazaki S., Nishi M., Iino M., Kangawa K. (2000) Junctophilins. A novel family of junctional membrane complex proteins. Mol. Cell 6, 11–22 [DOI] [PubMed] [Google Scholar]
- 14. Matsuda N., Kitami T., Suzuki T., Mizuno Y., Hattori N., Tanaka K. (2006) Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro. J. Biol. Chem. 281, 3204–3209 [DOI] [PubMed] [Google Scholar]
- 15. Takeshima H., Shimuta M., Komazaki S., Ohmi K., Nishi M., Iino M., Miyata A., Kangawa K. (1998) Mitsugumin29, a novel synaptophysin family member from the triad junction in skeletal muscle. Biochem. J. 331, 317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chew C. S. (1990) cAMP technologies, functional correlates in gastric parietal cells. Methods Enzymol. 191, 640–661 [DOI] [PubMed] [Google Scholar]
- 17. Yamazaki D., Tabara Y., Kita S., Hanada H., Komazaki S., Naitou D., Mishima A., Nishi M., Yamamura H., Yamamoto S., Kakizawa S., Miyachi H., Yamamoto S., Miyata T., Kawano Y., Kamide K., Ogihara T., Hata A., Umemura S., Soma M., Takahashi N., Imaizumi Y., Miki T., Iwamoto T., Takeshima H. (2011) TRIC-A channels in vascular smooth muscle contribute to blood pressure maintenance. Cell Metab. 14, 231–241 [DOI] [PubMed] [Google Scholar]
- 18. Fujii T., Takahashi Y., Ikari A., Morii M., Tabuchi Y., Tsukada K., Takeguchi N., Sakai H. (2009) Functional association between K+-Cl− cotransporter-4 and H+,K+-ATPase in the apical canalicular membrane of gastric parietal cells. J. Biol. Chem. 284, 619–629 [DOI] [PubMed] [Google Scholar]
- 19. Jaqaman K., Loerke D., Mettlen M., Kuwata H., Grinstein S., Schmid S. L., Danuser G. (2008) Robust single-particle tracking in live cell time-lapse sequences. Nat. Methods 5, 695–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jaqaman K., Kuwata H., Touret N., Collins R., Trimble W. S., Danuser G., Grinstein S. (2011) Cytoskeletal control of CD36 diffusion promotes its receptor and signaling function. Cell 146, 593–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takeshima H., Iino M., Takekura H., Nishi M., Kuno J., Minowa O., Takano H., Noda T. (1994) Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature 369, 556–559 [DOI] [PubMed] [Google Scholar]
- 22. Schultheis P. J., Clarke L. L., Meneton P., Harline M., Boivin G. P., Stemmermann G., Duffy J. J., Doetschman T., Miller M. L., Shull G. E. (1998) Targeted disruption of the murine Na+/H+ exchanger isoform 2 gene causes reduced viability of gastric parietal cells and loss of net acid secretion. J. Clin. Invest. 101, 1243–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sawaguchi A., Aoyama F., Ide S., Suganuma T. (2005) The cryofixation of isolated rat gastric mucosa provides new insights into the functional transformation of gastric parietal cells. An in vitro experimental model study. Arch. Histol. Cytol. 68, 151–160 [DOI] [PubMed] [Google Scholar]
- 24. Sawaguchi A., Aoyama F., Ohashi M., Ide S., Suganuma T. (2006) Ultrastructural transformation of gastric parietal cells reverting from the active to the resting state of acid secretion revealed in isolated rat gastric mucosa model processed by high pressure freezing. J. Electron Microsc. 55, 97–105 [DOI] [PubMed] [Google Scholar]
- 25. De Mey J., Moeremans M., Geuens G., Nuydens R., De Brabander M. (1981) High resolution light and electron microscopic localization of tubulin with the IGS (immuno-gold staining) method. Cell Biol. Int. Rep. 5, 889–899 [DOI] [PubMed] [Google Scholar]
- 26. Kaufhold M. A., Krabbenhöft A., Song P., Engelhardt R., Riederer B., Fährmann M., Klöcker N., Beil W., Manns M., Hagen S. J., Seidler U. (2008) Localization, trafficking, and significance for acid secretion of parietal cell Kir4.1 and KCNQ1 K+ channels. Gastroenterology 134, 1058–1069 [DOI] [PubMed] [Google Scholar]
- 27. Roth M. G. (2004) Phosphoinositides in constitutive membrane traffic. Physiol. Rev. 84, 699–730 [DOI] [PubMed] [Google Scholar]
- 28. Forte T. M., Machen T. E., Forte J. G. (1977) Ultrastructural changes in oxyntic cells associated with secretory function. A membrane-recycling hypothesis. Gastroenterology 73, 941–955 [PubMed] [Google Scholar]
- 29. Forte J. G., Zhu L. (2010) Apical recycling of the gastric parietal cell H,K-ATPase. Annu. Rev. Physiol. 72, 273–296 [DOI] [PubMed] [Google Scholar]
- 30. Sawaguchi A., McDonald K. L., Forte J. G. (2004) High pressure freezing of isolated gastric glands provides new insight into the fine structure and subcellular localization of H+/K+-ATPase in gastric parietal cells. J. Histochem. Cytochem. 52, 77–86 [DOI] [PubMed] [Google Scholar]
- 31. Weisleder N., Takeshima H., Ma J. (2009) Mitsugumin 53 (MG53) facilitates vesicle trafficking in striated muscle to contribute to cell membrane repair. Commun. Integr. Biol. 2, 225–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Corvera S., Czech M. P. (1998) Direct targets of phosphoinositide 3-kinase products in membrane traffic and signal transduction. Trends Cell Biol. 8, 442–446 [DOI] [PubMed] [Google Scholar]
- 33. Ishiki M., Klip A. (2005) Minireview. Recent developments in the regulation of glucose transporter-4 traffic. New signals, locations, and partners. Endocrinology 146, 5071–5078 [DOI] [PubMed] [Google Scholar]
- 34. Lang P. A., Schniepp R., Kirchhoff P., Socrates T., Sidani S. M., Geibel J. P. (2007) PI 3-kinase-dependent stimulation of gastric acid secretion by dexamethasone. Cell. Physiol. Biochem. 20, 527–534 [DOI] [PubMed] [Google Scholar]
- 35. Mettler S. E., Ghayouri S., Christensen G. P., Forte J. G. (2007) Modulatory role of phosphoinositide 3-kinase in gastric acid secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G532–G543 [DOI] [PubMed] [Google Scholar]
- 36. Black J. A., Forte T. M., Forte J. G. (1982) The effects of microfilament disrupting agents on HCl secretion and ultrastructure of piglet gastric oxyntic cells. Gastroenterology 83, 595–604 [PubMed] [Google Scholar]
- 37. Jain R. N., Al-Menhali A. A., Keeley T. M., Ren J., El-Zaatari M., Chen X., Merchant J. L., Ross T. S., Chew C. S., Samuelson L. C. (2008) Hip1r is expressed in gastric parietal cells and is required for tubulovesicle formation and cell survival in mice. J. Clin. Invest. 118, 2459–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee C. S., Yi J. S., Jung S. Y., Kim B. W., Lee N. R., Choo H. J., Jang S. Y., Han J., Chi S. G., Park M., Lee J. H., Ko Y. G. (2010) TRIM72 negatively regulates myogenesis via targeting insulin receptor substrate-1. Cell Death Differ. 17, 1254–1265 [DOI] [PubMed] [Google Scholar]
- 39. Itoh T., De Camilli P. (2006) BAR, F-BAR (EFC), and ENTH/ANTH domains in the regulation of membrane-cytosol interfaces and membrane curvature. Biochim. Biophys. Acta 1761, 897–912 [DOI] [PubMed] [Google Scholar]
- 40. Ko G., Paradise S., Chen H., Graham M., Vecchi M., Bianchi F., Cremona O., Di Fiore P. P., De Camilli P. (2010) Selective high level expression of epsin 3 in gastric parietal cells, where it is localized at endocytic sites of apical canaliculi. Proc. Natl. Acad. Sci. U.S.A. 107, 21511–21516 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.