

Background: Profilin1 is required for actin cytoskeletal modulation.
Results: Mice lacking Profilin1 in mesenchymal progenitor cells exhibit cleft sternum and delay in endochondral bone formation. Profilin1 inactivation in skeletal cells reduces motile function of skeletal cells.
Conclusion: Profilin1 regulates skeletal development and facilitates skeletal cell migration.
Significance: Our findings provide new insights into skeletal development based on cytoskeletal function.
Keywords: Bone, Bone Morphogenetic Protein (BMP), Cartilage, Cartilage Biology, Cytoskeleton, Development
Abstract
Bone development is a dynamic process that requires cell motility and morphological adaptation under the control of actin cytoskeleton. This actin cytoskeleton system is regulated by critical modulators including actin-binding proteins. Among them, profilin1 (Pfn1) is a key player to control actin fiber structure, and it is involved in a number of cellular activities such as migration. During the early phase of body development, skeletal stem cells and osteoblastic progenitor cells migrate to form initial rudiments for future skeletons. During this migration, these cells extend their process based on actin cytoskeletal rearrangement to locate themselves in an appropriate location within microenvironment. However, the role of Pfn1 in regulation of mesenchymal progenitor cells (MPCs) during skeletal development is incompletely understood. Here we examined the role of Pfn1 in skeletal development using a genetic ablation of Pfn1 in MPCs by using Prx1-Cre recombinase. We found that Pfn1 deficiency in MPCs caused complete cleft sternum. Notably, Pfn1-deficient mice exhibited an absence of trabecular bone in the marrow space of appendicular long bone. This phenotype is location-specific, as Pfn1 deficiency did not largely affect osteoblasts in cortical bone. Pfn1 deficiency also suppressed longitudinal growth of long bone. In vitro, Pfn1 deficiency induced retardation of osteoblastic cell migration. These observations revealed that Pfn1 is a critical molecule for the skeletal development, and this could be at least in part associated with the retardation of cell migration
Introduction
Skeletal development is a critical base for body plan and is essential for making bones for appropriate locomotive functions. Endochondral bone formation takes place during the skeletal development in vertebrates and plays a major role in the longitudinal growth of long bones. At the beginning of endochondral bone formation, mesenchymal progenitor cells (MPCs) condense and differentiate into chondrocytes to form cartilaginous rudiments. Subsequently, perichondrium differentiates, and osteoblasts produce bone collar. When the initial vascular invasion into the cartilage rudiments occurs, the primary ossification center is formed in developing bone. At this point, osteoblast precursors in the perichondrium migrate into developing marrow to form trabecular bone (1–4).
During such skeletal development, progenitor cells would form primordia of future bone. In this process these cells associate together by moving from their original sites to the location of each bone. When the cells become condensed within the primordia, the shape of these cells shifts from fibroblastic to cuboidal morphology. Then the cells begin to secrete extracellular matrix. The skeletal cells contact not only extracellular environment but also their neighboring cells. These physical features require cell movement, which is under the control of cytoskeletal actin dynamics (5).
Profilin1 (Pfn1)4 is an actin monomer-binding protein and has been implicated in many cellular activities including cell migration (6). Pfn1 general knock-out mice are lethal at the two-cell stage (7) and, therefore, are not suitable for functional studies of Pfn1 during skeletal development. Cartilage-specific Pfn1 conditional knock-out mice that were produced based on the use of type II collagen-Cre system reveal reduction in chondrocyte division, whereas development of skeletal elements that requires movement of progenitor cell is grossly normal in such mice (8). Thus, the role of Pfn1 in skeletal morphogenesis during developmental process of bone remains incompletely understood.
To obtain an insight into the role of Pfn1 in the early morphogenesis during skeletal development, we hypothesized that progenitor cells before the stage of cartilage production may require Pfn1 function. Therefore, we conditionally deleted Pfn1 in skeletal stem cells in the limb buds using Prx1-Cre system. Our results demonstrate that Pfn1 is a critical molecule for the sternum development and trabecular bone formation and that these defects caused by Pfn1 inactivation could be at least in part associated with the retardation of cell migration during endochondral bone formation.
EXPERIMENTAL PROCEDURES
Animals
Pfn1-floxed (Pfn1fl/fl) mice were generated previously (8). To obtain a conditional mutant mouse line lacking Pfn1 in mesenchymal cells of limb-bud origin, Pfn1fl/fl mice were crossed with Prx1-Cre transgenic mice (9). Genotypes of the progenies were examined by genomic PCR using the primer sets designed for Pfn1fl/fl (flox) allele, Pfn1-null (Δ) allele, and Prx1-Cre transgene (Cre). The experiments were approved by the animal welfare committee of Tokyo Medical and Dental University.
5-Bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) Staining
Whole bodies of newborn (P0) mice and embryonic day 11.5 (E11.5) to E18.5 mouse embryos were used for X-gal staining and skeletal preparation. For X-gal staining to detect LacZ activity, whole bodies of newborn mice and the embryos were skinned and eviscerated as much as possible. These specimens were fixed in 0.2% glutaraldehyde and in 1% paraformaldehyde for 1 h at room temperature. Whole mount LacZ activities of the mouse embryos and the new bone mice were visualized by incubating the prepared specimens with X-gal in PBS buffer containing K4Fe(CN)6 and K3Fe(CN)6 at 4 °C overnight (10).
Skeletal Preparation
For skeletal preparation, skinned and eviscerated mice and embryos were fixed by 100% ethanol. Skeletons were stained with Alcian blue and alizarin red for cartilaginous and mineralized regions, respectively, according to standard protocol (11).
Micro-CT Analyses
For micro-CT analysis, whole body or femora of newborn mice were fixed in 4% paraformaldehyde at 4 °C overnight and then stored in 70% ethanol. These samples were subjected to three-dimensional micro-CT analyses using MX-90 three-dimensional equipment (Medixtech, Tokyo) and Tri/3D-Bon software (Ratoc System Engineering). The x-ray source was set at 90 kV and at 90 μA, respectively.
Cell Culture
Osteoblastic MC3T3-E1 cells were cultured in the α-minimum Eagle's medium supplemented with 10% FBS at 37 °C under 5% CO2. Primary cells from E15.5 humerus/femora obtained by enzymatic digestion were cultured according to the protocol previously described (12). Briefly, the cells dissociated in the digestion medium (DMEM supplemented with 2% FBS, 0.3% collagenase, and 0.1% trypsin) for 2 h were plated at 1 × 104 cells/cm2 in the osteogenic medium (DMEM supplemented with 10% FBS, 5 mm β-glycerophosphate, and 100 μg/ml ascorbic acid).
Histological Analysis
Limbs, long bones, and thoraces dissected from newborn mice and embryos were fixed by 4% paraformaldehyde at 4 °C overnight. Fixed specimens were paraffin-embedded and sectioned at a 5-μm thickness. When necessary, bones were decalcified for 3 days before the paraffin embedding. Hematoxylin and eosin, tartrate-resistant acidic phosphatase, von Kossa, and Alcian blue staining were performed according to standard protocols. Alkaline phosphatase (ALP) activity was detected using BM Purple staining solution (Roche Diagnostics) according to the manufacturer's protocols.
Quantitative RT-PCR Analysis
Total RNA was isolated from limbs, humerus, and cultured cells using TRIzol regent (Invitrogen). First-strand cDNA synthesis was performed using a high capacity cDNA reverse transcription kit (Invitrogen Japan). Quantitative RT-PCR analysis was performed using ABI FAST SYBR Green Master Mix (Invitrogen) and the thermal-cycler/detection system “Step One” (Invitrogen).
siRNA Transfection
MC3T3-E1 osteoblast-like cells were plated on coverslips at the density of 2 × 103 cells/cm2. 24 h later the cells were transfected with 10 nm control or Pfn1 siRNAs (Invitrogen, #4390843 and #s71525) using RNAiMAX reagent (Invitrogen). The transfected cells were cultured at standard conditions for 48 h. To analyze the filamentous actin structure, the cells were fixed by 4% paraformaldehyde for 10 min at room temperature and permeabilized by 0.1% Triton X-1000. The cells were blocked with 1% BSA in PBS for 20 min followed by incubation with fluorescently labeled phalloidin (Invitrogen, #A12379) for 20 min at room temperature. Coverslips were treated with a mounting medium containing DAPI (Invitrogen, #P36931), and microscopic analysis was performed using fluorescent microscope (Olympus).
Proliferative activities of the MC3T3-E1 cells transfected with siRNAs were analyzed by MTT (3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) assay. MTT cell proliferation analyses were performed according to the standard method (13).
Migration Assay
Scratch cell migration assay was performed as described previously (14). Briefly, the siRNA-transfected MC3T3-E1 cells were trypsinized at 48 h after transfection and re-plated on 6 wells at 1 × 105 cells/cm2 density. The cells were incubated for 6 h and were scraped using a 200-μl pipette tip. Images were obtained at 0, 24, and 48 h after scratching.
Statistical Analysis
Values were expressed as the means ± S.E. Statistical significance of the differences of means was tested by two-way analysis of variance and by Student's t test.
RESULTS
Profilin1 Deficiency in Mesenchymal Progenitor Cells Induces Sternum Defect
To study the role of Pfn1 in early skeletal development, we planned to cross Pfn1-floxed mice (Pfn1fl/fl) (8) with Prx1-Cre transgenic mice (9). First, regional expression pattern of the Cre transgene activities was examined by crossing the Prx1-Cre transgenic mice with transgenic mice carrying LacZ cDNA in the locus of ROSA26 (15–17). Our data on X-gal staining for the double transgenic mice (Prx1-Cre+/−;ROSA26LacZ/+, hereafter Prx1-Cre;ROSA26R or Prx1·KO) indicated that the Cre transgene-mediated LacZ activity was positive in the limbs and limb-girdle mesenchyme at E11.5 (Fig. 1, A and B). This pattern of LacZ signals was also seen in neonatal (P0) mice (Fig. 1C) in higher magnification. The LacZ signals were observed in chondrocytes, osteoblasts, and periosteal cells in the long bones (Fig. 1, C and D). Cre-mediated recombination was further confirmed based on genomic PCR using DNA of the newborn humerus (Fig. 1E) and quantitative RT-PCR using the RNA prepared from embryonic limbs and sternum (Fig. 1F).
FIGURE 1.

Pfn1 is inactivated in limb and limb-girdle mesenchyme of cKO mice. X-gal staining of Prx1-Cre;ROSA26R mice at E11.5 (A and B) and P0 (C and D) is shown. LacZ activity was visualized in whole mount bodies of E11.5 (A) and P0 mouse (C). Histological sections of these specimens after dissecting forelimb (B) and phalangeal bone (D) were photographed under the microscope. LacZ activity was positive in mesenchyme of forelimbs and hind limbs at E11.5 (B) and was positive in the skeletal cells including chondrocytes and osteoblasts in phalange at P0 (D). Genotyping PCR was performed using genomic DNA isolated from control (ctrl) and Prx1·KO humerus (E); flox: amplified products corresponding to Pfn1fl/fl allele, Δ: Pfn1-null allele, Cre: Prx1-Cre transgene. Quantitative RT-PCR analysis of E16.5 limb and E18.5 sternum was performed for Pfn1 expression (F). Pfn1 expression was reduced by ∼70% in Prx1·KO limb and sternum (n = 3). PC, proliferative chondrocytes; HC, hypertrophic chondrocytes; OB, osteoblasts; Pe, perichondrium.
Then, we mated the Pfn1fl/fl mice with Prx1-Cre transgenic mice. Genotypes of the progenies at P0 indicated that mice were born at normal Mendelian ratio. The frequency of the mice carrying both Prx1-Cre transgene and homozygous Pfn1-floxed allele (Prx1-Cre;Pfn1fl/fl) was about 25% (from here on, we referred to these mice as conditional KO (cKO)). Strikingly, these newborn mice exhibited complete cleft sternum (Fig. 2, B and D) at high penetration ratio (16/17; 94%) as cKO phenotype and died shortly after birth. Although there was one exceptional cKO mouse escaping from complete cleft sternum, it still exhibited incomplete cleft sternum, and the partial fusion was only at xiphoid process (data not shown). These cKO mice also exhibited shortening and curving of the sternum (Fig. 2, A–D). Because no such phenotype was observed in control Pfn1fl/fl mice (0/22; hereafter, we designated these mice as control) (Fig. 2, A and C), we surmised that the phenotype was caused by Pfn1 deficiency in the sternum primordium in these mice. Positive X-gal staining was observed in the sternum in Prx1-Cre;ROSA26 mice at E11.5 (data not shown).
FIGURE 2.

Sternal abnormalities in cKO mice. Skeletal preparations of control and cKO mice at P0 (A–D), and embryos (G–L) were photographed. Arrowheads indicate sternal bars (G–J). High magnification pictures of P0 specimens indicate abnormality of cKO sternum compared with control (C and D; n = 5–6). Axial sections of P0 thorax were stained by Alcian blue and eosin (E and F; n = 3–4). st, sternum; rb, rib.
To examine whether the impairment of lung expansion may exist as a possible cause of perinatal death, we tested the floating capacity of the newborn lungs by putting them into the saline. Lungs dissected from control newborn mice floated in the saline (3/3; 100%). In contrast, cKO lung never floated (0/3; 0%). These results indicated that the lungs of cKO mouse failed to expand by air respiration after birth due to the cleft sternum.
Sternum develops from primordial bilateral sternal bars. Sternal bars move ventrally at first and then move medially to fuse at midline during E13.5–16.5 (18–20). To gain insights into the cause of impairment of sternal fusion in cKO mice, we examined morphological changes during the developmental process of the cKO sternum (E13.5 to E16.5 and P0) by using skeletal preparation. Morphological differences between the cKO and control mice were observed at E13.5 (Fig. 2, G and H). In cKO mice, the sternal bars were widely separated, whereas those in control mice were partially fused. At E14.5, the sternal bars in control mice were fused in the midline (Fig. 2I, arrowheads). In contrast, sternal bars in cKO mice were still separated from each other (Fig. 2J, arrowheads). Similar defects were observed in E16.5 and P0 mice (Fig. 2, L and B, respectively).
Histological sections of newborn sternum also indicated the defect of sternum bar fusion in cKO mice (Fig. 2, E and F). Differentiation of the chondrocytes did not seem to be severely affected as calcification of sternal segments was observed in skeletal preparation (Fig. 2, A–D) and in three-dimensional micro-CT analysis of the cKO and control mice (Fig. 3).
FIGURE 3.

Three-dimensional micro-CT analysis of P0 mouse. Lateral views of the control (A) and cKO (B) mice are shown. Note that calcified sternums were visible both in cKO and control mice, but cKO mouse sternum is hypoplastic and deformed (arrows).
Pfn1 Deficiency Causes Abnormal Endochondral Bone Formation
The analysis of sternum indicated apparent impairment of skeletal development in cKO mice. Therefore, we also analyzed long bones in limbs. To examine the effects of Pfn1 deficiency on the development of long bones, histological sections of humerus at E14.5, E15.5, and E16.5 for control and cKO background were examined (Fig. 4). Although the thickness of resting and proliferating chondrocyte layers did not seem to be affected, chondrocytes were sparse in cKO humerus compared with the control mice (Fig. 4, A–D). There was a certain heterogeneity in the density of the staining of Alcian blue in the bone, but these vary depending on animals, and no consistent trend of cKO effects in terms of the pattern of Alcian blue staining was observed in E14.5 humerus.
FIGURE 4.

Endochondral bone formation is affected in embryonic cKO humerus. Histological analysis of E14.5, E15.5, and E16.5 humerus of control and cKO mice is shown. Sections were von Kossa-stained with Alcian blue, and representative microscopic images are shown (A–F). Trabecular bone formation was absent in cKO mice throughout the period (B, D, and F). Sections were stained with ALP activity (G–L). ALP-positive cells (arrowheads) were detected in perichondrium but not in the bone-marrow space in cKO (N). n = 3. CB, cortical bone; TB, trabecular bone. Scale bar, 200 μm.
ALP-positive cells were observed in bone collar in E14.5 in both control and cKO (Fig. 4, G and H), and these cells continued to be present in E15.5 (Fig. 4, I and J) and E16.5 (Fig. 4, K and L). On E16.5, ALP-positive cells were on the periosteal regions in both control and cKO. However, ALP-positive cells were not observed in the marrow space in cKO bones even at E16.5 (Fig. 4L and inset), whereas the ALP-positive cells in control bones were observed not only in perichondrium but also in the marrow space by that time (Fig. 4K and inset).
An interesting feature in the limb bone of cKO mice at birth (P0) compared with control was the absence of trabecular bone (Fig. 5, A and B; trabecular bone (TB)). In contrast to the absence of trabecular bone, cortical bone was observed in the diaphysis in cKO mice (Fig. 5, A–D; cortical bone (CB), arrows). Primary and secondary trabecular bone structures as well as marrow were observed in the control long bone at birth (Fig. 5C; trabecular bone (TB); bone marrow (BM)). In contrast, the diaphyseal regions of cKO long bone at birth were mostly filled with hypertrophic cartilage (Fig. 5D; HC, hypertrophic cartilage), and trabecular bone was totally missing in the diaphysis of cKO mutants (Fig. 5D). Blood cells were localized in the marrow and adjacent to hypertrophic cartilage in control type (Fig. 5C, asterisk). However, the long bones in cKO mice contained less blood cells (Fig. 5D, asterisk). As hypertrophic cartilage and bone are resorbed by the multinucleated cells that are positive for tartrate-resistant acid phosphate, so called chondroclasts and osteoclasts, respectively, we stained the sections of long bones of control and cKO mutants at birth for tartrate-resistant acid phosphate activity. In control mice, tartrate-resistant acid phosphate-positive multinucleated cells were observed in the trabecular bone (Fig. 5E; osteoclast, arrowheads and inset). In cKO mutants, tartrate-resistant acid phosphate-positive multinucleated cells were observed along with the margin of hypertrophic cartilage that occupied the bone marrow space of long bone at birth (Fig. 5F; chondroclast, arrowheads and inset). These data indicate that at the time of birth, the appearance of multinucleated tartrate-resistant acid phosphate-positive osteoclasts or chondroclasts were still observed in the absence of Pfn1. In contrast to the retardation of trabecular bone development, cortical bones were observed both in cKO mutants and control at birth as shown by micro-CT examination (Fig. 5, G and H; arrowheads).
FIGURE 5.

Endochondral bone formation is affected in cKO humerus at P0. Histological sections of control (A and C) and cKO (B and D) humerus at P0 were stained with hematoxylin and eosin (HE). Low magnification images showed that trabecular bone formation is impaired in cKO humerus (B). The asterisk indicates the existence of cortical bone (B). High magnification images of P0 humerus is highlighted by a rectangle in A and B. Trabecular bone (TB) was indicated in the control (C), whereas it was absent in cKO (D). n = 5–6. Similar sections were stained with tartrate-resistant acid phosphate (TRAP) activity (E and F). Tartrate-resistant acid phosphate-positive cells were present both in control (E) and cKO (F). Multinucleated osteoclasts were present along with the margin of hypertrophic cartilage of the cKO bones (arrowheads). n = 3–5. Micro-CT images of control (G) and cKO (H) femur at P0 indicate the absence of trabecular bone structure in cKO bones (H), although cortical bone (arrowhead) and calcified hypertrophic chondrocyte layer was detected in cKO long bones (H). n = 3. PC, proliferative chondrocytes; HC, hypertrophic chondrocytes; CB, cortical bone; BM, bone marrow.
Pfn1 Is Required for Cell Adhesion and Migration in Skeletal Cells
To investigate whether the loss of Pfn1 affects the function of skeletal progenitor cells, cells obtained from long bones of E15.5 embryos were cultured. We found that the cells obtained from cKO embryos tended to fail to adhere and spread on the culture dish compared with the cells from control mice (Fig. 6, A–C). The effects of siRNA knockdown of Pfn1 expression were also examined in MC3T3-E1 cells. Efficient reduction was confirmed by quantitative RT-PCR and Western blotting (Fig. 7, A and B). Under this condition, actin cytoskeleton was stained using fluorescein-labeled phalloidin. The stress fibers consisting of actin filaments were reduced in Pfn1 siRNA-transfected (siPfn1) MC3T3-E1 cells, whereas control siRNA-transfected (siCtrl) cells maintained actin cytoskeleton and stress fibers (Figs. 7, C and D). These results suggest that Pfn1 activity is required for modulation of actin cytoskeleton in MC3T3-E1 cells.
FIGURE 6.
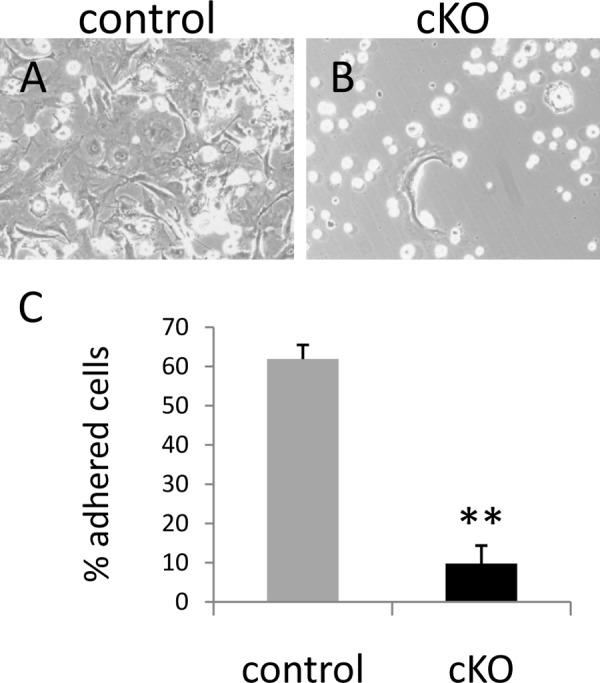
Pfn1 is required for cell spreading and/or cell adhesion in primary skeletal cells. Primary cultures of the skeletal cells obtained from control (A) and cKO (B) humerus and femur at E15.5 were photographed under the phase contrast microscopy. Note that cKO primary cells were incapable of adhering and/or spreading (B). Quantification of the ratios of adhered cells per plated cells indicates impaired adhesion/spreading of cKO primary cells was about 10% (C). Bars indicate the means ± S.E. Counted cells were above 25. **, p < 0.01 versus control cultures. The experiment was repeated twice with similar results.
FIGURE 7.

Pfn1 inactivation results in reduced actin cytoskeleton in MC3T3-E1 cells. MC3T3-E1 osteoblastic cells were transfected with siRNA for control sequence and for Pfn1 (siCtrl and siPfn1, respectively). The efficiency of the Pfn1 knockdown was confirmed by quantitative RT-PCR, showing ∼90% reduction (A; n = 3). Western blot analyses also confirmed Pfn1 knockdown (n = 4) (B). Reduced stress fibers in siPfn1 transfected cells were visualized by green-fluorescent labeled phalloidin (C and D). The transfected cells were labeled with the Block-iT Alexa Fluor Red Fluorescent Oligo (catalogue #14750-100, Invitrogen). The experiment was repeated three times with similar results.
We further examined the effects of Pfn-1 deficiency on cell motility. We found that Pfn1 knockdown reduced motile function of the cells based on scratch cell migration assay within 48 h after knockdown (Fig. 8, A and B). The reduced healing for the scratch wound in siPfn1 cells (Fig. 9) may be due to reduced motility and/or by reduced cell number. However, MTT cell proliferation assay revealed that a statistically significant difference was observed only after 72 h in culture with respect to cell proliferation in control and siPfn1-treated cells (Fig. 9).
FIGURE 8.
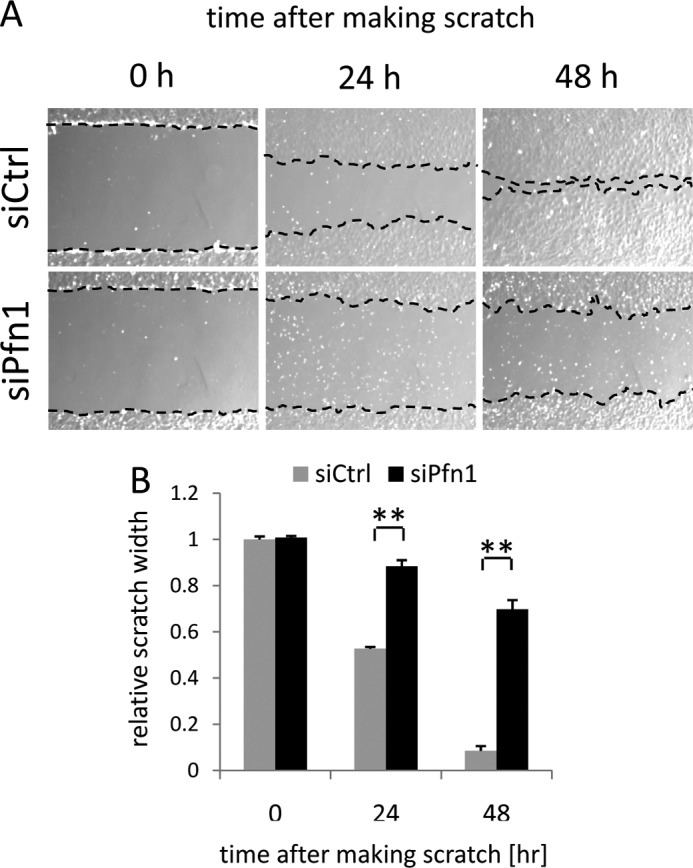
Pfn1 inactivation inhibits cell migration in MC3T3-E1 cells. Scratch cell migration assay was conducted with siCtrl and siPfn1-transfected cells. Phase contrast microscopic images were captured at 0, 24, and 48 h after scratching (A), and the width of closing scratch wound was measured for quantitation (B). Error bars indicate S.E. ; **, p < 0.01 (by Student's t test as a post hoc test for two-way analysis of variance). The experiment was repeated three times with similar results.
FIGURE 9.
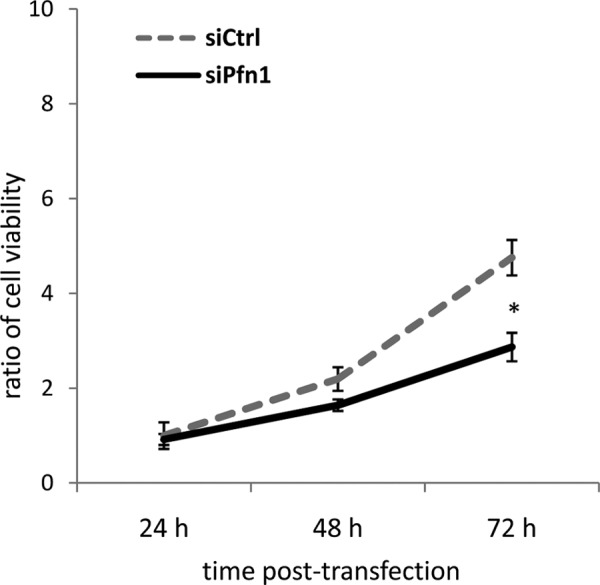
Pfn1 inactivation inhibits cell proliferation in MC3T3-E1 cells. Proliferative cell activity of the siCtrl and siPfn1 transfected MC3T3-E1 cells was determined by MTT cell proliferation assay. Error bars indicate S.E. (n = 6). *, p < 0.05 (by Student's t test as a post hoc test for two-way analysis of variance). The experiment was repeated twice with similar results.
DISCUSSION
This study demonstrates that Pfn1 in mesenchymal progenitor cells is required for sternum morphogenesis during development in vivo. Moreover, Pfn1 in mesenchymal progenitor cells is necessary for the development of trabecular bone during endochondral bone formation in newborn mice. Last, we found that Pfn1 regulates the actin cytoskeletal formation of osteoblastic cells and promotes the cell adhesion and cell migration in vitro. Earlier studies have demonstrated that Pfn1 is required for cellular functions associated with rapid actin cytoskeletal rearrangements (7, 8). However, the physiological role of Pfn1 in bone development in vivo has been largely unknown. This study provides the first in vivo evidence for the role of Pfn1 in sternum and trabecular bone development in vivo.
Genetic Requirement for Pfn1 in Sternum Development
Cleft sternum is a subject of surgical repair in human, whereas its genetics or etiology has not been known to date (21, 22). The sternum develops between E13.5 and E16.5 of mice embryos. The bilateral sternum bands known as sternal bars migrate ventrally and meet to fuse at midline (14, 15). Thus, the cleft sternum results from the defects of the rudiments in differentiation, growth, migration, and/or fusion (2). We observed that the connection to ribs and the segmentation of sternum in cKO mice were more or less similar to that of control. However, major sternum phenotype was splitting (failure in fusion or cleft), and the penetration rate was very high.
Our results showed that the cartilage and cortical bone developed in cKO sternum, suggesting that lack of Pfn1 did not prevent the differentiation of cKO chondrocytes and osteoblasts per se. Moreover, the bilateral sternal rudiments of cKO mice at E14.5-E16.5 were located far from midline, but the size of the rudiments was not largely different from control. Almost all cKO mice exhibited complete cleft sternum (16/17 mice), but some mutant mice exhibited incomplete cleft sternum (1/17 mice); its xiphoid process of the sternum was fused in the midline, suggesting a possibility that the cleft sternum is caused by the defects in the migration of the rudiments but is not caused by the defects in the fusion of the rudiments.
Although sternal malformations are sometimes observed in other types of mutant mouse models (18, 19), complete malformations of sternal defects are relatively rare. Interestingly, mice lacking filaminA, a widely expressed actin-binding protein, also have midline fusion defects manifesting as sternum abnormalities (23). Together with our data, actin cytoskeletal remodeling is suggested to be necessary for midline skeletal development. Whether our observation is related to human cleft sternum is still to be elucidated.
The Source of the Trabecular Osteoblasts
Although osteoblast differentiation was not affected in cKO mice, as ALP-positive cells were present in perichondrium at an appropriate stage of embryonic development, we did not observed any ALP-positive cells within the marrow space in cKO long bones at birth, whereas control mice showed ALP-positive cells in marrow space. The sources of the trabecular osteoblasts that form trabecular bone during endochondral bone formation have been controversial. Several hypotheses have been suggested that these osteoblasts may derive from perichondrium, chondrocytes on progenitors through the blood stream and/or from pericytes (2, 24–28). Consistent with previous findings (8), we found that Pfn1 did not inhibit the differentiation of chondrocytes per se and that cKO mice did not exhibit apparent indication for apoptotic defect in cartilage because hypertrophic cartilage were calcified normally and were replaced by bone-marrow space. However, trabecular bone had not been formed at least at birth. This indicates a delay over 4 days compared with control where trabecular bone formation was observed on E16.5. It appears that hypertrophic chondrocytes are unlikely to be the source of trabecular osteoblasts based on our observation.
The blood supply to the developing bone in cKO mice was observed on E16.5. If the progenitors are through the blood stream and/or if pericytes are the source of trabecular osteoblasts, it appears to be difficult to explain our observation. Thus, a possible alternative interpretation would be that osteoblast precursors in perichondrium may be the source. Several studies have suggested that osteoblasts and blood vessels are interdependent on each other for the invasion into developing bone. The inhibition of osteoblastogenesis in perichondrium by inactivation of Runx2, Osx, β-catenin, or Ihh showed the defect in vascular invasion and primary ossification (29–34). On the other hand, the inhibition of vascular invasion into developing bone also showed the defect in primary ossification. Our study demonstrated that the absence of trabecular osteoblasts in bone-marrow space was associated with the vascular in-growth into developing bone.
It is known that vasculogenesis is also an important factor that contributes to skeletogenesis and morphogenesis during development. Given that mesenchymal progenitor cells can also differentiate into endothelial cells, which also must migrate during limb development, we do not exclude the possibility that in addition to the effects on skeletal progenitor cell migration, Pfn-1 deficiency may also interfere with vasculogenesis during development and may indirectly affect skeletogenesis.
There was some delay in cortical bone formation. It might be possible that the delayed cortical bone formation observed in cKO mice is related to the slightly lower cell density in the E15.5 perichondrium of the cKO.
We also examined the expression of factors in the E15.5 bone based on quantitative RT-PCR to examine the effects of Pfn-1 deficiency on gene expression. We observed no significant differences in tartrate-resistant acid phosphate, Receptor Activation of NFκB, and VEGF expression levels (data not shown). Alph2-heparan sulfate glycoprotein, which is synthesized by hepatocytes, did not accumulate in developing bone at the stage when initial vascular invasion into developing bone occurs. Within the embryonic stage, osteocalcin was not expressed in bone. However, Alpha2-HS glycoprotein is also made in the lower hypertrophic zone of the growth plate (35). Similarly, osteocalcin and dentin matrix protein 1 as well as other proteins are made by the chondrocytes in this region (36). Some of these proteins may function in recruitment of osteoclasts.
In summary, we identify that Pfn1 is a novel molecule for sternal and trabecular bone development. The cellular motile function by actin cytoskeletal modulation would be involved in these events. Our observations provide a new insight into the understanding of sternum and trabecular development.
Footnotes
- Pfn1
- profilin1
- X-gal
- m5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside
- ALP
- alkaline phosphatase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- cKO
- conditional KO.
REFERENCES
- 1. Karsenty G., Wagner E. F. (2002) Reaching a genetic and molecular understanding of skeletal development. Dev. Cell 2, 389–406 [DOI] [PubMed] [Google Scholar]
- 2. Kronenberg H. M. (2007) The role of the perichondrium in fetal bone development. Ann. N.Y. Acad. Sci. 1116, 59–64 [DOI] [PubMed] [Google Scholar]
- 3. Karsenty G., Kronenberg H. M., Settembre C. (2009) Genetic control of bone formation. Annu. Rev. Cell Dev. Biol. 25, 629–648 [DOI] [PubMed] [Google Scholar]
- 4. Maes C., Kobayashi T., Selig M. K., Torrekens S., Roth S. I., Mackem S., Carmeliet G., Kronenberg H. M. (2010) Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev. Cell 19, 329–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Olson E. N., Nordheim A. (2010) Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 11, 353–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Witke W. (2004) The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 14, 461–469 [DOI] [PubMed] [Google Scholar]
- 7. Witke W., Sutherland J. D., Sharpe A., Arai M., Kwiatkowski D. J. (2001) Profilin I is essential for cell survival and cell division in early mouse development. Proc. Natl. Acad. Sci. U.S.A. 98, 3832–3836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Böttcher R. T., Wiesner S., Braun A., Wimmer R., Berna A., Elad N., Medalia O., Pfeifer A., Aszódi A., Costell M., Fässler R. (2009) Profilin 1 is required for abscission during late cytokinesis of chondrocytes. EMBO J. 28, 1157–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Logan M., Martin J. F., Nagy A., Lobe C., Olson E. N., Tabin C. J. (2002) Expression of Cre recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77–80 [DOI] [PubMed] [Google Scholar]
- 10. Kawaguchi J., Wilson V., Mee P. J. (2002) Visualization of whole-mount skeletal expression patterns of LacZ reporters using a tissue clearing protocol. Biotechniques 32, 66, 68–70, 72–73 [DOI] [PubMed] [Google Scholar]
- 11. McLeod M. J. (1980) Differential staining of cartilage and bone in whole mouse fetuses by Alcian blue and alizarin red S. Teratology 22, 299–301 [DOI] [PubMed] [Google Scholar]
- 12. Hill T. P., Später D., Taketo M. M., Birchmeier W., Hartmann C. (2005) Canonical Wnt/β-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 8, 727–738 [DOI] [PubMed] [Google Scholar]
- 13. Mosmann T. (1983) Rapid colorimetric assay for cellular growth and survival. Application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63 [DOI] [PubMed] [Google Scholar]
- 14. Liang C. C., Park A. Y., Guan J. L. (2007) In vitro scratch assay. A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2, 329–333 [DOI] [PubMed] [Google Scholar]
- 15. Soriano P. (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70–71 [DOI] [PubMed] [Google Scholar]
- 16. Durland J. L., Sferlazzo M., Logan M., Burke A. C. (2008) Visualizing the lateral somitic frontier in the Prx1Cre transgenic mouse. J. Anat. 212, 590–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiong J., Onal M., Jilka R. L., Weinstein R. S., Manolagas S. C., O'Brien C. A. (2011) Matrix-embedded cells control osteoclast formation. Nat. Med. 17, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. CHEN J. M. (1952) Studies on the morphogenesis of the mouse sternum. I. Normal embryonic development. J. Anat. 86, 373–386 [PMC free article] [PubMed] [Google Scholar]
- 19. CHEN J. M. (1952) Studies on the morphogenesis of the mouse sternum. II. Experiments on the origin of the sternum and its capacity for self-differentiation in vitro. J. Anat. 86, 387–401 [PMC free article] [PubMed] [Google Scholar]
- 20. Liu K. J., Arron J. R., Stankunas K., Crabtree G. R., Longaker M. T. (2007) Chemical rescue of cleft palate and midline defects in conditional GSK-3β mice. Nature 446, 79–82 [DOI] [PubMed] [Google Scholar]
- 21. Torre M., Rapuzzi G., Carlucci M., Pio L., Jasonni V. (2012) Phenotypic spectrum and management of sternal cleft: literature review and presentation of a new series. Eur. J. Cardiothorac. Surg. 41, 4–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kimura A., Inose H., Yano F., Fujita K., Ikeda T., Sato S., Iwasaki M., Jinno T., Ae K., Fukumoto S., Takeuchi Y., Itoh H., Imamura T., Kawaguchi H., Chung U. I., Martin J. F., Iseki S., Shinomiya K., Takeda S. (2010) Runx1 and Runx2 cooperate during sternal morphogenesis. Development 137, 1159–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hart A. W., Morgan J. E., Schneider J., West K., McKie L., Bhattacharya S., Jackson I. J., Cross S. H. (2006) Cardiac malformations and midline skeletal defects in mice lacking filamin A. Hum. Mol. Genet. 15, 2457–2467 [DOI] [PubMed] [Google Scholar]
- 24. Colnot C., Lu C., Hu D., Helms J. A. (2004) Distinguishing the contributions of the perichondrium, cartilage, and vascular endothelium to skeletal development. Dev. Biol. 269, 55–69 [DOI] [PubMed] [Google Scholar]
- 25. Galotto M., Campanile G., Robino G., Cancedda F. D., Bianco P., Cancedda R. (1994) Hypertrophic chondrocytes undergo further differentiation to osteoblast-like cells and participate in the initial bone formation in developing chick embryo. J. Bone Miner. Res. 9, 1239–1249 [DOI] [PubMed] [Google Scholar]
- 26. Mödder U. I., Khosla S. (2008) Skeletal stem/osteoprogenitor cells. Current concepts, alternate hypotheses, and relationship to the bone remodeling compartment. J. Cell Biochem. 103, 393–400 [DOI] [PubMed] [Google Scholar]
- 27. Roach H. I., Erenpreisa J. (1996) The phenotypic switch from chondrocytes to bone-forming cells involves asymmetric cell division and apoptosis. Connect. Tissue Res. 35, 85–91 [DOI] [PubMed] [Google Scholar]
- 28. Maes C., Carmeliet P., Moermans K., Stockmans I., Smets N., Collen D., Bouillon R., Carmeliet G. (2002) Impaired angiogenesis and endochondral bone formation in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Mech. Dev. 111, 61–73 [DOI] [PubMed] [Google Scholar]
- 29. Dy P., Wang W., Bhattaram P., Wang Q., Wang L., Ballock R. T., Lefebvre V. (2012) Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev. Cell 22, 597–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Komori T., Yagi H., Nomura S., Yamaguchi A., Sasaki K., Deguchi K., Shimizu Y., Bronson R. T., Gao Y. H., Inada M., Sato M., Okamoto R., Kitamura Y., Yoshiki S., Kishimoto T. (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89, 755–764 [DOI] [PubMed] [Google Scholar]
- 31. Otto F., Thornell A. P., Crompton T., Denzel A., Gilmour K. C., Rosewell I. R., Stamp G. W., Beddington R. S., Mundlos S., Olsen B. R., Selby P. B., Owen M. J. (1997) Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89, 765–771 [DOI] [PubMed] [Google Scholar]
- 32. Colnot C., de la Fuente L., Huang S., Hu D., Lu C., St-Jacques B., Helms J. A. (2005) Indian hedgehog synchronizes skeletal angiogenesis and perichondrial maturation with cartilage development. Development 132, 1057–1067 [DOI] [PubMed] [Google Scholar]
- 33. St-Jacques B., Hammerschmidt M., McMahon A. P. (1999) Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nakashima K., Zhou X., Kunkel G., Zhang Z., Deng J. M., Behringer R. R., de Crombrugghe B. (2002) The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108, 17–29 [DOI] [PubMed] [Google Scholar]
- 35. Yang F., Schwartz Z., Swain L. D., Lee C. C., Bowman B. H., Boyan B. D. (1991) Alpha 2-HS-glycoprotein. Expression in chondrocytes and augmentation of alkaline phosphatase and phospholipase A2 activity. Bone 12, 7–15 [DOI] [PubMed] [Google Scholar]
- 36. Aizawa T., Kon T., Einhorn T. A., Gerstenfeld L. C. (2001) Induction of apoptosis in chondrocytes by tumor necrosis factor α. J. Orthop. Res. 19, 785–796 [DOI] [PubMed] [Google Scholar]