

Background: Cyclotides are a family of plant-expressed pesticidal cyclic peptides.
Results: A broad range of cyclotides specifically interact with membranes containing phosphatidylethanolamine (PE)-phospholipids.
Conclusion: Cyclotide bioactivity correlates with an ability to target, insert into, and disrupt lipid membranes containing PE-phospholipids.
Significance: Cyclotides constitute a new lipid-binding protein family that has potential as a scaffold to target tumor cells.
Keywords: Drug Design, Fluorescence, Lipid-binding Protein, Membrane Bilayer, Phospholipid Vesicle, Cyclic Peptides, Peptide-Membrane Interactions, Anti-HIV Peptide, Membrane Permeabilization, Phosphatidylethanolamine-binding Domain
Abstract
Cyclotides are bioactive cyclic peptides isolated from plants that are characterized by a topologically complex structure and exceptional resistance to enzymatic or thermal degradation. With their sequence diversity, ultra-stable core structural motif, and range of bioactivities, cyclotides are regarded as a combinatorial peptide template with potential applications in drug design. The mode of action of cyclotides remains elusive, but all reported biological activities are consistent with a mechanism involving membrane interactions. In this study, a diverse set of cyclotides from the two major subfamilies, Möbius and bracelet, and an all-d mirror image form, were examined to determine their mode of action. Their lipid selectivity and membrane affinity were determined, as were their toxicities against a range of targets (red blood cells, bacteria, and HIV particles). Although they had different membrane-binding affinities, all of the tested cyclotides targeted membranes through binding to phospholipids containing phosphatidylethanolamine headgroups. Furthermore, the biological potency of the tested cyclotides broadly correlated with their ability to target and disrupt cell membranes. The finding that a broad range of cyclotides target a specific lipid suggests their categorization as a new lipid-binding protein family. Knowledge of their membrane specificity has the potential to assist in the design of novel drugs based on the cyclotide framework, perhaps allowing the targeting of peptide drugs to specific cell types.
Introduction
Cyclotides (1) are an intriguing family of plant-derived bioactive peptides that are characterized by a head-to-tail macrocyclic structure and three disulfide bonds arranged in a cystine knot (Fig. 1). The cystine knot occupies the peptide core and is important for stability, whereas the backbone segments between the Cys residues, designated as loops, have their side chains exposed and are likely to be involved in the bioactivity of cyclotides. The unique cyclic and knotted topology of cyclotides gives them exceptional resistance to high temperatures, chaotropic agents, and proteolytic enzymes that would digest most peptides (2).
FIGURE 1.

Cyclotide structural information. Three-dimensional structure (Protein Data Bank code 1nb1) and sequence of the prototypic cyclotide kB1 are shown. Cyclotides are characterized by a cystine knot motif formed by three disulfide bonds, shown in yellow. The six Cys residues are labeled I–VI, and the residues between adjacent Cys residues are designated as loops 1–6. The direction of the peptide chain N-C is shown with a black arrow. G1 is highlighted as the starting point of the numbering scheme.
The majority of cyclotides discovered so far have been found in plants belonging to the Violaceae (violet) or Rubiaceae (coffee) families. Individual plants express suites of many different cyclotides that vary in quantity and distribution within the plant. To date, more than 250 cyclotides have been isolated and categorized into two main subfamilies, Möbius and bracelet (3). Cyclotides belonging to the Möbius subfamily have a cis-Pro residue in loop 5 that is responsible for a conceptual twist in the peptide backbone, whereas bracelet peptides comprise a backbone ring composed solely of trans-peptide bonds (1). A selection of cyclotide sequences belonging to the Möbius and bracelet subfamilies is given in Table 1. Apart from the six conserved Cys residues, a conserved Glu in loop 1 and a conserved Asn/Asp in loop 6, cyclotides have large sequence diversity.
TABLE 1.
Sequences of selected cyclotides belonging to the Möbius and bracelet subfamilies

Two macrocyclic peptides isolated from dormant seeds of Momordica cochinchinensis and with powerful trypsin inhibitory activities, MCoTI-I and MCoTI-II (4), have been categorized as a third subfamily of cyclotides, the trypsin inhibitor subfamily (5). Although having limited sequence similarity when compared with the members of the other two subfamilies, trypsin inhibitors have the cyclic cystine knot structure, which defines them as cyclotides.
Although the natural function of cyclotides is thought to be host defense based on their pesticidal activities (6), several pharmaceutically relevant bioactivities have also been reported for members of this family, including uterotonic (7), anti-HIV (8), anti-cancer (9), and antimicrobial activities (10). Their stable structure, along with a range of different biological activities and tolerance for amino acid substitution, has inspired the use of cyclotides for drug design applications (5). In particular, their native pharmacological activities might be used in the development of uterotonic or antimicrobial agents, or the cyclotide scaffold could in principle be used as an inert framework to stabilize linear bioactive peptide epitopes (11). Indeed, the use of cyclotides as a scaffold for drug design has been explored both using synthetic chemistry (12, 13) and through biosynthesis of a genetically encoded cyclotide combinatorial library expressed inside Escherichia coli (14). The bioengineering of such libraries enables the rapid screening and the selection of new sequences with specific biological activities and high stability (15). More recently, a library displayed at the E. coli surface using a linearized cyclotide scaffold was also reported (16). This strategy provides a new alternative for high throughput screening using flow cytometry and a stable scaffold (16).
With hundreds of cyclotides identified so far, understanding their mechanism(s) of action is of much interest to assist with rational drug design efforts and to decrease potentially toxic effects associated with their native insecticidal functions. As an individual plant can express a suite of different cyclotides (17, 18), understanding the differences and similarities in the modes of action of a broad selection of cyclotides could also help unravel reasons for the biosynthetic production of a multiplicity of cyclotides in a single plant.
The bioactivities of cyclotides seem to broadly correlate with their ability to target and disrupt cell membranes, an observation based on biological assay data and biophysical studies with model membranes. Bioassays show that cyclotides belonging to both Möbius and bracelet subfamilies have cytotoxic (9) and hemolytic (19) properties. Studies with model membranes show that kalata B1 (kB1)6 (20, 21), the prototypic Möbius cyclotide, and cycloviolacin O2 (cyO2) (22), the prototypic bracelet cyclotide, can bind and disrupt lipid bilayers. In addition, the majority of cyclotides have a surface-exposed patch of hydrophobic residues that has been suggested to be required for their bioactivities (23, 24). In summary, single examples of a prototypic bracelet and a prototypic Möbius cyclotide have a common feature in their mode of action, in that cell membranes seem to be the main target.
Although they have essentially identical structures, the biological potency of different cyclotides varies (19, 23) and depends on the target organism (25). For instance, kB1 does not show antimicrobial activity, whereas cyO2 has antimicrobial activity against some Gram-negative bacteria (26). Therefore, as different cyclotides have different hydrophobic and electrostatic properties, it is important to evaluate the lipid specificity, the membrane affinity, and the membrane-binding mechanism.
In this study, the membrane-binding properties of a diverse selection of native cyclotides (see Table 1) were determined. Studies with model membranes were correlated with cyclotide bioactivities against different target organisms.
EXPERIMENTAL PROCEDURES
Peptide Extraction
The cyclotides kB1, kB2, kB5, kB6, kB7, kB8, and kB9 were isolated from the above ground parts of Oldenlandia affinis (18); cyO2 was isolated from Viola odorata leaves (17), and tricyclon A (tcA) was isolated from Viola tricolor flowers (27). Extraction, isolation, identification, and purification followed methods described previously (17, 18). The isolated cyclotides had ≥95% purity, as confirmed by analytical reverse phase-HPLC and MS. Peptide concentrations were determined before each assay by absorbance at 280 nm (20). Unless otherwise stated, all peptide solutions were prepared in 10 mm HEPES buffer, pH 7.4, containing 150 mm NaCl.
To reduce kB1, 10 mm tris(2-carboxyethyl)phosphine was added to 300 μm of kB1 and incubated at 55 °C for 1 h. To reduce cyO2, 10 mm tris(2-carboxyethyl)phosphine was added to 300 μm of cyO2 and incubated at 55 °C for 4 h. The reduced form of the peptides was confirmed by reverse phase-HPLC.
Peptide Synthesis
All-d-kB2 was synthesized using manual solid-phase peptide synthesis as described previously for all-d-kB1 (28). The purity of the peptide (≥95%) was evaluated using analytical reverse phase-HPLC, and the folding was confirmed by 1H NMR spectroscopy as before (29).
Preparation of Lipid Vesicles for Peptide/Membrane Studies
Synthetic lipids (palmitoyloleoylphosphatidylcholine (POPC), palmitoyloleoylphosphatidylglycerol (POPG), palmitoyloleoylphosphatidylethanolamine (POPE), palmitoyloleoylphosphatidylserine, dipalmitoylglycerophosphatidylcholine (DPPC), dipalmitoylglycerophosphatidylethanolamine (DPPE), and cholesterol (Chol)) and extracted sphingomyelin (SM) from porcine brain, which contains 50% C(18:0), 21% C(24:1), 2% C(16:0), 5% C(20:0), 7% C(22:0), and 5% C (24:0)) were obtained from Avanti Polar Lipids and used to prepare model membranes.
Large unilamellar vesicles (LUVs) with a diameter of 100 nm or small unilamellar vesicles (SUVs) with a diameter of 50 nm were prepared from lipid films by freeze-thaw fracturing and sizing by extrusion (30). Briefly, lipid films were prepared from lipids dissolved in chloroform. The solvent was evaporated with a N2 stream, and the lipid film was left under vacuum overnight. Buffer was added to the lipid film, and the suspension was vortexed, freeze-thawed eight times, and extruded 21 times through a polycarbonate membrane with a 100- or 50-nm pore size diameter to obtain LUVs or SUVs, respectively. LUVs were used in fluorescence studies, and SUVs were prepared for surface plasmon resonance (SPR) studies (31). Unless otherwise stated, all of the lipid systems were prepared in pH 7.4 buffer containing 10 mm HEPES and 150 mm NaCl.
Cyclotide Interaction with Lipid Membranes Followed by SPR
The membrane specificity of native cyclotides was evaluated with SPR following protocols previously detailed (21, 31). L1 sensor chips and a Biacore 3000 instrument were used. Briefly, all solutions were freshly prepared and filtered (0.22 μm pore size) before use. An SUV suspension (1 mm lipid) was injected over an L1 chip surface (2 μl/min, 2400-s contact time) reaching a stable steady state, suggesting coverage of the chip surface and a stable lipid bilayer surface. Loosely bound SUVs were removed with a short pulse of 10 mm NaOH (50 μl/min, 36 s). Cyclotide solutions were injected over the lipid surfaces (5 μl/min, 180 s), and the dissociation was monitored for 600 s. The chip surface was regenerated as described previously (31). A range of lipid mixtures was used to delineate the role of lipid composition in binding. Unless otherwise stated, the temperature was maintained at 25 °C throughout the assays.
Peptide-to-lipid molar ratio (P/L) was calculated to evaluate the binding ability of the tested cyclotides. The amount of lipid deposited at the chip surface was determined by converting the response units (RU), at the end of lipid deposition, into pg·mm−2 (1 RU = 1 pg·mm−2) (32) and into moles by considering the average mass of the lipid mixture. The amount of peptide bound to the lipid bilayer was calculated at the end of association phase (170 s after peptide injection starts) by converting the RU into pg·mm−2 and then into moles (21, 28). P/L versus injected peptide concentration was plotted and fitted with a nonlinear regression equation dose-response binding with variable slope, P/L = (P/Lmax × [peptide]H)/(KdH + [peptide]), in which P/Lmax is the maximum binding; Kd is the peptide concentration needed to achieve half-maximum binding at equilibrium, and H is the Hill slope.
Cyclotide Fluorescence Studies
All of the native cyclotides included in this study have intrinsic fluorescence due to the presence of a Tyr and/or a Trp residue in their sequence (see Table 1). The fluorescence of Trp and Tyr are sensitive to the environment; therefore, the intrinsic steady-state fluorescence properties of the different cyclotides were studied. Cyclotides containing Trp residues were excited at 280 nm, whereas cyclotides containing only Tyr as aromatic residues were excited at 275 nm.
The interaction of the native cyclotides with model membranes was followed by changes in fluorescence emission spectra or changes in the fluorescence anisotropy (33). Specifically, 25 μm peptide was titrated with stock of POPC or POPC/POPE (80:20) LUV suspensions. An LS50B fluorescence spectrophotometer (PerkinElmer Life Sciences) was used to follow fluorescence emission and excitation spectra, whereas the fluorimeter FluoroMax-4 (from Jobin Yvon) was used for measurements with polarized light. Partition curves obtained with mean anisotropy (〈r〉) were fitted as described previously (33).
Membrane Dipolar Potential Changes in the Presence of Different Cyclotides
200 μm LUVs composed with POPC or POPC/POPE (80:20) and 4 μm 4-[2-[6-(dioctylamino)-2-naphthalenyl]ethenyl]-1-(3-sulfopropyl)-pyridinium (di-8-ANEPPS) dye were prepared in HEPES buffer to evaluate membrane dipole potential as described previously (34). The fluorescence excitation spectra were scanned in the presence and absence of different cyclotides (10, 25, and 50 μm were tested).
Vesicle Aggregation
Vesicle aggregation induced by cyclotides was followed by absorbance at 436 nm as described previously (35). 25 μm of each cyclotide was added to 100 μm POPC or POPC/POPE (80:20) LUVs. The absorbance was followed for 30 min.
Membrane Integrity as Monitored by Leakage Studies
The extension of vesicle leakage induced by the cyclotides was monitored and quantified by carboxyfluorescein (CF) dequenching, as detailed previously (21). Permeabilization of POPC, POPC/Chol/SM (27:33:40 molar ratio), POPC/POPG (80:20), POPC/POPE (90:10), POPC/POPE/Chol/SM (17:10:33:40), and POPC/POPE/POPG (60:10:20) lipid mixtures were compared for the selected native cyclotides. Briefly, varying peptide concentrations (2-fold starting with 10 μm) were incubated with vesicles (5 μm total lipid concentration) during 10 min. The fluorescence intensity of each solution was measured (λexcitation = 489 nm/λemission = 515 nm) to quantify the CF release, and the percentage of leakage was calculated as before (20).
Data analysis was performed with Graphpad Prism Version 5.0d. The peptide concentration needed to achieve 50% of vesicle leakage (LC50) and the standard error of the best fit were determined fitting a nonlinear regression equation dose response binding with variable slope, in which the maximum response was constrained to 100% leakage as follows: % leakage = (100 × [peptide]H)/(LC50H + [peptide]), in which H is the Hill slope. To evaluate if the membrane leakage efficiencies induced by the tested cyclotides are statistically different, LC50 obtained by curve fitting was compared two by two with a t test for the lipid mixtures tested. Differences were considered statistically significant when p < 0.05.
Membrane Integrity Monitored by Confocal Microscopy with Giant Unilamellar Vesicles
Membrane leakage induced by the cyclotides kB1, kB2, kB7, and kB8 and cyO2 was evaluated with giant unilamellar vesicles (GUVs) and confocal microscopy (36, 37). GUVs composed of POPC or POPC/POPE encapsulating fluorescent dyes were prepared by electroformation as described previously (36). The dyes tetramethylrhodamine (TR)-dextran (∼3 kDa, 2 μm), Alexa Fluor647-dextran (∼10 kDa, 5 μm), and Oregon Green 488-dextran (∼70 kDa, 2 μm) were encapsulated in the vesicles to evaluate the size of a potential membrane pore, whereas 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine (1% mol/mol) was used to follow membrane integrity (37). The excitation wavelengths were 543 nm (HeNe laser; for 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine or TR-dextran), 488 nm (Ar laser; for Oregon Green 488-dextran), and 633 nm (HeNe laser, for Alexa Fluor647-dextran). Micrographs were recorded after peptide addition, and the fluorescence of the dyes was followed versus time.
Hemolytic Activity
Hemolysis induced by the native cyclotides kB1, kB2, kB6, kB7, kB8, and kB9, cyO2, and tcA was compared following protocols previously described (25). The all-d-kB2 and the reduced forms of kB1 and cyO2 were also compared. All peptide solutions were assayed in triplicate with 2-fold dilution concentrations, starting with 50 μm, and prepared in HEPES buffer, pH 7.4, containing 150 mm NaCl.
Anti-HIV Studies
The virucidal activities of kB1, kB2, and tcA were compared using a HLA-DR viral capture method (21, 38). Two HIV-1 particles strains were compared (NL4.3, clade B, CXCR4 co-receptor using strain, and clade A, CCR5 co-receptor using strain). The experiment was repeated three times.
Antimicrobial Assays
Bacterial growth inhibition induced by the various cyclotides was evaluated to examine their antimicrobial activity. Growth of Gram-negative E. coli (ATCC25922) and Gram-positive Staphylococcus aureus (ATCC25923) was tested using the broth dilution method (39); bacterial suspensions (2 × 105 cfu/ml E. coli or 2 × 106 cfu/ml S. aureus) were incubated with the peptides (series of 2-fold peptide concentrations, starting with 100 μm) and compared with controls following protocols previously described (40). The assay was done with Luria Broth (LB). The peptide concentration to inhibit 50% of bacterial growth was defined as the minimal inhibitory concentration (MIC50). The experiment was repeated three times.
RESULTS
Cyclotide-Membrane Binding Affinity Is Dependent on Lipid Composition
Cell membranes had been implicated as the main target of the few cyclotides that have been tested so far for membrane binding. In this study, a broad selection of cyclotides belonging to the Möbius and bracelet subfamilies was studied. The cyclotides were selected based on their diversity and abundance in the selected plants, and they included the prototypic Möbius cyclotide kB1, the prototypic bracelet, cyO2, and tcA, a cyclotide with a notably low hemolytic activity (27). Interactions of cyclotides with model membranes were evaluated using SPR to gain insights into their mechanism of action. Various lipid mixtures that represent different membrane phase properties, charges, or phospholipid headgroups were compared for each cyclotide.
PC-containing phospholipids are the most common form of phospholipid in eukaryotic membranes (41). Thus, membranes composed of POPC, which forms zwitterionic bilayers with liquid disordered phase properties at 25 °C, along with model membranes of increased complexity, including POPC/POPG (80:20 molar ratio) and POPC/Chol/SM (27:33:40), were included. At 25 °C, POPC/Chol/SM (27:33:40) membranes are in a homogeneous liquid-ordered phase (42). Phospholipids with a PG headgroup are negatively charged and were included to evaluate the effects of charge. The effects of PE-phospholipids, which are also common in eukaryotic membranes (41) and previously shown to be required for membrane targeting by kB1 (21), were also evaluated via the inclusion of POPC/POPE (90:10) and POPC/POPE (80:20) mixtures. These mixtures are in the liquid-disordered phase under the conditions studied (43). The presence of the negatively charged POPG and the effect of Chol/SM in membranes containing PE was further studied with POPC/POPE/POPG (60:20:20) and POPC/POPE/Chol/SM (17:10:33:40) mixtures. In the latter mixture, the overall proportions of Chol and SM were kept the same as in the POPC/Chol/SM (27:33:40) mixture. Based on previous studies with POPE, Chol, and SM (44), we assumed that the addition of 10% POPE to the POPC/Chol/SM mixture would not change the overall fluidity of the lipid bilayer, i.e. this lipid system will be in a homogeneous liquid-ordered phase. This assumption is based on the fluorescence anisotropies of DPH and TMA-DPH being comparable for vesicles composed of POPC/Chol/SM and POPE/palmitoyloleoylphosphatidylserine/Chol/SM at the same molar fractions (44).
All of the tested cyclotides had identical specificity, i.e. they showed a preference for PE-containing phospholipids over other lipids, but they did differ in their affinity for the membranes (Fig. 2 and Table 2). For example, all of the tested cyclotides showed weak binding to POPC membranes but had markedly increased affinity with increasing amounts of PE (Fig. 2, A–C). Plotting the P/L as a function of the amount of POPE in liquid disordered membranes (i.e. POPC, POPC/POPE (90:10), and POPC/POPE (80:20), Fig. 2D) showed a linear correlation with unit slope, suggesting 1:1 stoichiometry between the peptide and PE.
FIGURE 2.

Membrane binding of cyclotides requires PE-phospholipids. The membrane binding activity of selected cyclotides was monitored by SPR. The peptide-to-lipid ratio (P/L mol/mol) at the end of peptide injection was calculated to normalize the response to the total amount of lipid deposited on the chip and to the amount of peptide that binds to the lipid bilayer (1 RU = 1 pg·mm−2 of peptide or lipid). P/L was plotted versus peptide concentration, and the curves were fitted with a nonlinear regression equation, dose-response binding with Hill slope (see Table 2 for fitting parameters). Binding of cyclotides with membranes comprising different lipid mixtures is as follows. A, POPC; B, POPC/POPE (90:10 molar ratio); C, POPC/POPE (80:20); E, POPC/POPG (80:20); F, POPC/POPE/POPG (60:20:20); G, POPC/Chol/SM (27:33:40), and H, POPC/POPE/Chol/SM (17:10:33:40). D shows the correlation between the P/L and the fraction of PE in the membrane when POPC, POPC/POPE (90:10) and POPC/POPE (80:20) membranes are compared. Cyclotides belonging to the Möbius subfamily and cyO2 are represented at a fixed peptide concentration close to the determined Kd value, and the slope of the plots are as follows: 1.01 ± 0.22 for kB1, 0.97 ± 0.03 for kB2, 1.08 ± 0.02 for kB6, 1.09 ± 0.14 for kB7, and 1.11 ± 0.00 for cyO2, suggesting that these cyclotides have a 1:1 stoichiometry binding to PE-phospholipids when in a fluid membrane.
TABLE 2.
Affinity of cyclotides for lipid membranes
The parameters were obtained by fitting the binding response (P/L) versus the peptide concentration injected over the lipid bilayer (see Fig. 2). P/L max is the maximum binding response, and Kd is the peptide concentration needed to achieve half-maximum binding at equilibrium. Although peptide-lipid binding events are more complex than Langmuir binding, one-site specific binding with Hill slope equation was used to fit the parameters and to have a quantitative comparison of the SPR response.
| Peptide | POPC/POPE (90:10) |
POPC/POPE (80:20) |
POPC/POPE/POPG (60:20:20) |
POPC/POPE/Chol/SM (17:10:33:40) |
POPC/Chol/SM (27:33:40)a |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| P/L max | Kd | P/L max | Kd | P/L max | Kd | P/L max | Kd | P/L max | Kd(app) | |
| μm | μm | μm | μm | μm | ||||||
| kB1 | 0.18 ± 0.01 | 73 ± 9 | 0.24 ± 0.01 | 24 ± 0.5 | 0.29 ± 0.01 | 18 ± 1 | 0.11 ± 0.003 | 29 ± 2 | 0.11 | 2893 |
| kB2 | 0.18 ± 0.03 | 19 ± 4 | 0.22 ± 0.01 | 16 ± 1 | 0.31 ± 0.01 | 15 ± 1 | 0.12 ± 0.004 | 13 ± 1 | 0.12 | 1586 |
| kB6 | 0.23 ± 0.02 | 25 ± 3 | 0.31 ± 0.02 | 22 ± 2 | 0.35 ± 0.01 | 10 ± 1 | 0.13 ± 0.01 | 16 ± 2 | 0.13 | 594 |
| kB7 | 0.20 ± 0.02 | 66 ± 15 | 0.34 ± 0.01 | 47 ± 2 | 0.37 ± 0.01 | 15 ± 1 | 0.13 ± 0.01 | 57 ± 6 | 0.13 | 1846 |
| kB5 | 0.12 ± 0.003 | 6.6 ± 4 | 0.34b | 143 ± 42 | 0.28b | 18 ± 4 | 0.05 ± 0.003 | 11 ± 2 | 0.05 | 129 |
| kB8 | 0.17b | 19 ± 3 | 0.34b | 190 ± 39 | 0.28b | 10 ± 2 | 0.14b | 112± 4 | 0.14 | 983 |
| kB9 | 0.17b | 41± 7 | 0.10 ± 0.003 | 13 ± 1 | 0.08 ± 0.005 | 32 ± 5 | 0.08 | 143 | ||
| cyO2 | 0.17 ± 0.003 | 8.2 ± 0.4 | 0.34 ± 0.01 | 8.6 ± 0.6 | 0.28 ± 0.004 | 3.8 ± 0.5 | 0.14 ± 0.002 | 24 ± 1 | 0.14 | 1283 |
| tcA | 0.15 ± 0.01 | 41 ± 6 | 0.34b | 178 ± 12 | 0.24 ± 0.01 | 35 ± 4 | 0.08 ± 0.017 | 88 ± 48 | 0.08 | 700 |
a The binding of cyclotides to membranes lacking PE-phospholipids is weak; therefore, the curves do not reach a plateau in the concentration range tested, the P/Lmax cannot be defined, and nor can Kd value. Nevertheless to illustrate the weak affinity to membranes lacking PE, binding to POPC/Chol/SM is compared with POPC/POPE/Chol/SM assuming the same P/Lmax for both lipid mixtures (which possess identical fluidity properties at 25 °C). As this fit implies extrapolations, the error associated with the Kd is high, and therefore the fitted values are referred to as apparent Kd (Kd (app)).
b The curve did not reach a plateau in the concentration range tested; therefore, the P/Lmax was constrained to be equal to the value obtained with cyO2 which is also a bracelet cyclotide.
Inspection of Fig. 2, C and F, and Table 2 shows that the presence of the negatively charged phospholipid POPG in POPE-containing membranes (POPC/POPG/POPE (60:20:20)) had varying effects on the affinity of cyclotides for the membrane relative to the globally neutral POPC/POPE (80:20); POPG by itself did not improve the binding for the membrane (see POPC versus POPC/POPG, Fig. 2, A and E).
The importance of PE-phospholipids to binding was also evident when POPC/Chol/SM and POPC/POPE/Chol/SM (Fig. 2, G and H) mixtures were compared for all of the cyclotides tested, revealing that cyclotides also bind to liquid ordered membranes containing PE-phospholipids. Overall, the studies on the binding of the cyclotides to the tested lipid systems (Fig. 2 and Table 2) revealed that although the binding affinity is dependent on the specific peptide and lipid composition, all cyclotides have increased affinity for membranes containing PE lipids.
The all-d-enantiomer of kB2 was also included to evaluate possible effects of chirality. d-kB2 bound to membranes containing PE-phospholipids, but with a lower affinity than the mirror image native form (Fig. 3A), consistent with previous studies on d-kB1 (28). Disulfide bond-reduced kB1 and cyO2 did not bind to membranes, even when PE-phospholipids were present (Fig. 3B), suggesting that the three-dimensional conformation of cyclotides is important for the membrane binding.
FIGURE 3.

Effects of chirality and reduction of cyclotides on membrane binding. A, 40 μm l-kB2 and its d-isomer were injected for 180 s over POPC/POPE (80:20) lipid surfaces deposited on an L1 chip. Dissociation was monitored for 600 s after the injection was stopped. Sensorgrams obtained show that for the same amount of lipid on the surface, d-kB2 has a lower amount of peptide bound to the membrane than its mirror image isomer. Dissociation rates are identical. B, reduced kB1 and cyO2 were tested for their membrane-binding ability to POPC/POPE (80:20) model membranes. P/L was plotted versus peptide concentration. Reduced peptides have weak binding relative to their oxidized native forms.
Trp/Tyr fluorescence studies (anisotropy and emission spectral shift) confirmed the importance of PE-phospholipids for the membrane binding of cyclotides and provided further evidence that they insert into membranes. For example, cyO2, which has an exposed Trp residue, has an increase in its Trp fluorescence anisotropy with a concomitant emission spectrum blue shift upon titration with POPC/POPE vesicles but not to POPC vesicles (Fig. 4, A and B). The partition coefficient was calculated and confirmed that cyO2 has high affinity for POPC/POPE membranes but only weak affinity for POPC (Fig. 4B). This observation showed that the Trp residue inserts into the POPC/POPE lipid environment. Although the Tyr fluorescence is not as sensitive as Trp fluorescence to the environment, identical conclusions were obtained when the fluorescence anisotropy of the Tyr residues in kB8 was followed (Fig. 4B).
FIGURE 4.

Fluorescence properties of cyclotides in lipids. A, normalized fluorescence emission spectra of 25 μm cyO2 in buffer or in the presence of 2 mm POPC or 2 mm POPC/POPE (80:20) LUV suspensions. B, mean fluorescence anisotropy of cyO2 or kB8 upon titration with POPC or POPC/POPE (80:20). The Trp residue in cyO2 or the Tyr residues in kB8 become inserted in a more rigid environment upon titration with lipid. The partition coefficient, Kp, was calculated using equations previously described (33) and show that both peptides have large partition to POPC/POPE membranes and a weak insertion into POPC membranes. The Kp in POPC cannot be accurately determined as the maximum 〈r〉 is not well defined in the lipid concentration range tested.
The intrinsic fluorescence signal of the Möbius cyclotides did not change upon titration with lipid vesicles. It appears that this is because the fluorescence quantum yield of the Trp residue is weak and blue-shifted due to intramolecular quenching and therefore is not sensitive to changes surrounding the indole group in this subfamily of cyclotides.
Cyclotides Bind Weakly to Membranes Having a Solid-ordered Phase
Although PE-phospholipids are clearly important for membrane targeting by cyclotides, membrane fluidity appears to modulate the binding affinity. At a similar POPE content, the membrane-binding affinity differs depending on whether the membrane displays a liquid-ordered phase (POPC/POPE/Chol/SM (17:10:33:40)) or a fluid-disordered phase (POPC/POPE (90:10)) (see Table 2), suggesting that the environment surrounding the PE-phospholipids modulates the insertion and the affinity. The influence of ordered domains was further studied with 5% POPE and kB1. Fig. 5A shows that 5% POPE is enough to improve the binding of kB1 relative to POPC membranes, with further improvement if ordered domains (see POPC/POPE/Chol and POPC/POPE/Chol/SM in Fig. 5A) are also present.
FIGURE 5.

Importance of the PE-phospholipid environment for cyclotide binding. A, SPR sensorgrams obtained at 25 °C upon injection of 30 μm of kB1 for 180 s over POPC, POPC/POPE (90:5), POPC/POPE/Chol (62:5:33), and POPC/POPE/Chol/SM (22:5:33:40) at 25 °C. Dissociation was followed for 600 s. The response units were converted to P/L to normalize the response to the total amount of lipid deposited in the chip and to the number of peptide moles that bind to the lipid bilayer. Comparison of POPC versus POPC/POPE (95:5) reveals that 5% of POPE is enough to improve the binding of kB1 to the membrane. Including 33% of Chol further improves the affinity, and the dissociation rate is decreased. The introduction of 40% SM decreases the dissociation rate of kB1 from the lipid membrane. B, SPR sensorgrams obtained at 25 °C upon injection of 100 μm of kB1 or (C) cyO2 for 180 s over DPPC, DPPC/DPPE (80:20), or POPC/POPE (80:20) deposited on L1 chip. Dissociation was monitored for 600 s after the injection stopped. The sensorgrams show only weak binding to the gel-like DPPC bilayer. Although an increase in affinity is seen with DPPC/DPPE membranes, the amount of peptide binding to this mixture in the gel phase is much lower than for POPC/POPE in the fluid-disordered phase. D, binding of kB1 to DPPC/POPE (80:20) at 25 °C and 40 °C. P/L is plotted versus peptide concentration. An increase in binding with temperature confirms that kB1 prefers more fluid membranes over solid membranes.
To evaluate if insertion into the membrane hydrophobic core is important for cyclotide-membrane affinity, we evaluated the binding of cyclotides to membranes in the solid-ordered (gel) phase, where the lipid packing is very high. DPPC and DPPC/DPPE (80:20), which form bilayers in the solid-ordered phase at 25 °C, were studied (Fig. 5, B and C). Stronger binding to DPPC/DPPE than DPPC is evident, but when compared with a membrane in the liquid-disordered phase containing the same amount of PE-phospholipid (POPC/POPE (80:20)), a significant decrease in the binding is evident, as seen for kB1 and cyO2.
To further confirm the importance of membrane fluidity, binding of kB1 to DPPC/POPE was compared at 25 °C (solid-ordered phase) and 40 °C (liquid-disordered phase) (45). Increasing the temperature makes DPPC/POPE less rigid, and an increase in the binding of kB1 occurs (Fig. 5D), confirming that the insertion in the membrane, rather than just adsorption at the phospholipid headgroup region, is important for cyclotide-membrane affinity.
Cyclotides Induce Vesicle Aggregation and Alterations in the Membrane Dipolar Potential
The membrane surface is mainly stabilized by repulsive electrostatic forces, van der Waals attractions, and a repulsive hydration shell between approaching membrane surfaces (46, 47). When these forces are destabilized by peptides or proteins, distinct membrane surfaces can be brought into close proximity, resulting in vesicle aggregation (46, 47). Thus, in principle, vesicle aggregation efficiency can be followed to evaluate if cyclotides affect membrane surface stability. Here, their ability to aggregate pure POPC or POPC/POPE vesicle dispersions was examined (Fig. 6, A and B). None of the cyclotides tested aggregate POPC vesicles, but most of them aggregate POPC/POPE vesicles. This result further supports the ability of cyclotides to bind PE-containing membranes and reveals that cyclotides are able to disturb membrane surface properties. Nevertheless, the aggregation induction is dependent on the peptide. For the same bulk peptide concentration added to POPC/POPE lipid vesicles, the extent of aggregation follows the trend cyO2 > kB1 ∼ kB7 ∼ tcA > kB5 > kB2 ∼ kB6. No aggregation was induced by kB8. This trend is not the same as observed for the membrane affinity (Table 2), suggesting that effects exerted at the membrane surface are not only dependent on the amount of peptide bound to the membrane.
FIGURE 6.

Alterations in membrane properties caused by cyclotides. A and B, vesicle aggregation was followed via absorbance variation at 436 nm; 25 μm peptide was added to lipid vesicles at a lipid concentration of 100 μm. An increase in the absorbance indicates vesicle aggregation. A, absorbance variation of POPC or POPC/POPE (80:20) vesicle suspensions upon addition of kB1. B, comparison of absorbance variation of POPC/POPE (80:20) vesicle suspensions upon addition of tested cyclotides. C and D, changes in membrane dipolar potential can be sensed by the spectral shift obtained in the fluorescence excitation spectra of di-8-ANEPPS dye. The excitation spectrum of di-8-ANEPPS-labeled vesicles (200 μm final lipid concentration containing 4 μm of di-8-ANEPPS dye) was subtracted from the spectrum obtained in the presence of peptide and normalized to integrated areas to reflect the spectral shift. C, di-8-ANEPPS fluorescence difference obtained with POPC/POPE (80:20) or with POPC in the presence of kB1. A blue shift is evident for POPC/POPE (80:20), revealing changes in the membrane dipolar potential, which are more pronounced with increased peptide concentration (i.e. 10 versus 25 versus 50 μm). No change in the dipolar potential of POPC was sensed even with 50 μm kB1. D, fluorescence difference obtained with POPC/POPE (80:20) in the presence of 50 μm tested native cyclotide. A blue shift indicates an increase in the dipolar potential, and a red shift indicates a decrease.
The membrane dipolar potential changes upon insertion of peptides into the hydrophobic membrane core, and this change can be detected with the dye di-8-ANEPPS (34). The change in dipolar potential was dependent on the peptide and its concentration (Fig. 6, C and D). Interestingly, cyclotides belonging to the Möbius subfamily showed a weaker shift in the differential spectra than bracelet cyclotides. All cyclotides induced a decrease in the dipolar potential, except kB1, which increased the dipolar potential. In agreement with membrane binding studies, none of the peptides tested induced changes in the dipolar potential of membranes lacking-POPE (see kB1 example in Fig. 6C).
Permeabilization of Model Membranes Induced by Cyclotides
The ability of peptides to induce permeabilization of phospholipids vesicles has been correlated with their ability to disrupt the cell membranes of target organisms (48, 49). Therefore, the ability of cyclotides to disrupt model membranes was evaluated with lipid systems of varying composition.
In the concentration range tested, none of the cyclotides were able to permeabilize POPE-deficient vesicles (Fig. 7). By contrast, when POPE is present, all of the cyclotides were able to permeabilize the membrane; the leakage efficiency was dependent on the lipid mixture and on the peptide, as quantified by the LC50 (Table 3). Overall it is clear that cyO2 has the highest permeabilization efficiency and tcA the lowest.
FIGURE 7.

Membrane leakage induced by cyclotides. The percent leakage is plotted as a function of peptide concentration for a set of six cyclotides. The percent leakage was calculated by measuring the fluorescence (λexcitation = 489 nm/λemission = 515 nm) of CF release after a 10-min incubation of the cyclotide with lipid vesicles (5 μm lipid concentration). The leakage from vesicles made with six lipid mixtures is compared. The leakage curves were fitted with a nonlinear regression equation, dose-response binding with variable slope using Graphpad Prism Version 5.0d. LC50 values are given in Table 3.
TABLE 3.
LC50 (μm) for cyclotide induction of vesicle leakage
The peptide concentration needed to achieve 50% leakage (LC50) and the standard error were determined by fitting a non-linear regression equation (dose-response binding with variable slope), in which the maximum response was constrained to 100% leakage; see data and fitting in Fig. 7.
| Peptide | POPC/POPE (90:10) | POPC/POPE/POPG (70:10:20) | POPC/POPE/Chol/SM (17:10:33:40 ) |
|---|---|---|---|
| kB1 | 1.06 ± 0.04 | 5.77 ± 0.70 | 0.65 ± 0.04 |
| kB2 | 1.22 ± 0.12 | 2.94 ± 0.29 | 0.24 ± 0.01 |
| kB6 | 3.02 ± 0.83 | 22.45 ± 1.30 | 0.83 ± 0.17 |
| kB7 | 2.79 ± 0.01 | 6.86 ± 0.29 | 2.61 ± 0.08 |
| kB5 | 2.07 ± 0.16 | 2.47 ± 0.12 | 0.46 ± 0.01 |
| kB8 | 0.90 ± 0.08 | 0.89 ± 0.05 | 0.77 ± 0.04 |
| cyO2 | 0.24 ± 0.01 | 0.28 ± 0.01 | 0.21 ± 0.01 |
| tcA | 7.70 ± 0.38 | 18.50 ± 2.81 | 4.98 ± 0.21 |
From a comparison of the ability of each cyclotide to permeabilize vesicles containing an identical percentage (10%) of POPE, but different fluidity or charge, it is clear that kB1, kB2, kB6, kB5, and tcA permeabilize liquid-ordered membranes (POPC/POPE/Chol/SM) with higher efficiency than liquid-disordered membranes (POPC/POPE), whereas kB7, kB8, and cyO2 do not distinguish between them. The effect of negatively charged phospholipids differs for different peptides. In particular, kB1, kB2, kB6, kB7, and tcA have a lesser ability to permeabilize POPC/POPE/POPG than POPC/POPE, whereas the leakage inducing efficiency of cyO2, kB8, and kB5 does not differ significantly in the presence of negatively charged phospholipids.
To provide insights into the mode of permeabilization, vesicle leakage was further evaluated using GUVs and confocal microscopy. The leakage efficiency exerted by selected native cyclotides (kB1, kB2, kB7, kB8 and cyO2) on POPC and POPC/POPE (80:20) GUVs was compared. Negligible dye leakage was observed in vesicles lacking PE. In contrast, dye leakage was evident in PE-rich membranes after incubation with any of the cyclotides tested (Fig. 8 and supplemental Movies 1–6). The different dyes, possessing distinct sizes, leak from the vesicles concomitantly (see kB1 and cyO2 examples in Fig. 8, A–D, and supplemental Movies 1–6), suggesting that if a pore is formed it is large enough to let the largest dye escape. Interestingly, after addition of cyO2, the green dye seems to co-localize within the membrane (see Fig. 8, C and D). This might be due to electrostatic attractions between the cationic cyO2, bound to the membrane, and the anionic dye. To evaluate the membrane integrity, the dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine was used to label the lipid bilayer (Fig. 8, E and F, and supplemental Movie 6). After incubation with kB1, the vesicle is permeabilized prior to disruption of the membrane. This effect is in agreement with electrophysiological measurements with kB1, in which conductive pores were detected prior to membrane disruption (20).
FIGURE 8.

Leakage from POPC/POPE GUVs induced by cyclotides and monitored using confocal microscopy. A–D, GUVs loaded with TR-dextran (red, 3 kDa), Alexa Fluor647-dextran (blue, 10 kDa), and Oregon Green 488-dextran (green, 70 kDa) were followed after addition of 30 μm kB1 (A and B) or 30 μm cyO2 (C and D). A and C, micrographs show the merge fluorescence after peptide addition (at 2700, 3170, or 3600 s for kB1, and 2000, 2500, or 3000 s for cyO2). B and D, average fluorescence intensity was determined in the time course in each micrograph using Zeiss LSM 510 META software. The fluorescence of the three dyes was normalized, which shows that the three dyes follow the same trend, suggesting that they escape the vesicles at the same time. E, GUVs, in which the membrane was labeled with the membrane dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine (red) and loaded with Alexa Fluor647-dextran (blue, 10 kDa) and Oregon Green 488-dextran (green, 70 kDa), are shown after addition of 30 μm kB1 at 2700, 3170, or 3600 s. F, average fluorescence in each micrograph was plotted as a function of time and shows that the dyes escape from the vesicles without the GUV membranes bursting.
Hemolytic Properties of Cyclotides
The hemolytic activity of the cyclotides was compared at 37 and 4 °C (Fig. 9A). At 37 °C, tcA and kB8 have weak hemolytic activities, and cyO2 is the most potent cyclotide. Interestingly, the all-d-kB2 is hemolytic but less so than the native kB2. This result is in agreement with previous studies with d-kB1 and its native form (28). At 4 °C, no hemolytic activity was found for any of the cyclotides in the concentration range tested. Melittin was included as a positive control, and although a 2-fold decrease in its hemolytic efficiency was observed at 4 °C (HC50 = 1.2 μm at 37 °C versus 2.2 μm at 4 °C), melittin is still hemolytic at this temperature.
FIGURE 9.

Bioactivities of cyclotides. A, hemolytic dose-response curves were obtained using 2-fold dilutions starting with 50 μm peptide. Peptide solutions were tested in triplicate with a 0.25% (v/v) suspension of human RBCs and incubated for 1 h at 37 or at 4 °C. The peptide concentration needed to cause 50% of hemolysis (HC50) was determined by fitting a sigmoidal dose-response curve. Melittin was included as a positive control. None of the cyclotides tested induced hemolysis at 4 °C. B, activity of cyclotides against E. coli. The survival percentage is plotted as function of peptide concentration. For clarity, only kB1, kB2, and cyO2 are shown. The errors bars represent the S.E. None of the cyclotides tested showed antimicrobial activity except cyO2; the MIC50 for cyO2 as determined with a nonlinear regression analysis is 6.8 ± 0.4 μm. C, HIV virucidal activity of cyclotides. The ability of kB1, kB2, and cyO2 to inactivate HIV-1 NL4.3, or HIV-1 Clade A is shown. The errors bars represent the S.E. The concentration of cyclotide needed to cause a 50% reduction in viral infectivity (VC50), compared with untreated virus, was calculated by a nonlinear regression analysis and compared with curves previously obtained for kB1 (21).
Antimicrobial Activity of Cyclotides
Growth inhibition of E. coli (Gram-negative) and S. aureus (Gram-positive) bacteria was tested in the presence of the cyclotides. No antimicrobial activity against Gram-negative (E. coli) or Gram-positive (S. aureus) was detected for any of the cyclotides tested except cyO2 agianst E. coli, which has a MIC50 of 6.8 ± 0.4 μm in LB (Fig. 9B).
Anti-HIV Studies
It was previously reported that kB1 and a membrane-active mutant are able to target and disrupt the HIV virus-enveloped membrane as determined using a HLA-DR viral capture method (21). The membrane of HIV particles is very rich in PE, Chol, and SM and is assumed to be in liquid-ordered phase, being referred to as a raft-like membrane (50, 51). Here, we found that kB2 had a higher binding affinity and caused more leakage in model membranes rich in PE, Chol, and SM than kB1 (see Tables 2 and 3). By contrast, tcA has lower membrane affinity than kB1 for those model membranes. To evaluate if an HIV membrane-targeting mechanism can also be considered for the other cyclotides, the virucidal activities of kB2 and tcA were compared with the results previously obtained with kB1 (21) and quantified using the VC50 (Fig. 9C). kB2 has more potent virucidal activity (VC50 = 0.25 ± 0.05 μm for NL4.3 and 0.42 ± 0.11 μm for clade A) than kB1 (VC50 = 1.7 ± 0.4 μm for NL4.3 and 4.5 ± 0.3 μm for clade A), whereas tcA has the lowest activity (5.0 ± 1.6 μm for NL4.3 and 10.3 ± 1.2 μm against clade A). Together, these results are consistent with the proposed HIV cytoprotective mechanism of cyclotides being dependent on membrane-binding affinity (21), and it involves targeting the HIV-particle membrane envelope and disrupting it.
DISCUSSION
In this study, we compared a selection of cyclotides isolated from O. affinis, V. odorata, and V. tricolor with respect to their affinity for model membranes and their toxicity against mammalian RBCs, bacteria, and HIV particles. All of the cyclotides tested were specific for membranes containing PE-phospholipids over the other lipids tested and caused perturbations that eventually led to membrane disruption. Their affinity for model membranes correlated with whole-cell toxicities. As well as providing a fundamental understanding of cyclotide-membrane interactions, the results have the potential to assist in the design of pharmaceutically active cyclotides to target specific cell types and thus enhance medical applications of cyclotides.
All Tested Cyclotides Selectively Target, Insert into, and Permeabilize Membranes Containing PE-phospholipids
Fluorescence and SPR studies with model membranes revealed that cyclotides are able to bind, insert into, and disrupt model lipid membranes by a mechanism dependent on initial peptide-lipid recognition and subsequent hydrophobic interactions. Specifically, all of the cyclotides bound to and permeabilized membranes containing PE-phospholipids but showed only very weak binding to, or permeabilization of, membranes lacking PE. Membrane fluidity was found to further modulate binding affinity; cyclotides had higher affinity for membranes displaying liquid-disordered or liquid-ordered phases than for solid-ordered phases, even when the same proportion of PE was present. We propose that the enhanced affinity for fluid membranes reflects a greater ability of cyclotides to insert into these membranes compared with solid membranes.
Hemolytic Potency Correlates with Cyclotide-Membrane Binding, Insertion, and Permeabilization
Hemolytic assays at 37 °C showed that cyclotides interact with and disrupt RBCs, with the hemolytic efficiency having the following order: cyO2 > kB2 ∼ kB6 > kB5 ∼ kB1 > kB9 > kB7 > kB8 > tcA. This trend is in agreement with previous studies in which a few cyclotides were examined for other biological activities (52–54). The relative hemolytic activity correlates with the relative leakage efficiency trend in POPC/POPE/Chol/SM model membranes (Fig. 10), consistent with the ability of cyclotides to insert in the hydrophobic core and disrupt the RBC membrane, which includes phosphatidylcholine phospholipids, Chol, SM, and a small amount of PE-phospholipids in the outer leaflet (41, 55).
FIGURE 10.

Correlation between hemolytic activity and vesicle leakage efficiency. The correlation between the relative leakage efficiency of POPC/POPE/Chol/SM (17:10:33:40) and the hemolytic activity of the tested cyclotides is shown. LC50 and HC50 were normalized relative to kB1.
Although RBCs only have a small amount of PE-containing phospholipids (∼5%) exposed in the outer leaflet (55), we found that this amount was sufficient to facilitate the binding of kB1 to model membranes, especially when Chol is present (see Fig. 5A). Furthermore, it appears that kB1 is able to induce the outward movement of PE-phospholipids, thus exposing more PE and self-promoting its binding (21).
The deviation from linearity for kB2 and cyO2 on the correlation between membrane leakage and hemolytic efficiency shown in Fig. 10 suggests that as well as targeting PE-phospholipids in RBCs, other membrane components/properties (e.g. the membrane asymmetry) not represented in the POPC/POPE/Chol/SM model can further improve the hemolytic efficiency of cyclotides.
The influence of membrane fluidity was demonstrated by hemolysis assays at 4 °C, where the cell membrane is less fluid than at 37 °C (56), and all of the cyclotides were unable to disrupt RBC membranes. In agreement with studies in model membranes in the solid-ordered phase, cyclotides have low affinity for very rigid membranes, even when PE-phospholipids are in the bilayer.
The importance of insertion into membranes for the biological effects of cyclotides was further demonstrated by comparing l- and d-isomers. Although d-kB1 (28) and d-kB2 were active and had affinity for PE-containing membranes, they were less hemolytic and had correspondingly lower affinity for model membranes than the cognate l-peptides. As there are no differences in charge, hydrophobicity, or three-dimensional structure (apart from mirror symmetry) between d-kB1 and the native enantiomer (28), the lower membrane affinity for the d-enantiomers can only be explained by the membrane chiral environment. As the PE headgroup does not have a chiral center, the chiral preference can be explained by the asymmetry at the glycerol skeleton immediately adjacent to the hydrophobic acyl chain, supporting the proposal that cyclotides insert in membranes rather than just associating loosely with the headgroup region.
Membrane-binding Affinity Also Correlates with Other Bioactivities
The studies on hemolytic activity and model membrane binding confirmed the role of PE and peptide insertion, but we were also interested to determine whether the proposed mechanism dependent on membrane-binding could explain other biological activities of cyclotides. Antimicrobial and anti-HIV activities of selected cyclotides were examined. Antimicrobial peptides are generally positively charged and typically have high affinity for negatively charged bacterial outer membranes. Although often referred to in the literature as antimicrobial peptides, most natural cyclotides do not match the typical profile of antimicrobial peptides. Indeed, of the peptides included in this study, cyO2 was the only one shown to have significant antimicrobial activity, in agreement with a previous report (26). Nevertheless, its activity is restricted to Gram-negative bacteria, and its antimicrobial potency is lower than its hemolytic activity, suggesting that cyO2 has limited value as an antimicrobial agent.
E. coli possesses an outer membrane with a lipopolysaccharide (LPS) layer exposed in the outer leaflet and a inner leaflet rich in PE-phospholipids (57), but this inner leaflet is only accessible after overcoming the anionic LPS layer. The lack of potent antimicrobial activity of cyclotides is in agreement with their lack of ability to target and disrupt negatively charged membranes in the absence of PE-phospholipids and with the inability to target LPS, as shown with kB1 (21). cyO2 is the most positively charged cyclotide tested and the one with highest affinity for lipid membranes, which would explain an improved ability to target the LPS layer and consequently access the inner leaflet rich in PE to kill E. coli.
Gram-positive bacteria have a thick peptidoglycan layer as a protective shell (57), which makes it even more difficult to access the PE-phospholipids located in the cytoplasmic membrane, thus explaining the lack of activity of all of the tested cyclotides against S. aureus.
The anti-HIV effect of kB1 was recently shown to correlate with its ability to target and disrupt the membrane of the HIV particle (21), which is very rich in PE, Chol, and SM (51). The higher virucidal activity of kB2 and lower activity of tcA against HIV particles, relative to kB1, suggest a similar HIV virucidal mechanism, dependent on viral membrane targeting.
Importance of the “Bioactive Patch” and Hydrophobic Properties at the Surface of Cyclotides for Their Membrane-binding Affinity and Biological Potencies
To develop a more detailed molecular interpretation of the proposed conserved mechanism for cyclotide bioactivities (hemolytic, HIV, antimicrobial, and insecticidal), we focused on the molecular surfaces of cyclotides. The potency of kB1 against RBCs and insect cells has been shown to be dependent on both a bioactive patch and hydrophobic patch at the surface of this molecule, as demonstrated in an Ala scan (58) and confirmed with a Lys scan (25). More recently, it was shown that both the bioactive and hydrophobic patches at the surface of kB1 are required for membrane targeting and insertion (21), and when these patches are compromised kB1 lacks ability to target membranes and becomes inactive. Based on NMR studies (21, 24), it was determined that the bioactive patch is involved in the specific targeting of PE headgroups, whereas the hydrophobic patch is required for insertion into the hydrophobic core of the membrane.
This proposed mechanism of activity suggested for kB1 is extended here and shown to be common to all tested cyclotides. Because of the extensive sequence variability of cyclotides, the same specificity and mechanism of action was not expected, and the conserved specificity for PE-phospholipids was initially surprising. However, Fig. 11 shows that the bioactive patch is well conserved across the cyclotide family, consistent with it being responsible for the binding to the PE headgroup not only for kB1 but also across the cyclotide family. Although not directly involved in recognition of the PE headgroup, the hydrophobic patch is also a requirement for the membrane activity of kB1 (21). Albeit with different sizes and compositions, all of the cyclotides in this study have a hydrophobic patch.
FIGURE 11.

Surface representation of selected cyclotides. Surface representations of the cyclotides included in this study are shown in two views. The “bioactive face” previously identified for kB1 (25, 58) is shown in red, i.e. residues in loop 1 (GET for kB1), the two first residues in loop 3 (NT for kB1), and the Lys/Arg in loop 6. Hydrophobic residues (Ala, Ile, Leu, Met, Phe, Pro, Trp, Val, and Tyr) are shown in green. The three-dimensional structure of kB1 is shown inside the box to define the orientation of the cyclotide surfaces.
We propose that subtle differences in the bioactive and hydrophobic patches, as well as the location of charges on the surface of cyclotides, can explain the observed trends in the affinity and effects induced in the membrane stability and biological potency. For instance, kB1 and kB2 have the same residues in the bioactive patch, but the overall hydrophobicity of kB2 is higher, which correlates with higher membrane binding affinity and hemolytic activity. Among the tested cyclotides, tcA has the lowest membrane affinity, the weakest vesicle leakage efficiency, and is the least hemolytic cyclotide; these observations correlate with the hydrophobic residues being more dispersed, instead of being located in a single patch, and also with the lack of an Arg/Lys in loop 6, shown to be important for the activity of kB1 (58). At the other extreme, cyO2 has the highest membrane affinity, high leakage efficiency, and induces more membrane surface destabilization than any other cyclotide, correlating with the highest hemolytic activity among the cyclotides tested. CyO2 has its bioactive and hydrophobic patches close together, but it also has two Lys residues located in a distinct part of the molecule that can establish extra contacts (e.g. electrostatic attractions or H-bonds) with phospholipid headgroups.
The bioactive patch is centered on a conserved Glu residue in loop 1, and we propose that the geometry of this patch facilitates an ionic interaction between the ammonium group of PE and the carboxylate of the Glu. In addition, the side chain of the Lys/Arg in loop 6 can establish an interaction with the PE-phosphate group. The hydrophobic side chains protruding from cyclotides are likely to stabilize peptide-membrane interactions by extra nonspecific peptide-membrane interactions with the hydrophobic core region. A proposed membrane binding mechanism is shown in Fig. 12, highlighting initial ionic interactions with PE followed by membrane insertion.
FIGURE 12.
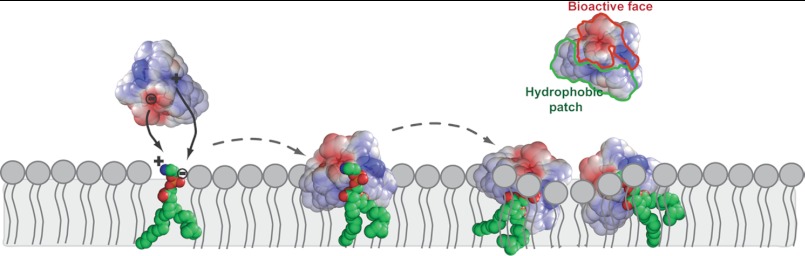
Schematic representation of proposed mechanism for the interaction of cyclotides with lipid bilayers. The interaction of cyclotides with the lipid membrane is initiated by targeting PE-phospholipids, followed by insertion in the lipid membrane. KB1 is shown with the electrostatic surface represented. In the first step, the bioactive face interacts and establishes electrostatic attractions with the PE headgroup (21). The hydrophobic patch inserts in hydrophobic core of the membrane (24). Upon insertion of several peptide molecules, the membrane is overloaded, and local membrane disturbances may occur, leading to membrane disruption.
The importance of the bioactive patch and of the Glu residue in particular for the recognition of the PE headgroup is supported by studies on naturally occurring kB12 and a synthetic kB1_E7D mutant; both peptides have an Asp in place of the Glu and are strikingly inactive against RBCs (59). Although they have the same overall structure and enzymatic and thermal stability as kB1, kB12 and kB1_E7D have negligible hemolytic activity (59), which correlates with the inability of kB1_E7D to bind to PE-phospholipids in membranes (21). Studies by Göransson and co-workers (60), in which the Glu residue of cyO2 was chemically modified by esterification, further support the importance of the Glu residue for the recognition of PE headgroups. Modification of the Glu residue caused a 48-fold decrease in the cytotoxic potency of cyO2 (60). A correlation between cytotoxicity and an ability to recognize PE headgroups is shown by a 50-fold higher leakage potency against PE-rich membranes (i.e. E. coli polar lipid extract that has an ∼67% PE-phospholipids weight ratio) than against PE-lacking membranes (i.e. 1,2-dioleoyl-sn-glycero-3-phosphocholine/1,2-dioleoyl-sn-glycero-3-phosphate mixture) (61). However, esterified cyO2 does not distinguish between PE-containing and PE-lacking lipid membranes (61). Overall, the modified cyclotides and native kB12 show that the Glu residue is essential for the biological effects of cyclotides, consistent with a proposed role in the PE targeting.
As noted in the Introduction, cyclotides belonging to the trypsin inhibitor subfamily are distinguished from Möbius and bracelet cyclotides, not only by their lack of sequence homology but also by distinct bioactivities. In particular, MCoTI-II is not cytotoxic and does not bind to model membranes (62). These data support the idea that the trypsin inhibitor subfamily is distinct from the other two subfamilies and in particular lacks the bioactive patch and a hydrophobic patch on its surface. Thus, we emphasize that the findings reported here relating to a common mechanism of action apply only to the Möbius and bracelet cyclotide subfamilies.
Cinnamycin and duramycin are two lantibiotic peptides produced by Gram-positive bacteria also reported to have specificity for PE-phospholipids (63). In addition to PE headgroup recognition, hydrophobic interactions with the phospholipid acyl chains are required for efficient membrane targeting (64). Cinnamycin and duramycin possess a binding pocket that fits a PE headgroup, and the binding is stabilized by an ion-pair interaction between the ammonium group of PE and the carboxylate of Asp-15. A Phe side chain stabilizes the peptide membrane binding by anchoring to the hydrophobic core of the membrane bilayer (63). The membrane binding modes of these lantibiotics have similarities to the cyclotide-membrane binding mode, as binding to PE headgroup and hydrophobic interactions are both required.
Cyclotides, a New Lipid-binding Protein Family
The fact that cyclotides, either isolated from the same plant species and belonging to the same subfamily (e.g. kB1 and kB2 from O. affinis) or from multiple species and belonging to different subfamilies (e.g. kB2 from O. affinis versus cyO2 from V. odorata), specifically recognize a phospholipid headgroup, prompts the suggestion that they can now be categorized as a new lipid-binding protein family. There are many conserved lipid-binding domains in the eukaryotic proteome, particularly in proteins involved in signal transduction and in membrane trafficking (65). A comparison of different lipid-binding domains reveals that they all have a pocket for the recognition of a unique lipid headgroup, for which electrostatic interactions are important, and in some cases the surrounding membrane is required for additional contacts, which is modulated by a hydrophobic patch that inserts in the membrane core (65). These features are consistent with cyclotides having a patch on their surface that recognizes PE headgroups and the hydrophobic part of the molecule inserting into and causing membrane destabilization. A PE-binding protein (PEBP) family, an evolutionarily conserved group of proteins that occurs in all taxa from bacteria to plants and animals, has been identified (66); although cyclotides do not resemble PEBP members (typically proteins ∼25 kDa in size), they might have communal roles. Interestingly, proteins belonging to PEBP family members expressed in plants seem to regulate the transition from the vegetative to the reproductive phase and are involved in modulation of plant architecture and flowering (66). Remarkably, two PEBP members with high homology have been shown to have opposite functions in Arabidopsis thaliana; one of them activates flowering and the other represses it, and their activities can be exchanged by swapping a single amino acid (67, 68).
Here, we show that Möbius and bracelet cyclotides, in addition to having a communal three-dimensional structure, have another communal feature, namely specific lipid binding. The physiological role of cyclotides has been suggested to be as insecticidal agents (69) with a mode of action involving membrane disruption (70), and this is consistent with multiple cyclotides having communal lipid recognition. The observation that one plant produces many cyclotides is puzzling in light of these findings if insecticidal activity is the sole function of cyclotides. Thus, the findings support the suggestion that cyclotides probably also have other, as yet undiscovered, functions.
Synthetically modified cyclotides have been engineered with various introduced functions relevant in drug design. Some of these applications of cyclotides for drug design might benefit from the new knowledge reported here on their PE-binding properties. PE-phospholipids have pivotal roles in membrane structure and function (71). Although mainly located in the inner leaflet in a typical mammalian cell, PE-phospholipids are more extensively externalized and/or overexpressed in apoptotic cells (72) and also on the surface of tumor vascular endothelium (73). In fact, PE has been suggested as a broad tumor marker common to many malignancies (73). Therefore, cyclotides have potential application as tools for tumor imaging, as observed with other PE-binding peptides such as duramycin and cinnamycin (73). An advantage of using cyclotides over these molecules is that in addition to being tools to target PE-rich cells, they can also be used as scaffolds for intracellular delivery (62, 74) of active epitopes based on their amenability to chemical synthesis.
Supplementary Material
Acknowledgments
We thank Dr. Quentin Kaas (Institute for Molecular Bioscience, The University of Queensland) for help in the modeling of the interaction of kB1 with PE. We also thank Priscilla Tacore for synthesis of all-d-kalata B2.
This work was supported in part by Australian Research Council Grant DP0880105 and by Marie Curie International Outgoing Fellowship PIOF-GA-2008-220318 (to S. T. H.) within the 7th European Community Framework Program.

This article contains supplemental Movies 1–6.
- kB
- kalata B
- tcA
- tricyclon A
- cyO2
- cycloviolacin O2
- PE
- phosphatidylethanolamine
- POPC
- palmitoyloleoylphosphatidylcholine
- POPG
- palmitoyloleoylphosphatidylglycerol
- POPE
- palmitoyloleoylphosphatidylethanolamine
- DPPC
- dipalmitoylglycerophosphatidylcholine
- DPPE
- dipalmitoylglycerophosphatidylethanolamine
- Chol
- cholesterol
- SM
- sphingomyelin
- LUV
- large unilamellar vesicle
- SUV
- small unilamellar vesicle
- SPR
- surface plasmon resonance
- P/L
- peptide-to-lipid molar ratio
- RU
- response units
- di-8-ANEPPS
- 4-[2-[6-(dioctylamino)-2-naphthalenyl]ethenyl]-1-(3-sulfopropyl)-pyridinium
- CF
- carboxyfluorescein
- GUV
- giant unilamellar vesicle
- TR
- tetramethylrhodamine
- MIC
- minimal inhibitory concentration
- PEBP
- PE-binding protein.
REFERENCES
- 1. Craik D. J., Daly N. L., Bond T., Waine C. (1999) Plant cyclotides. A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J. Mol. Biol. 294, 1327–1336 [DOI] [PubMed] [Google Scholar]
- 2. Colgrave M. L., Craik D. J. (2004) Thermal, chemical, and enzymatic stability of the cyclotide kalata B1. The importance of the cyclic cystine knot. Biochemistry 43, 5965–5975 [DOI] [PubMed] [Google Scholar]
- 3. Wang C. K., Kaas Q., Chiche L., Craik D. J. (2008) CyBase. A database of cyclic protein sequences and structures, with applications in protein discovery and engineering. Nucleic Acids Res. 36, D206–D210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hernandez J. F., Gagnon J., Chiche L., Nguyen T. M., Andrieu J. P., Heitz A., Trinh Hong T., Pham T. T., Le Nguyen D. (2000) Squash trypsin inhibitors from Momordica cochinchinensis exhibit an atypical macrocyclic structure. Biochemistry 39, 5722–5730 [DOI] [PubMed] [Google Scholar]
- 5. Craik D. J., Cemazar M., Daly N. L. (2006) The cyclotides and related macrocyclic peptides as scaffolds in drug design. Curr. Opin. Drug Discov. Dev. 9, 251–260 [PubMed] [Google Scholar]
- 6. Jennings C., West J., Waine C., Craik D., Anderson M. (2001) Biosynthesis and insecticidal properties of plant cyclotides. The cyclic knotted proteins from Oldenlandia affinis. Proc. Natl. Acad. Sci. U.S.A. 98, 10614–10619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gran L. (1973) On the effect of a polypeptide isolated from “Kalata-Kalata” (Oldenlandia affinis DC) on the oestrogen dominated uterus. Acta Pharmacol. Toxicol. 33, 400–408 [DOI] [PubMed] [Google Scholar]
- 8. Gustafson K. R., Sowder R. C., Henderson L. E., Parsons I. C., Kashman Y., Cardellina J. H., McMahon J. B., Buckheit R. W., Pannell L. K., Boyd M. R. (1994) Novel HIV inhibitory macrocyclic peptides from the tropical tree, Chassalia parvifolia. J. Am. Chem. Soc. 116, 9337–9338 [Google Scholar]
- 9. Lindholm P., Göransson U., Johansson S., Claeson P., Gullbo J., Larsson R., Bohlin L., Backlund A. (2002) Cyclotides. A novel type of cytotoxic agents. Mol. Cancer Ther. 1, 365–369 [PubMed] [Google Scholar]
- 10. Tam J. P., Lu Y. A., Yang J. L., Chiu K. W. (1999) An unusual structural motif of antimicrobial peptides containing end-to-end macrocycle and cystine-knot disulfides. Proc. Natl. Acad. Sci. U.S.A. 96, 8913–8918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Henriques S. T., Craik D. J. (2010) Cyclotides as templates in drug design. Drug Discov. Today 15, 57–64 [DOI] [PubMed] [Google Scholar]
- 12. Gunasekera S., Foley F. M., Clark R. J., Sando L., Fabri L. J., Craik D. J., Daly N. L. (2008) Engineering stabilized vascular endothelial growth factor-A antagonists. Synthesis, structural characterization, and bioactivity of grafted analogues of cyclotides. J. Med. Chem. 51, 7697–7704 [DOI] [PubMed] [Google Scholar]
- 13. Chan L. Y., Gunasekera S., Henriques S. T., Worth N. F., Le S. J., Clark R. J., Campbell J. H., Craik D. J., Daly N. L. (2011) Engineering pro-angiogenic peptides using stable, disulfide-rich cyclic scaffolds. Blood 118, 6709–6717 [DOI] [PubMed] [Google Scholar]
- 14. Austin J., Wang W., Puttamadappa S., Shekhtman A., Camarero J. A. (2009) Biosynthesis and biological screening of a genetically encoded library based on the cyclotide MCoTI-I. ChemBioChem. 10, 2663–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gould A., Ji Y., Aboye T. L., Camarero J. A. (2011) Cyclotides, a novel ultrastable polypeptide scaffold for drug discovery. Curr. Pharm. Des. 17, 4294–4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Getz J. A., Rice J. J., Daugherty P. S. (2011) Protease-resistant peptide ligands from a knottin scaffold library. ACS Chem. Biol. 6, 837–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ireland D. C., Colgrave M. L., Craik D. J. (2006) A novel suite of cyclotides from Viola odorata. Sequence variation and the implications for structure, function, and stability. Biochem. J. 400, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Plan M. R., Göransson U., Clark R. J., Daly N. L., Colgrave M. L., Craik D. J. (2007) The cyclotide fingerprint in Oldenlandia affinis. Elucidation of chemically modified, linear, and novel macrocyclic peptides. ChemBioChem. 8, 1001–1011 [DOI] [PubMed] [Google Scholar]
- 19. Wang C. K., Colgrave M. L., Gustafson K. R., Ireland D. C., Goransson U., Craik D. J. (2008) Anti-HIV cyclotides from the Chinese medicinal herb Viola yedoensis. J. Nat. Prod. 71, 47–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang Y. H., Colgrave M. L., Daly N. L., Keleshian A., Martinac B., Craik D. J. (2009) The biological activity of the prototypic cyclotide kalata b1 is modulated by the formation of multimeric pores. J. Biol. Chem. 284, 20699–20707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Henriques S. T., Huang Y. H., Rosengren K. J., Franquelim H. G., Carvalho F. A., Johnson A., Sonza S., Tachedjian G., Castanho M. A., Daly N. L., Craik D. J. (2011) Decoding the membrane activity of the cyclotide kalata B1. The importance of phosphatidylethanolamine phospholipids and lipid organization on hemolytic and anti-HIV activities. J. Biol. Chem. 286, 24231–24241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Svangård E., Burman R., Gunasekera S., Lövborg H., Gullbo J., Göransson U. (2007) Mechanism of action of cytotoxic cyclotides. Cycloviolacin O2 disrupts lipid membranes. J. Nat. Prod. 70, 643–647 [DOI] [PubMed] [Google Scholar]
- 23. Ireland D. C., Wang C. K., Wilson J. A., Gustafson K. R., Craik D. J. (2008) Cyclotides as natural anti-HIV agents. Biopolymers 90, 51–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shenkarev Z. O., Nadezhdin K. D., Sobol V. A., Sobol A. G., Skjeldal L., Arseniev A. S. (2006) Conformation and mode of membrane interaction in cyclotides. Spatial structure of kalata B1 bound to a dodecylphosphocholine micelle. FEBS J. 273, 2658–2672 [DOI] [PubMed] [Google Scholar]
- 25. Huang Y. H., Colgrave M. L., Clark R. J., Kotze A. C., Craik D. J. (2010) Lysine-scanning mutagenesis reveals an amendable face of the cyclotide kalata B1 for the optimization of nematocidal activity. J. Biol. Chem. 285, 10797–10805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pränting M., Lööv C., Burman R., Göransson U., Andersson D. I. (2010) The cyclotide cycloviolacin O2 from Viola odorata has potent bactericidal activity against Gram-negative bacteria. J. Antimicrob. Chemother. 65, 1964–1971 [DOI] [PubMed] [Google Scholar]
- 27. Mulvenna J. P., Sando L., Craik D. J. (2005) Processing of a 22-kDa precursor protein to produce the circular protein tricyclon A. Structure 13, 691–701 [DOI] [PubMed] [Google Scholar]
- 28. Sando L., Henriques S. T., Foley F., Simonsen S. M., Daly N. L., Hall K. N., Gustafson K. R., Aguilar M. I., Craik D. J. (2011) A synthetic mirror image of kalata B1 reveals that cyclotide activity is independent of a protein receptor. ChemBioChem. 12, 2456–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Daly N. L., Clark R. J., Craik D. J. (2003) Disulfide folding pathways of cystine knot proteins. Tying the knot within the circular backbone of the cyclotides. J. Biol. Chem. 278, 6314–6322 [DOI] [PubMed] [Google Scholar]
- 30. Mayer L. D., Hope M. J., Cullis P. R. (1986) Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta 858, 161–168 [DOI] [PubMed] [Google Scholar]
- 31. Henriques S. T., Pattenden L. K., Aguilar M. I., Castanho M. A. (2008) PrP(106–126) does not interact with membranes under physiological conditions. Biophys. J. 95, 1877–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cooper M. A. (2003) Label-free screening of bio-molecular interactions. Anal. Bioanal. Chem. 377, 834–842 [DOI] [PubMed] [Google Scholar]
- 33. Santos N. C., Prieto M., Castanho M. A. (2003) Quantifying molecular partition into model systems of biomembranes. An emphasis on optical spectroscopic methods. Biochim. Biophys. Acta 1612, 123–135 [DOI] [PubMed] [Google Scholar]
- 34. Henriques S. T., Pattenden L. K., Aguilar M. I., Castanho M. A. (2009) The toxicity of prion protein fragment PrP(106–126) is not mediated by membrane permeabilization as shown by an M112W substitution. Biochemistry 48, 4198–4208 [DOI] [PubMed] [Google Scholar]
- 35. Henriques S. T., Castanho M. A. (2004) Consequences of nonlytic membrane perturbation to the translocation of the cell penetrating peptide pep-1 in lipidic vesicles. Biochemistry 43, 9716–9724 [DOI] [PubMed] [Google Scholar]
- 36. Henriques S. T., Quintas A., Bagatolli L. A., Homblé F., Castanho M. A. (2007) Energy-independent translocation of cell-penetrating peptides occurs without formation of pores. A biophysical study with pep-1. Mol. Membr. Biol. 24, 282–293 [DOI] [PubMed] [Google Scholar]
- 37. Ambroggio E. E., Separovic F., Bowie J. H., Fidelio G. D., Bagatolli L. A. (2005) Direct visualization of membrane leakage induced by the antibiotic peptides. Maculatin, citropin, and aurein. Biophys. J. 89, 1874–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Telwatte S., Moore K., Johnson A., Tyssen D., Sterjovski J., Aldunate M., Gorry P. R., Ramsland P. A., Lewis G. R., Paull J. R., Sonza S., Tachedjian G. (2011) Virucidal activity of the dendrimer microbicide SPL7013 against HIV-1. Antiviral Res. 90, 195–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wiegand I., Hilpert K., Hancock R. E. (2008) Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163–175 [DOI] [PubMed] [Google Scholar]
- 40. Alves C. S., Melo M. N., Franquelim H. G., Ferre R., Planas M., Feliu L., Bardají E., Kowalczyk W., Andreu D., Santos N. C., Fernandes M. X., Castanho M. A. (2010) Escherichia coli cell surface perturbation and disruption induced by antimicrobial peptides BP100 and pepR. J. Biol. Chem. 285, 27536–27544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Daleke D. L., Huestis W. H. (1989) Erythrocyte morphology reflects the transbilayer distribution of incorporated phospholipids. J. Cell Biol. 108, 1375–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pokorny A., Yandek L. E., Elegbede A. I., Hinderliter A., Almeida P. F. (2006) Temperature and composition dependence of the interaction of delta-lysin with ternary mixtures of sphingomyelin/cholesterol/POPC. Biophys. J. 91, 2184–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Domènech O., Redondo L., Picas L., Morros A., Montero M. T., Hernández-Borrell J. (2007) Atomic force microscopy characterization of supported planar bilayers that mimic the mitochondrial inner membrane. J. Mol. Recognit. 20, 546–553 [DOI] [PubMed] [Google Scholar]
- 44. Cheng H. T., Megha, London E. (2009) Preparation and properties of asymmetric vesicles that mimic cell membranes. Effect upon lipid raft formation and transmembrane helix orientation. J. Biol. Chem. 284, 6079–6092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jaworsky M., Mendelsohn R. (1985) Fourier-transform infrared studies of CaATPase partitioning in phospholipid mixtures of 1,2-dipalmitoylphosphatidylcholine-d62 with 1-palmitoyl-2-oleoylphosphatidylethanolamine and 1-stearoyl-2-oleoylphosphatidylcholine. Biochemistry 24, 3422–3428 [DOI] [PubMed] [Google Scholar]
- 46. Mulgrew-Nesbitt A., Diraviyam K., Wang J., Singh S., Murray P., Li Z., Rogers L., Mirkovic N., Murray D. (2006) The role of electrostatics in protein-membrane interactions. Biochim. Biophys. Acta 1761, 812–826 [DOI] [PubMed] [Google Scholar]
- 47. Persson D., Thorén P. E., Nordén B. (2001) Penetratin-induced aggregation and subsequent dissociation of negatively charged phospholipid vesicles. FEBS Lett. 505, 307–312 [DOI] [PubMed] [Google Scholar]
- 48. Coccia C., Rinaldi A. C., Luca V., Barra D., Bozzi A., Di Giulio A., Veerman E. C., Mangoni M. L. (2011) Membrane interaction and antibacterial properties of two mildly cationic peptide diastereomers, bombinins H2 and H4, isolated from Bombina skin. Eur. Biophys. J. 40, 577–588 [DOI] [PubMed] [Google Scholar]
- 49. Gregory S. M., Cavenaugh A., Journigan V., Pokorny A., Almeida P. F. (2008) A quantitative model for the all-or-none permeabilization of phospholipid vesicles by the antimicrobial peptide cecropin A. Biophys. J. 94, 1667–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aloia R. C., Tian H., Jensen F. C. (1993) Lipid composition and fluidity of the human immunodeficiency virus envelope and host cell plasma membranes. Proc. Natl. Acad. Sci. U.S.A. 90, 5181–5185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brügger B., Glass B., Haberkant P., Leibrecht I., Wieland F. T., Kräusslich H. G. (2006) The HIV lipidome. A raft with an unusual composition. Proc. Natl. Acad. Sci. U.S.A. 103, 2641–2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Colgrave M. L., Kotze A. C., Huang Y. H., O'Grady J., Simonsen S. M., Craik D. J. (2008) Cyclotides. Natural, circular plant peptides that possess significant activity against gastrointestinal nematode parasites of sheep. Biochemistry 47, 5581–5589 [DOI] [PubMed] [Google Scholar]
- 53. Daly N. L., Clark R. J., Plan M. R., Craik D. J. (2006) Kalata B8, a novel antiviral circular protein, exhibits conformational flexibility in the cystine knot motif. Biochem. J. 393, 619–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Plan M. R., Saska I., Cagauan A. G., Craik D. J. (2008) Backbone cyclized peptides from plants show molluscicidal activity against the rice pest Pomacea canaliculata (golden apple snail). J. Agric. Food Chem. 56, 5237–5241 [DOI] [PubMed] [Google Scholar]
- 55. Daleke D. L. (2008) Regulation of phospholipid asymmetry in the erythrocyte membrane. Curr. Opin. Hematol. 15, 191–195 [DOI] [PubMed] [Google Scholar]
- 56. Cho K. H., Wang H. S., Kim Y. K. (2010) Temperature-dependent hemolytic activity of membrane pore-forming peptide toxin, tolaasin. J. Pept. Sci. 16, 85–90 [DOI] [PubMed] [Google Scholar]
- 57. Huijbregts R. P., de Kroon A. I., de Kruijff B. (2000) Topology and transport of membrane lipids in bacteria. Biochim. Biophys. Acta 1469, 43–61 [DOI] [PubMed] [Google Scholar]
- 58. Simonsen S. M., Sando L., Rosengren K. J., Wang C. K., Colgrave M. L., Daly N. L., Craik D. J. (2008) Alanine-scanning mutagenesis of the prototypic cyclotide reveals a cluster of residues essential for bioactivity. J. Biol. Chem. 283, 9805–9813 [DOI] [PubMed] [Google Scholar]
- 59. Wang C. K., Clark R. J., Harvey P. J., Rosengren K. J., Cemazar M., Craik D. J. (2011) The role of conserved Glu residue on cyclotide stability and activity. A structural and functional study of kalata B12, a naturally occurring Glu to Asp mutant. Biochemistry 50, 4077–4086 [DOI] [PubMed] [Google Scholar]
- 60. Herrmann A., Svangård E., Claeson P., Gullbo J., Bohlin L., Göransson U. (2006) Key role of glutamic acid for the cytotoxic activity of the cyclotide cycloviolacin O2. Cell. Mol. Life Sci. 63, 235–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Burman R., Strömstedt A. A., Malmsten M., Göransson U. (2011) Cyclotide-membrane interactions. Defining factors of membrane binding, depletion, and disruption. Biochim. Biophys. Acta 1808, 2665–2673 [DOI] [PubMed] [Google Scholar]
- 62. Cascales L., Henriques S. T., Kerr M. C., Huang Y. H., Sweet M. J., Daly N. L., Craik D. J. (2011) Identification and characterization of a new family of cell-penetrating peptides. Cyclic cell-penetrating peptides. J. Biol. Chem. 286, 36932–36943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhao M. (2011) Lantibiotics as probes for phosphatidylethanolamine. Amino Acids 41, 1071–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Machaidze G., Ziegler A., Seelig J. (2002) Specific binding of Ro 09-0198 (cinnamycin) to phosphatidylethanolamine. A thermodynamic analysis. Biochemistry 41, 1965–1971 [DOI] [PubMed] [Google Scholar]
- 65. Hurley J. H. (2006) Membrane binding domains. Biochim. Biophys. Acta 1761, 805–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Karlgren A., Gyllenstrand N., Källman T., Sundström J. F., Moore D., Lascoux M., Lagercrantz U. (2011) Evolution of the PEBP gene family in plants. Functional diversification in seed plant evolution. Plant Physiol. 156, 1967–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hanzawa Y., Money T., Bradley D. (2005) A single amino acid converts a repressor to an activator of flowering. Proc. Natl. Acad. Sci. U.S.A. 102, 7748–7753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pin P. A., Benlloch R., Bonnet D., Wremerth-Weich E., Kraft T., Gielen J. J., Nilsson O. (2010) An antagonistic pair of FT homologs mediates the control of flowering time in sugar beet. Science 330, 1397–1400 [DOI] [PubMed] [Google Scholar]
- 69. Jennings C. V., Rosengren K. J., Daly N. L., Plan M., Stevens J., Scanlon M. J., Waine C., Norman D. G., Anderson M. A., Craik D. J. (2005) Isolation, solution structure, and insecticidal activity of kalata B2, a circular protein with a twist. Do Möbius strips exist in nature? Biochemistry 44, 851–860 [DOI] [PubMed] [Google Scholar]
- 70. Barbeta B. L., Marshall A. T., Gillon A. D., Craik D. J., Anderson M. A. (2008) Plant cyclotides disrupt epithelial cells in the midgut of lepidopteran larvae. Proc. Natl. Acad. Sci. U.S.A. 105, 1221–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. van Meer G., Voelker D. R., Feigenson G. W. (2008) Membrane lipids. Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Williamson P., Schlegel R. A. (2002) Transbilayer phospholipid movement and the clearance of apoptotic cells. Biochim. Biophys. Acta 1585, 53–63 [DOI] [PubMed] [Google Scholar]
- 73. Stafford J. H., Thorpe P. E. (2011) Increased exposure of phosphatidylethanolamine on the surface of tumor vascular endothelium. Neoplasia 13, 299–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Contreras J., Elnagar A. Y., Hamm-Alvarez S. F., Camarero J. A. (2011) Cellular uptake of cyclotide MCoTI-I follows multiple endocytic pathways. J. Control. Release 155, 134–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.