

Background: Viral scaffolding proteins interact with coat proteins to drive procapsid assembly.
Results: Amino acid substitutions in the turn and between the helices of the coat protein-binding domain of scaffolding protein block procapsid assembly.
Conclusion: The orientation of helices in the scaffolding helix turn helix domain is critical for procapsid assembly.
Significance: Understanding scaffolding/coat protein interactions illuminates the mechanism of assembly of many large viruses.
Keywords: Bacteriophage, Protein Assembly, Protein-Protein Interactions, Virus Assembly, Virus Structure, Circular Dichroism, Electrostatics, Procapsid Assembly, Prophage, Scaffolding Protein
Abstract
Many viruses encode scaffolding and coat proteins that co-assemble to form procapsids, which are transient precursor structures leading to progeny virions. In bacteriophage P22, the association of scaffolding and coat proteins is mediated mainly by ionic interactions. The coat protein-binding domain of scaffolding protein is a helix turn helix structure near the C terminus with a high number of charged surface residues. Residues Arg-293 and Lys-296 are particularly important for coat protein binding. The two helices contact each other through hydrophobic side chains. In this study, substitution of the residues of the interface between the helices, and the residues in the β-turn, by aspartic acid was used examine the importance of the conformation of the domain in coat binding. These replacements strongly affected the ability of the scaffolding protein to interact with coat protein. The severity of the defect in the association of scaffolding protein to coat protein was dependent on location, with substitutions at residues in the turn and helix 2 causing the most significant effects. Substituting aspartic acid for hydrophobic interface residues dramatically perturbs the stability of the structure, but similar substitutions in the turn had much less effect on the integrity of this domain, as determined by circular dichroism. We propose that the binding of scaffolding protein to coat protein is dependent on angle of the β-turn and the orientation of the charged surface on helix 2. Surprisingly, formation of the highly complex procapsid structure depends on a relatively simple interaction.
Introduction
Viral capsid formation is an example of a complex macromolecular assembly process during which different protein components interact to form a defined stable structure. Many virions are built by first assembling intermediate structures called procapsids into which nucleic acid is subsequently packaged, and in many of these a scaffolding protein is required to assemble properly shaped procapsids. Scaffolding proteins act as molecular chaperones for assembly and are called “scaffolding” proteins because they are not present in the mature virion but are present in procapsids (1–3). Most large dsDNA viruses such as bacteriophages P22, Φ29, P2, SPP1, T7, T4, and λ and the herpesviruses have scaffolding proteins that are stably present within the interior of the procapsid but are released from these particles at or before the time of nucleic acid packaging. Among such viruses, scaffolding proteins of P2 (4, 5), T4 (6, 7), λ (8, 9), and the herpesviruses (10–13) undergo proteolytic cleavage upon maturation of the capsid, whereas those of P22 (14), T7 (15), and Φ29 (16) exit the capsid intact and are reused in additional rounds of assembly. There are variations on this theme; for example, the very small single-stranded DNA bacteriophage ΦX174, employs both internal and external scaffolding proteins for proper assembly of procapsid shells (17), and in the large double-stranded DNA phages HK97 and T5 the N-terminal domain of the major capsid protein is thought to perform the scaffolding function (18). The detailed mechanism(s) by which scaffolding proteins function in capsid shell assembly remains very poorly understood. The scaffolding-like δ domain of the HK97 major capsid protein and the internal scaffolding protein of ΦX174 both appear to function by directing a switch of the major capsid protein domains during assembly (19–21). It is not known if the free scaffolding proteins of the large viruses may use a similar mechanism; however, Suhanovsky and Teschke (22) have argued that phage P22 scaffolding protein also affects conformational switching in the major capsid protein during assembly.
The in vitro and in vivo assembly of bacteriophage P22 procapsids has been the subject of a number of studies, but much still remains to be learned about the role of the P22 scaffolding protein in this process. The P22 capsid shell is composed of one type of major capsid protein called coat protein, which assembles into a capsid structure with T = 7 icosahedral symmetry (23, 24). After its assembly, the P22 procapsid undergoes a maturation process during which all of the internal scaffolding protein molecules are released intact, holes in the coat protein shell close, and the shell becomes thinner and expands about 11% in diameter (24–28). These structural changes are mediated by a conformational change in coat protein (24, 25, 27, 29). P22 procapsids made during a typical infection contain about 250–300 molecules of scaffolding protein (2, 14, 25, 30, 31), but there can be as few as 60 depending on assembly conditions (31–34). Without scaffolding protein, coat protein polymerizes in vivo and in vitro into aberrant forms such as empty, closed T = 7 or T = 4 shells and spiral type “monster” structures (35–38). Scaffolding protein is also required for incorporation of the portal protein ring (32, 39, 40) and at least one of the three minor proteins that are essential for subsequent DNA ejection during infection (39–41).
The association between P22 coat protein and scaffolding protein is regulated by electrostatic interactions (42, 43). P22 scaffolding protein is comprised of 303 amino acids and is an elongated, highly flexible molecule mainly composed of α-helices (44–47). Its coat protein-binding domain is within residues 280–296 near the C terminus (39–41, 48, 49). A high resolution NMR structure of the C-terminal domain of the P22 scaffolding protein has been determined, and forms a helix turn helix (HTH)5 with a highly charged surface (50). Cortines et al. (49) showed that the side chains of just two amino acid residues in the HTH, Arg-293 and Lys-296, are critical for coat binding. Substitutions of these two positively charged amino acids by alanine abolished binding to coat protein. Changes in the other charged HTH surface residues have only a modest impact on the coat protein/scaffolding protein interaction. Although electrostatic interactions, via the two critical amino acids in the scaffolding protein HTH, are a primary factor responsible for this interaction, the exact shape of the HTH might also be crucial. The scaffolding protein is folded so that the N terminus, which also plays a role in assembly (22), is positioned in the immediate vicinity of the C terminus, and it is not a rigid molecule because it requires flexibility of some domains to function properly (51).
The importance of the HTH fold/shape to scaffolding protein activity has not been studied. Here, we investigate the roles of the hydrophobic core or zipper of the C-terminal HTH and the residues in the turn at the “top” of the HTH in the structural stability of the HTH and in the ability to bind coat protein. We provide evidence that only a single charged substitution in the hydrophobic core is sufficient to destabilize the HTH structure, and that destabilization of the HTH is largely correlated with assembly activity. We find charged substitutions in the turn do not cause major structural effects in the HTH. Nevertheless, coat binding is severely affected. In aggregate, our findings indicate that the spatial arrangement of the two helices with respect to one another is a crucial aspect of scaffolding protein activity.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis and Protein Purification
The N-terminal His6-tagged scaffolding protein gene (gp8) and the N terminally truncated scaffolding protein (Δ1–237) gene were constructed in pet15b (Novagen). These plasmids were used as the template used for site-directed mutagenesis (51). The primers used to generate the variants in this study are listed in Table 1. The mutations were introduced by PCR (polymerase chain reaction), and then digested with DpnI. Mutated plasmids were transformed by electroporation into Escherichia coli DH5α cells. All plasmid changes were confirmed by the University of Connecticut and University of Utah sequencing facilities. The plasmids containing the introduced mutations were transformed in E. coli BL21 (Stratagene). The cells were grown in 1 liter of LB broth; 100 μg/ml of ampicillin at 37 °C until midlog phase, and expression of the protein was induced with 1 mm isopropyl β-d-1-thiogalactopyranoside. Four hours after induction the cells were centrifuged at 4300 × g for 10 min at 4 °C. Pelleted cells were frozen at −20 °C. The scaffolding proteins were purified as previously described (51). Briefly, the frozen pellet was resuspended in 30 ml of lysis buffer (20 mm NaH2PO4, 300 mm NaCl, 5 mm imidazole, 10 mm phenylmethylsulfonyl fluoride). The thawed sample was sonicated at 38 amps, 45-s pulse, and 1-min pause, with a total processing time of 9 min using a Misonix sonicator 4000 with a standard tip. Sonicated samples were cleared of cell debris by centrifugation at 15,000 × g for 10 min at 4 °C. The cleared supernatant was loaded on a 15-ml Talon® (Clontech) column and eluted using an imidazole step gradient. The purity of samples was confirmed with 10% SDS-PAGE. Protein concentrations were determined by absorbance at 280 nm with ϵ = 17,420 m−1 cm−1 for full-length scaffolding protein, ϵ = 1,490 m−1 cm−1 for the Δ1–237 scaffolding protein, ϵ = 5,500 m−1 cm−1 for the Δ1–237/T265W/Y292D, and ϵ = 6990 m−1 cm−1 ml/g for the other Δ1–237/T265W variants of scaffolding protein.
TABLE 1.
Oligonucleotides used in this study
| Name/substitution | Oligonucleotide (5′ → 3′)a |
|---|---|
| N272D | gatgtcagcgcagcagataaagatgccattc |
| I276D | gcagcaaataaagatgccgatcgtaaacaaatggatgc |
| M280D | gatgccattcgtaaacaagatgatgctgctgcgagcaag |
| A283D | caaatggatgctgatgcgagcaagggag |
| A284D | taaacaaatggatgctgctgatagcaagggagatgtggaaac |
| G287D | ctgctgcgagcaaggatgatgtggaaacctac |
| V289D | gagcaagggagatgatgaaacctaccgcaag |
| T291D | caagggagatgtggaagattaccgcaagctaaaggc |
| Y292D | gggagatgtggaaaccgaccgcaagctaaaggc |
| L295D | gatgtggaaacctaccgcaaggataaggcaaaacttaaaggaatc |
| L299D | ccgcaagctaaaggcaaaagataaaggaatccgataatag |
| T265W | gctgaccagcctatttggggtgatgtcagcgc |
| A | tgcgcacgccgcctcgatcgccgatggtcgctttgacgatgtctattaagacccactttcacatt |
| B | cgcggtcagcgacatccattttcgcgaatccggagtataaaaatgctaagcacttgtctcctg |
| C | cttttagctaacgaagctacccggcagtagcgctataagccaaggacggttaagacccactttcacatt |
| D | catttaatttgctccagatgcaaggaattgccgcagcatggctacggtgctaagcacttgtctcctg |
| E | cttttagctaacgaagctacccggcagtagcgctataagccaaggacggcaccgtagccatgctgcggcaattccttgcatctggagcaaattaaatg |
a Only one strand shown in cases where double-strand oligonucleotides were used.
Analysis of in Vivo Scaffolding Protein Function
P22 prophages carrying scaffolding protein point mutations were created by phage λ Red mediated recombineering of strains UB-1757 and UB-1790 (Salmonella enterica LT2 leuA−414, r−, sup°, which carry a P22 15−ΔSC302::KanR 13−amH101 and P22 ΔsieA-1, 15−ΔSC302::KanR 13−amH101 prophage, respectively). Strain UB-1757 construction was reported by Cortines et al. (49) and UB-1790 was constructed in this work. Normal P22 prophage induction results in virions that are deficient in tailspikes because Mnt and/or Arc repressors bound to their operators in the Immunity I (ImmI) region lower transcription of the tailspike gene (52–54). We therefore deleted the entire ImmI region by the recombineering methodology described in Cortines et al. (49) and Warming et al. (55). The galK gene of strain UB-1757 was first replaced by the PCR amplified, tetracycline resistance encoding TetRA cassette from S. enterica strain TH2788 (56) using primers A and B (Table 1). The 5′-tails of these primers allowed the amplified DNA to replace the native galK gene of UB-1757 by homologous recombination to create strain UB-1777. The ImmI region of the UB-1777 P22 prophage was then replaced by a functional E. coli galK gene amplified from plasmid pGalK (55) with primers C and D (Table 1). Recombinational integration of this amplicon via its 3′-primer tails replaced P22 bp 13,333–16,157 with the galK gene, and the resulting strain (UB-1794) was able to utilize galactose. Finally, replacement of the galK gene of UB-1794 by the synthetic 98-bp double-stranded oligonucleotide E (Table 1), which contained the P22 sequence from bp 13,284–13,333 joined to bp 16,158–16,206), resulted in strain UB-1790 (gal−) whose prophage now carries the ΔSieA-1 deletion that removes P22 at bp 13,334–16,157 (20 bp transcriptionally downstream of gene 16 and 45 upstream of gene 9 remain intact; the deletion removes the mnt and arc genes and their operators). The P22 ΔSieA-1, 15−ΔSC302::KanR, 13−amH101 phage released after mitomycin C induction of UB-1790 makes plaques efficiently on amber mutant suppressing hosts, and its virions are not tailspike deficient as measured by tailspike band intensity in Coomassie Brilliant Blue staining of SDS-PAGE of purified virions and the failure of added tailspike protein to increase the titer. Selections for gal+ and gal− bacteria during the above manipulations required growth on galactose as the sole carbon source or growth on rich medium in the presence of 2-deoxygalactose, respectively, as described by Warming et al. (55).
Prophages carrying scaffolding protein point mutations were also created by galK recombineering (above) using PCR-amplified DNA from the above mutant scaffolding protein expression plasmids to replace the homologous region in the prophages of bacterial strains UB-1838 or UB-1962 in which the galK gene replaces codons 266–303 of the scaffolding protein gene in UB-1757 or UB-1790, respectively. Most of the prophage mutant scaffolding protein genes encode the native proline at position 259; the V289D, Y292D, L295D, and I320D prophages have an His at this position, but this change is known not to affect scaffolding protein function in vivo (49, 57). Prophages were induced to lytic growth by the addition of Mitomycin C or carbadox (0.5 or 1 μg/ml, respectively). Cultures were shaken at 37 °C for 3–5 h and shaken with 0.05 volumes of chloroform to cause lysis because the nonsense mutation in gene 13 blocks normal lysis (58). Phage titers of the resulting lysates were determined with the indicator strain DB7004 (59) on LB plates that contained 10 mm Na citrate after the addition of excess tailspike protein; the gene 15 deletion mutant parental prophage plaques inefficiently in the absence of a divalent cation chelator such as citrate (60). Virions are tailspike deficient after UB-1757 prophage induction and so required the addition of excess tailspike protein for accurate titer determinations (49, 52–54). For examination of procapsid-like particles after induction, cells were concentrated 50-fold, the concentrated cells were shaken with chloroform to cause lysis, treated briefly with DNase I to reduce viscosity, cell debris was removed by centrifugation, and phage-related particles were purified by CsCl step gradient centrifugation as previously described (26). The procapsid fraction was analyzed by SDS-PAGE and particle-agarose gel electrophoresis (described below).
In Vitro Procapsid Assembly
Monomeric coat proteins were generated according to previously published methods (51, 61, 62). Assembly reactions for kinetic measurements were initiated by mixing 0.5 mg/ml of coat protein and 0.5 mg/ml of scaffolding protein in 10 mm NaH2PO4, pH 7.6, and 45 mm NaCl (42). These assembly reactions were monitored every second for 16 min at 20 °C by light scattering in a SLM Aminco Bowman 2 spectrofluorometer at excitation and emission wavelengths of 500 nm using band passes of 4 nm.
End point assembly salt titration reactions were initiated with the same concentrations of monomeric coat protein and scaffolding proteins as the kinetic reactions except with variable concentrations of NaCl (0, 5, 15, 30, 45, 60, 75, and 100 mm). Change of the NaCl concentration from 0 to 100 mm NaCl does not greatly affect scaffolding/scaffolding interactions (43). These assembly reactions were allowed to incubate for 2 h at room temperature and an aliquot of each reaction was loaded on 1% agarose gel using TAE buffer (40 mm Tris acetate, 1 mm EDTA). Gel bands were visualized with Coomassie Blue staining (63, 64).
Shell-refilling Reaction
Empty procapsid shells were isolated as previously described (31, 62, 64). The shell refilling reaction followed the procedure of Cortines et al. (49). A mixture of 0.2 mg/ml shells and 0.33 mg/ml of scaffolding protein were mixed in buffer (10 mm NaH2PO4, pH 7.6, 45 mm NaCl) and incubated overnight at room temperature. The refilled procapsids were separated from unassembled scaffolding protein as follows: 200-μl aliquots of the samples were layered on a 7-ml cushion of 20% sucrose in 10 mm NaH2PO4, pH 7.6, 45 mm NaCl and centrifuged in Sorvall® S50T rotor at 40,000 × g for 90 min at 20 °C. At the concentration and temperature used in this experiment, the scaffolding protein is >90% monomeric (45). After centrifugation, the supernatant was removed and the pellet was dissolved in 50 μl of 10 mm NaH2PO4, pH 7.6, 45 mm NaCl. The protein contents of the resuspended pellet were then assessed in a 12.5% SDS-PAGE and visualized with Coomassie Blue staining (49). The band intensities were measured using a Bio-Rad imager and Image One software.
Electron Microscopy
Negative stained grids of assembled procapsids were prepared as previously published (51). Briefly, aliquots (4 μl) of assembly reaction samples were placed on 300-mesh carbon formvar copper grids and allowed to adsorb for 1 min. Grids were washed with deionized water and then stained 1% uranyl acetate for 1 min. Excess stain was removed by blotting with filter paper, and the grid was air dried. The prepared grids were viewed in a Fei Technai Biotwin transmission electron microscope at 80 kV and magnification of ×68,000.
Circular Dichroism Spectroscopy
CD spectra of full-length and N terminally truncated scaffolding protein variants (Δ1–237) were measured in an Applied Photophysics π*-180. CD spectra were measured from 260 to 200 nm with 2-nm slits at 1-nm increments for 15 s/nm at 7 °C. Full-length scaffolding protein samples were measured at a concentration of 0.1 mg/ml in 10 mm NaH2PO4, pH 7.6, using a path length of 1 mm, whereas the N terminally truncated scaffolding protein variants were measured at a concentration of 0.5 mg/ml in 20 mm NaH2PO4, pH 7.6, 150 mm NaCl, using a path length of 0.1 mm. Thermal stability of the N terminally truncated scaffolding protein variants was measured at 222 nm with 2-nm slits at over temperature range of 4 to 85 °C ramping at 0.3 min/°C with 20 s averaging/°C. Samples were measured using a path length of 1 mm. The CD signals were converted to mean residue ellipticity using the concentrations measured by absorbance at 280 nm.
RESULTS
Design of Mutations That Affect the Zipper Core and Turn of the Scaffolding Protein Coat-binding Domain
For analysis of the in vitro activity of scaffolding protein, amino acid changes were made in a pET15b vector carrying the scaffolding protein gene that adds a His6 tag (MGSHHHHHHSSGLVPRGSH-) to the amino terminus. This tag does not affect scaffolding protein function in vivo, nor does it affect the Tm of scaffolding protein in vitro (49). For ease of discussion we refer to the histidine-tagged but otherwise wild-type protein as WT. Changes in scaffolding protein were made that were projected to alter the fold of the HTH, and in vitro assembly activity was assessed. These changes were also moved into phage chromosomes to determine their effect on in vivo scaffolding protein function. Due to its small size, aspartic acid replacement of nonpolar residues is expected to only induce distortions in the structure due to a solvation penalty in a hydrophobic environment, and not impose steric restrictions. Thus, most of the changes analyzed here are changes of hydrophobic zipper residues to aspartic acid.
Analysis of the NMR structure of the scaffolding protein HTH shows that Ile-276, Met-280, Ala-283, Tyr-292, Leu-295, and Leu-299 are the residues whose side chains form the hydrophobic zipper (Fig. 1) (50). The turn/loop residues are Ala-284 to Ala-288 (50). Even though residues Asn-272 and Ile-302 are not part of the hydrophobic zipper, they are adjacent to the N-terminal end of helix 1 and C-terminal end of helix 2, respectively, and a charged mutation at the periphery of the HTH could affect hydrophobic interactions of the residues within the zipper. The turn is stabilized by i, i+3 hydrogen bond interaction of the main chain CO group of Ala-284 and main chain NH group of Gly-287, thereby classifying it as a type 1 β-turn (50, 65). Residues Val-289, Glu-290, and Thr-291 are at the N terminus of helix 2, and are not part of the hydrophobic core of the zipper. These residues could influence the angle by which the two helices are oriented (Fig. 1). Therefore, they were included in this study. Charged or hydrophilic residues Ser-285, Lys-286, Asp-288, and Glu-290 were adjacent to or in the turn. Charge changes in the last three were previously shown not to affect scaffolding function (49) and we show here that S285D and I302D scaffolding proteins have full function in vivo (Table 2). Thus, these hydrophilic residues were not included in the remainder of the study presented here (Fig. 1). Although Val-289 is technically the most N-terminal residue in helix 2, because its side chain appears not to participate in the zipper, we consider it with A284D and G287D as part of the turn region.
FIGURE 1.

Structure of the C-terminal coat protein-binding domain of phage P22 scaffolding protein. The NMR structure of the C-terminal HTH domain (PDB entry 2GP8) (50) was modeled using PyMOL (PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC). a, the labeled residues in (i) helix 1, (ii) the β1-turn, and (iii) helix 2 were individually substituted with aspartic acid. b, the primary sequence and secondary structure of the C-terminal domain. The residues in orange represent substituted residues.
TABLE 2.
In vivo functionality of altered P22 scaffolding proteins
| Mutanta | Average induction titerb | Bacterial strainc |
|---|---|---|
| Wild-type | 3 × 1010 | UB-1757 |
| Wild-type | 9 × 1010 | UB-1790 |
| N272D | 1 × 1011 | UB-1971 |
| I276D* | 1 × 107 | UB-1968 |
| M280D* | 5 × 1010 | UB-1780 |
| A283D* | 6 × 103 | UB-1787 |
| A284D† | 6 × 103 | UB-1779 |
| S285D† | 2 × 1010 | UB-1810 |
| G287D† | <105 | UB-1813 |
| G287P† | × 108 | UB-1811 |
| G287A† | 1 × 1010 | UB-1799 |
| V289D | <105 | UB-1808 |
| V289A | 7 × 109 | UB-1784 |
| T291D | 4 × 1010 | UB-1783 |
| Y292D* | 1 × 104 | UB-1788 |
| L295D* | <104 | UB-1901 |
| L299D* | <104 | UB-1972 |
| I302D | 1 × 1011 | UB-1970 |
| N272D, I267R, | <104 | UB-2018 |
| L299D |
a Scaffolding protein changes are shown. Asterisks (*) mark the amino acids whose side chains are participants in the HTH zipper of hydrophobic core of the scaffolding protein, and daggers (†) mark those in the β1-turn.
b Average of two or more determinations as described under “Experimental Procedures.”
c Mutations were constructed in a P22 15−ΔSC302::KanR, 13−amH101 prophage (except UB-1968, UB-1970–2, UB-1790, UB-1901, and UB-3018, which were in P22 ΔsieA-1, 15−ΔSC302::KanR, 13−amH101) as described under “Experimental Procedures.”
Scaffolding Proteins with Altered Hydrophobic Zipper or β1-Turn Are Defective in Vivo
Mutant P22 prophages in S. enterica serovar Typhimurium LT2 encoding scaffolding proteins with single amino acid changes that alter the hydrophobic zipper core or β1-turn were constructed and induced to lytic growth as described under “Experimental Procedures.” In addition to the scaffolding gene mutations, the prophage genotype was either 15−ΔSC302::KanR, 13−amH101 or ΔSieA-1, 15−ΔSC302::KanR, 13−amH101. These phages are both fully capable of lytic growth, and the genotypes were chosen for technical ease of mutation construction and control of lysis (see “Experimental Procedures”). Placing the scaffolding changes in prophages allows analysis of the phenotypes of mutations that are lethal to lytic growth. The numbers of viable phage particles produced by these inductions are shown in Table 2. Most of the scaffolding protein changes in the hydrophobic zipper core (I276D, A283D, Y292D, L295D, and L299D) gave less than 0.1% the normal WT phage yield, indicating that they drastically reduce overall scaffolding function. Among the zipper core mutants, only M280D gave normal yields of phage progeny; however, infectious particle production was slowed by the M280D mutation (data not shown). We conclude that the zipper core is quite sensitive to amino acid changes, much more so than the surface of the HTH domain (49) and so the proper fold of this domain is likely important in scaffolding protein function. We also tested N272D and I302D mutants, which are near the distal ends of the HTH, and, as is indicated in Table 2, their in vivo virion production was not impaired.
The in vivo functionality of scaffolding proteins with changes in the β1-turn (residues 284–289) between the two helices was also tested. We previously showed that scaffolding proteins with charge alteration mutations K286E, D288R, and T291D are fully functional (49), and we show here that S285D and G287A proteins are also functional (Table 2). On the other hand, A284D, G287P, G287D, and V289D mutants gave low phage yields, indicating that these scaffolding proteins have impaired function. Thus, some but not all changes in the turn inactivate scaffolding protein.
To understand the nature of the defect in the above mutants in more detail, prophages with the defective A283D, A284D, G287D, V289D, Y292D, and L295D scaffolding proteins were induced to lytic growth and large phage-related protein assemblies were isolated and analyzed by agarose gel and SDS-PAGE electrophoresis (see “Experimental Procedures”). The phage-related particles from all six inductions included the accumulation of a band in agarose gels that co-migrates with authentic procapsids and procapsids from which scaffolding protein has been removed and a much less intense, faster migrating band (data not shown). Both of these bands contain coat protein but no scaffolding protein (data not shown), and we interpret them to be empty T = 7 and T = 4 shells, respectively. An infection by gene 2 minus P22 phage is blocked in DNA packaging and accumulates procapsids, and particles from P22 2− amN16 (nonsense in codon 193 of gene 2 (30)) induction gave a band, on the agarose gel that was two to three times as intense as band corresponding to T = 7 sized particles produced by the scaffolding protein mutants. As expected, these 2− procapsids contain both coat and scaffolding proteins (data not shown). Infections with scaffolding protein null mutations result in 40–45% of the coat protein being assembled into empty T = 7 spherical PC-sized shells (35); the rest accumulates in larger spiral structures and a smaller number of T = 4 shells. Thus, the particle types and amounts produced by the defective scaffolding protein point mutant phages are similar to those expected from a scaffold null mutant. We suggest that this indicates that these mutant scaffolding proteins are unable to interact with coat protein in vivo. To understand these defects in more detail we pursued the analysis of these mutant proteins in vitro as described below.
In Vitro Procapsid Assembly with Mutant Scaffolding Proteins
Experimental assembly conditions are much more easily controlled in vitro and have proven to be sensitive to subtle differences in the ability of mutant scaffolding protein to interact with coat protein (49, 51). Therefore, we purified WT and mutant histidine-tagged scaffolding proteins as described under “Experimental Procedures” and studied their ability to assemble in vitro with coat protein to form “procapsids.” These procapsid-like particles, which have the correct shape and size, and contain coat and scaffolding proteins in the amounts expected for authentic procapsids, do not contain portal or ejection proteins; however, we refer to them as procapsids or PCs for ease of discussion. PC assembly reactions were performed by mixing coat protein and scaffolding protein at concentrations and conditions previously found to be optimal (49, 51). Assembly competence was monitored using two assays (details under “Experimental Procedures”). In the first assay, the kinetics of PC assembly at 45 mm NaCl (see below) were followed by light scattering at 500 nm (44, 66) (Fig. 2a). With light scattering, the activity of the scaffolding variants that affect nucleation of PC assembly can be assessed in a short duration experiment; however, this assay not does reveal if variants are able to nucleate slowly and form PCs when the reaction is allowed to approach its end point or whether the structures of the product are correctly formed. The second assay is an end point measurement in which the relative affinity of different scaffolding proteins for monomeric coat protein during assembly is monitored using salt titration (42) (Fig. 3). In this assay, coat protein and scaffolding protein are mixed with increasing concentrations of NaCl and incubated at room temperature for 2 h. Because the interaction between the proteins is largely electrostatic, assembly is sensitive to the ionic conditions of the solution. In low NaCl, the affinity between coat and scaffolding proteins is strong, leading to the nucleation of many particles, which ultimately leads to exhaustion of coat protein and formation of partial capsids. Partial procapsids appear as bowl-like structures in electron micrographs, depending on the orientation of the structure on the grid (42). As the NaCl concentration in the reaction is increased, the strength of the electrostatic interaction decreases. Fewer nuclei are formed and reactants are not exhausted, so that complete PCs form (42). Thus, the relative affinity of the scaffolding and coat protein interaction can be indirectly assessed by comparing the concentrations of NaCl required to shift the assembly reaction from production of partial capsids to fully formed PCs; a higher required NaCl concentration indicates tighter binding (42).
FIGURE 2.

Kinetic analysis of in vitro procapsid assembly. a, monomeric coat protein (0.5 mg/ml) was mixed with scaffolding protein variants (0.5 mg/ml) in buffer. The progress of procapsid polymerization was monitored by light scattering. Reactions for each of the scaffolding protein mutants are shown as follows: (i) mutations in helix 1, (ii) mutations in the β1-turn and its periphery, and (iii) mutations in helix 2. b, micrographs of selected in vitro assembled procapsids made with different scaffolding protein variants in 45 mm NaCl as follows: (i) WT scaffolding protein, (ii) N272D, (iii) M280D, and (iv) T291D scaffolding proteins. No partial procapsids are readily evident in these micrographs because normal PCs are generated in 45 mm NaCl. The arrows in b (iii) point to spiral structures. The scale bar represents 100 nm. All micrographs were taken at ×68,000 magnification.
FIGURE 3.

Salt titration assay to assess coat binding affinity of mutant scaffolding proteins. Each panel shows a native agarose electrophoresis gel of an in vitro procapsid assembly reaction. The gels are run with the cathode (+) at the bottom. Samples were prepared by mixing monomeric coat protein (0.5 mg/ml) and scaffolding protein (0.5 mg/ml) at increasing NaCl concentrations from left to right (0, 5, 15, 30, 45, 60, 75, and 100 mm) as indicated by the triangle above the gel. The reactions were incubated for 2 h at room temperature. The indicated labels above the gels are lanes with only monomeric coat protein (MC), in vivo assembled procapsids (PC), and each mutant scaffolding protein (s). The small amount of material that migrates to the position of PC in the assembly reactions with A283D, A284D, G287D, Y292D, or L295D scaffolding proteins is due to uncontrolled assembly of coat protein, and is not generated by scaffolding protein mediated assembly.
The in vitro assembly kinetics of helix 1 zipper core mutants N272D, I276D, M280D, and A283D were assessed over 16 min using light scattering (Fig. 2a). All four are less functional than the WT scaffolding protein and show a progressive decrease in the rate of assembly as the substitution approaches the turn (Fig. 2a). Only scaffolding protein N272D assembled at more than 50% the WT rate. The A283D variant protein did not assemble PCs at all by light scattering. The relative affinity of scaffolding proteins N272D, I276D, M280D, and A283D, measured by the salt titration assay, also weakens as the substitution moves toward the turn, with only N272D scaffolding protein having an affinity similar to WT scaffolding protein (Fig. 3). When the assembly reactions were allowed to proceed to their end point, PC-like products were observed for variants I276D and M280D (Fig. 3); however, for these two mutants the amount of product is decreased, which is consistent with the results of the kinetic assay (Fig. 2a) and indicate that these substitutions affect the ability of scaffolding protein to catalyze assembly. The A283D variant shows no kinetics of assembly, and in the end point assay, the material observed in the position of PC is due to uncontrolled assembly of coat protein. A small amount of uncontrolled assembly of coat protein always occurs during incubation in assembly conditions (67).
Electron micrographs (EMs) of N272D assembled particles show mostly properly assembled PCs similar to those made with WT scaffolding protein (Fig. 2b). On the other hand, the assembly products observed in the salt titration of M280D (at NaCl concentrations: 0, 5, 15, and 30 mm) showed production of fully formed PCs and partial or “half” procapsids (Fig. 3), and EMs of the assembly reaction of M280D with 45 mm NaCl show a mixture of what appear to be proper PCs and a higher proportion of spiral structures (capsids that do not close so the coat protein polymer forms spiral shapes) than the other mutant proteins (Fig. 2b). These results suggest that the M280D protein binds coat protein rather weakly and that this substitution may cause a moderate defect in placement of the 5-fold vertices during coat protein polymerization and likely is the reason for its delay in in vivo assembly (35).
The assembly kinetics of helix 2 zipper core mutants Y292D, L295D, and L299D also show a strong negative effect on function (Fig. 2a and Table 3). The Y292D and L299D scaffolding protein have similar light scattering intensity curves. The Y292D scaffolding protein ultimately is incapable of supporting assembly. On the other hand, the L299D mutant was able to assemble PCs with a reasonable yield at the reaction end point (Fig. 3). The L295D scaffolding protein was incapable of assembly even at long incubation times. These results strongly support the notion that the hydrophobic core of the HTH is very important for productive interaction with coat protein.
TABLE 3.
Properties of altered P22 scaffolding proteins
| Mutanta | In vivo functionalityb | Assembly kineticsc | Affinity by salt titrationd | Shell refillinge | HTH foldedf |
|---|---|---|---|---|---|
| Wild type | + | Fast | Strong | + | Yes |
| N272D | + | Med | Med | + | Yes |
| I276D | − | Slow | Med | + | No |
| M280D | + | Slow | Weak | − | No |
| A283D | − | Very slow | None | − | No |
| A284D | − | Very slow | None | − | Yes |
| G287D | − | Slow | None | − | Yes |
| V289D | − | Slow | Weak | − | Yes |
| T291D | + | Med | Strong | + | Yes |
| Y292D | − | Slow | None | − | No |
| L295D | − | Very slow | None | − | No |
| L299D | − | Slow | Med | +/− | No |
| N272D, I267R, L299D | − | Very slow | Med | +/− | No |
a Scaffolding protein changes are shown.
b “−“ denotes ≤1% wild-type phage yield; summarized from Table 2.
c Assembly over the first 16 min after mixing scaffolding and coat proteins (Fig. 2a). Assembly at >40% WT rate is called medium (“med”); assembly at 5–40% the WT rate is called “slow”; and <5% is called “very slow.”
d Relative scaffolding protein affinity for coat protein deduced from the concentration of NaCl required to shift the assembly reaction from production of partial capsids to fully formed PCs (Figs. 3 and 7b).
e Ability of scaffolding protein to enter and bind to coat protein within emptied procapsid shells (Figs. 4 and 7c).
f Summarized from CD spectra of Fig. 5.
As mentioned above, amino acid substitutions in the β1-turn between the two HTH helices gave mixed in vivo results, with charge change alterations S285D, K286E, and D288R allowing plaque formation and substitutions A284D, G287D, and V289D blocking plaque formation. The A284D, G287D, and V289D proteins also do not cause significant PC assembly in the kinetic assay (Fig. 2a). However, when allowed to reach end point, V289D gave a limited yield of PCs. We also examined the in vitro assembly of the T291D scaffolding protein, even though it is sufficiently functional to allow plaque formation in vivo. It gave a moderate rate of assembly in 16 min, albeit with somewhat slower kinetics than the WT scaffolding protein. EMs show that this variant assembles apparently proper PCs (Fig. 2b). The T291D scaffolding protein has a similar relative affinity for coat protein as WT scaffolding protein in the salt titration assembly assay (Fig. 3). The in vitro assembly results are summarized in Table 3, and overall they indicate that, in addition to the two previously identified critical surface amino acids (49), proper formation of the HTH fold is important for scaffolding protein function.
Binding of Scaffolding Protein Variants to Procapsid Shells
Because the ability of most of the scaffolding protein zipper core variants to catalyze coat protein polymerization was less robust than WT scaffolding protein, we asked if this effect was due to elimination of interaction with coat protein, or if interaction is possible but nucleation of assembly was specifically affected. The empty PC shell refilling assay of Greene et al. (68) measures binding to pre-assembled, unexpanded T = 7 coat protein shells and so can identify scaffolding protein variants that are able bind to coat protein whether or not they can nucleate PC assembly. In this reaction scaffolding protein spontaneously re-enters the shell, presumably through the holes in the hexon centers (24, 25) and binds to purified PC shells from which scaffolding protein has been removed by guanidine HCl treatment.
Empty PC shells were prepared, and the ability of mutant scaffolding proteins to bind to them was measured as described under “Experimental Procedures.” The refilled PCs were purified by centrifugation through a 20% sucrose cushion to separate them from free unassembled scaffolding protein. The pelleted material was resuspended in sample buffer and the proteins present were analyzed by SDS-PAGE. The presence of scaffolding protein in the pelleted shells indicates the ability to interact with the assembled coat protein shell (69). Scaffolding protein does not pellet in the conditions used here (Fig. 4, lane WT − shells). WT, N272D, I276D, T291D, and L299D scaffolding proteins were able to bind to the interior of shells (Fig. 4). About the same amount of N272D and T291D variants bound to coat protein shells as the WT scaffolding protein (119 and 93%, respectively). The I276D and L299D mutant proteins were able to bind to coat protein shells, but with reduced efficiency of 67 and 61%, respectively. Among the remaining mutant scaffolding proteins M280D, A283D, G287D, and L295D bound shells very weakly (<15%) and no binding was detected for the A284D, V289D, and Y292D proteins. These results show that assembly kinetics and coat binding activity are correlated and none of the mutants specifically affects only nucleation or coat binding, emphasizing the essential role of the hydrophobic core of the HTH domain turn in the scaffolding protein binding to coat protein.
FIGURE 4.
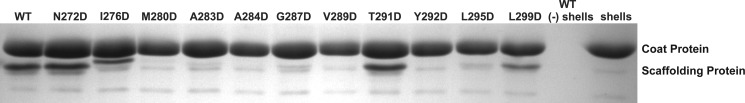
Refilling of empty coat protein shells with mutant P22 scaffolding proteins. A mixture of 0.33 mg/ml of empty coat protein shells, 0.2 mg/ml of scaffolding protein, and buffer were incubated overnight at room temperature. The reactions were layered on 20% sucrose cushions and centrifuged to remove unbound scaffolding protein; the proteins in the pellet, which contained the refilled shells, were visualized with 12.5% SDS-PAGE. The lanes are labeled with the scaffolding protein used in the assay. The lane labeled WT (−) shells shows scaffolding protein incubated in buffer with no shells added does not pellet and the lane labeled shells shows the pelleting of the shells without added scaffolding protein.
Secondary Structure C Terminus Helix Turn Helix Domain Variants
Our data thus far show that substitutions in the core and the turn of the coat binding HTH domain strongly affect the PC assembly activity of the scaffolding protein. We hypothesize that this is due to disruption of the secondary structure of this domain. Because scaffolding protein is a highly helical protein, subtle changes to the structure of the HTH are unlikely to be observed in the context of the entire 303-amino acid protein. Therefore, we generated a series of plasmid constructs that express the C-terminal 66-residue fragment of scaffolding protein (238–303), carry various HTH amino acid changes, and have an N-terminal His6 tag (6Xhis-Δ1–237). In these constructs, residue Thr-265 (outside the HTH) is replaced by a tryptophan to allow protein quantification by absorbance at 280 nm. This smaller fragment, which has coat protein assembly nucleation activity (50), allows specific detection of the secondary structure of the HTH coat-binding domain.
Circular dichroism spectroscopy (CD) was used to compare the secondary structure of the various mutated 6Xhis-Δ1–237 proteins in solution. In the far UV, CD spectra of highly helical domains have double minima at 222 and 208 nm as well as a positive signal near 193 nm, whereas random coil proteins have low ellipticity above 210 nm and a strong negative band near 195 nm (70). The CD spectra of WT-6Xhis-Δ1–237 and 6Xhis-Δ1–237/T265W fragments were similar, and were consistent with these peptides having both α-helical (negative peak at ∼222 nm) and random coil character (negative peak at ∼200 nm) (Fig. 5a). The NMR structure of this fragment shows amino acids 269–303 folded into the HTH domain and amino acids 238–268 as random coil (50). In addition, the T265W substitution does not have a significant effect on the secondary structure. Among the truncated proteins with amino acid changes in helix 1, only the N272D variant, which has a WT-like assembly activity in the full-length construct, shows a WT-like far-UV CD spectrum (Fig. 5a). The other substitutions in helix 1 (I276D, M280D, and A283D) each cause the spectrum to shift significantly toward random coil, with a deep minimum at about 200 nm. All three of the aspartic acid substitutions in the hydrophobic core in helix 2 (Tyr-292, Leu-295, and Leu-299) cause significant unfolding of the HTH, giving a strong negative signal at ∼200 nm (Fig. 5c). The replacement of leucines 295 or 299 with aspartic acid also gives a very strong random coil CD signal. These changes, like the zipper substitutions in helix 1, likely result in a solvation penalty that destabilizes the interaction between the two helices. Substitution of those residues adjacent to or within the turn of the HTH that inactivate the ability of the scaffolding protein to bind coat (A284D, G287D, V289D) did not alter the helical structure relative to the WT-6Xhis-Δ1–237 protein (Fig. 5b), and T291D retained a secondary structure similar to WT, as is expected because it is nearly WT in function in the full-length context (Fig. 5c). Because other charge change substitutions in or near the turn (S285D, K286E, D288R, E290K) do not affect activity, these data indicate that it is not likely the surface charge that is important. Rather, we suggest that some changes in the turn at positions Ala-284, Gly-287, and Val-289 affect the relative orientation of the helices so that they no longer interact well with the coat protein. None of the full-length scaffolding protein mutants studied showed changes in their CD spectra compared with WT protein (data not shown), suggesting that structural changes in these mutants are restricted to the C-terminal domain and do not significantly affect the fold of the whole molecule.
FIGURE 5.

Circular dichroism spectra of mutant scaffolding proteins. The secondary structure composition of each variant was determined at a protein concentration of 0.5 mg/ml. The panels show CD spectra of the indicated aspartic acid-substituted 6Xhis-Δ1–237 truncated scaffolding proteins as follows: (a) in helix 1, (b) in and adjacent to the β1-turn, and (c) in helix 2 along with the WT 6Xhis-Δ1–237 protein.
Thermal Denaturation of 6Xhis-Δ1–237/T265W Variant Proteins
Some of the 6Xhis-Δ1–237 fragment variants, primarily those at the turn, had a secondary structure similar to WT as determined by CD analysis, but no assembly activity. To understand the effect of the substitutions on the stability of the HTH, we determined their thermal stabilities by measuring CD ellipticity at 222 nm as a function of temperature. A decrease in negative ellipticity at 222 nm indicates unfolding of the helices of the HTH. The hydrophobic core substitutions in both helix 1 and 2 (Ile-276, Met-280, Leu-295, Leu-299) exhibited a low signal at 222 nm and negligible change in that signal, from 5 to 80 °C, indicative of a highly unfolded initial state (Fig. 6, a and c). The negligible change in the signal signifies that the hydrophobic residues between helices 1 and 2 do not interact and that these helices are mostly random coil in these mutant proteins. At the beginning of helix 1, the N272D fragment is shifted to a slightly higher melting point than WT, indicating that N272D has a somewhat more stable structure (Fig. 6a). Variants V289D, A284D, G287D, and T291D (in or near the turn) all have a have slightly lower melting point than the WT fragment (Fig. 6, b and c), suggesting that replacement with aspartic acid in this region only moderately destabilizes the HTH domain. Thus, the assembly defect in scaffolding proteins with turn substitutions is not due to a lowered HTH stability.
FIGURE 6.

Thermal stability of mutant scaffolding protein HTH domains. The CD signal of 6Xhis-Δ1–237 scaffolding protein variants at 222 nm was monitored with increasing temperature. The samples contained 0.5 mg/ml of protein in 10 mm Na2HPO4 buffer. Denaturation curves for proteins with substitutions in (a) helix 1, (b) in and adjacent to the β1-turn, and (c) helix 2 are compared with the WT 6Xhis-Δ1–237 scaffolding protein.
Salt Bridge Replacement of Hydrophobic Interactions
In a previous study, we showed that replacement of the arginine at position 303 with alanine affected shell refilling, decreasing it by about 50% compared with WT scaffolding protein (49). There is a plausible salt bridge between Asn-272 and Arg-303 that could contribute to the stability of the C-terminal HTH. Indeed, when Asn-272 is changed to an aspartic acid, a small increase in the stability of the 6Xhis-Δ1–237 fragment was observed (Fig. 6a). Thus, we hypothesized that replacement of a hydrophobic pair in the core with a salt bridge might not have a helix destabilizing effect. A variant with three charged substitutions (N272D/I276R/L299D) was generated in the full-length histidine-tagged scaffolding protein to determine whether replacement of hydrophobic interactions with salt bridges could maintain the structure of the C-terminal HTH and its activity. This construct might, in theory, form two salt bridges: N272D:R303 and I276R:L299D. These substitutions were chosen because the orientations of the WT residue side chains seem sterically favorable for ionic interactions. No in vitro PC assembly was observed with the triple mutant protein in the kinetic assay (Fig. 7a). Salt titration end point assembly experiments showed that the affinity of the triple mutant scaffolding protein is weaker than WT, and it produced some procapsids but fewer than WT (Fig. 7a). The shell refilling assay of this variant showed a much lower binding affinity than WT (Fig. 7a). CD experiments with the 6Xhis-Δ1–237 triple mutant showed a highly unfolded protein (Fig. 7b), and in vivo, this triple mutant was unable to produce functional virions (Table 2). These data indicate that introduction of the potential I276R/L299D salt bridge does not compensate for the destabilization of the HTH when an aspartic acid is introduced at residue Leu-299.
FIGURE 7.

Replacement of hydrophobic interactions in the HTH core with salt bridges. a, activity assay of full-length N272D/I276R/L299D scaffolding protein. (i) the effect of the N272D/I276R/L299D mutations on the in vitro assembly as measured by light scattering. Samples of 0.5 mg/ml (— —) WT or (solid line) N272D/I276R/L299D scaffolding proteins were mixed with 0.5 mg/ml of coat protein and monitored for 16 min. As negative control, assembly of 0.5 mg/ml of the (— —) coat protein with no scaffolding protein was also measured. (ii) salt titration of PC assembly at various concentrations of NaCl (0, 5, 15, 30, 45, 60, 75, and 100 mm). Lanes with monomeric coat protein (MC), in vivo assembled procapsids (PC), and each scaffolding protein are indicated. (iii) shell refilling assay of the ability of scaffolding protein to bind empty procapsid shells. Solutions of 0.33 mg/ml of procapsid shells and 0.22 mg/ml of scaffolding protein variants were incubated overnight at room temperature. Refilled procapsids were purified by centrifugation through a 20% sucrose cushion and the proteins in the pellet were visualized with 12.5% SDS-PAGE. b, CD measurements of WT and N272D/I276R/L299D mutant 6Xhis-Δ1–237/T265W scaffolding proteins fragments: (i) CD spectra of 0.5 mg/ml of WT (squares) and N272D/I276R/L299D (circles) fragments. (ii) Measurement of molar ellipticity signal at 222 nm with increasing temperature showing WT (squares) and N272D/I276R/L299D (circles).
DISCUSSION
The Structure of the Scaffolding Protein C-terminal HTH Is Maintained by Its Hydrophobic Zipper
We previously showed that only two surface residues, Arg-293 and Lys-296, of the P22 scaffolding protein HTH coat-binding domain are critical to coat protein binding. In this report we sought to understand why the scaffolding protein needs the entire HTH domain for this interaction if only two surface amino acids are required. Table 3 and Fig. 8 summarize the properties of the mutant scaffolding proteins with changes in the zipper core and turn of the HTH that were studied in this report. Changes just outside the bottom of the zipper (N272D and I302D, green in Fig. 8) did not block the in vivo scaffolding protein function. The N272D protein was analyzed and found to contain the HTH fold by CD analysis, to be fully functional in the coat binding (shell refilling) assay, and to be only moderately negatively affected in its ability to cause PC assembly. On the other hand changing any of the HTH hydrophobic zipper residues (Ile-276, Met-280, Ala-283, Tyr-292, Leu-295, or Leu-299, yellow in Fig. 8) to a hydrophilic aspartate cause a strong decrease in all the in vitro activities we measured. The strong decrease in the measured HTH secondary structure with aspartic acid substitutions of any of the zipper residues indicates that the hydrophobic interactions among these side chains are indeed central to maintaining the structure and relative orientation of the two helices. Very severe defects were found when changes were made in the zipper residues at the top and center of the HTH helices (Ala-283, Y292D, and L295D), and more moderate defects were present when changes were made in the lower portion of the helices (I276D and L299D). These data demonstrate that single amino acid perturbations in helices 1 and 2 can significantly destabilize the whole HTH domain, and thereby abolish the coat binding capability of scaffolding protein. The fact that proteins with alterations in the lower part of the HTH retain weak function suggests that maintenance of the HTH structure is most critical at the top and middle, and that perhaps “fraying” near the bottom is weakly tolerated. Our previous observations showed that the most important coat-binding residues are surface residues Arg-293 and Lys-296 (49) (red in Fig. 8), which also reside on the upper and middle parts of the HTH. This agrees well with the conclusion reached here that the structure of this part of the HTH is most important in maintaining coat-binding ability.
FIGURE 8.

Scaffolding protein amino acid residues important for coat protein interaction. Opposite side views of the coat protein-binding HTH domain of scaffolding protein showing the informative residues analyzed in this report as space-filling spheres on a ribbon diagram frame (the two important surface residues, red; the three important turn residues, blue; and important zipper core residues, yellow; nonessential residues are at the bottom of HTH, green (figure created with PyMOL)).
Scaffolding protein mutant M280D is unusual compared with the other altered proteins studied here. It is located near the top of helix 1, but unlike the other HTH core zipper mutants, it still catalyzes assembly of PCs in vivo because normal numbers of phage particles are slowly produced (Table 2). It similarly causes assembly of PCs in vitro, albeit less efficiently than WT scaffolding protein (Figs. 2 and 3). However, CD analysis showed a substantial decrease in helical structure in the M280D protein even though methionine is a strong helix former (71, 72). The puzzle is why the M280D scaffolding protein is unable to bind empty PC shells. Helix 1 does not have any major outwardly facing residue that is required for interaction with coat protein (49). Helix 2 probably retains some of its helical character, allowing limited interaction of the M280D scaffolding protein with the monomeric coat protein, which can still be induced to undergo conformational switching during assembly. We hypothesize that scaffolding protein M280D is unable to interact with empty PC shells because the scaffolding protein-binding site is in a fixed conformation that can no longer accommodate the structural alterations in scaffolding protein caused by this amino acid substitution. This result might suggest that the scaffolding protein-binding region of monomeric coat has some flexibility that could accommodate a moderate change in scaffolding protein HTH structure at this location, but the binding region in polymerized coat protein may be more rigid.
β1-Turn Size and Shape Are Important for Coat Interactions
Substitutions within the turn (A284D, G287D, and V289D, blue in Fig. 8) strongly affect the interaction of the scaffolding protein with coat protein. However, CD spectra of these three mutant proteins show HTH secondary structural features that are similar to WT protein, although the helix structures are slightly less thermally stable in the mutants. There are several possible explanations for this finding that include the following. (i) Because Ala-284 and Gly-287 contribute to the hydrogen bond that pulls the turn into a tight conformation, changes in these residues could destabilize the interaction of the two helices and/or change the shape of the turn and angle between the two helices. Additionally, a bulkier amino acid (aspartic acid in this case) could affect the angle of the turn due to the limit of rotamer preferences caused by steric hindrance with neighboring residues. Changes in the turn region to amino acids with bulkier side chains might sterically hinder the binding to coat protein. (ii) Val-289 could make a specific hydrophobic contact with coat protein that is required for this interaction (50). The V289D variant can weakly nucleate assembly of coat proteins and binding to procapsid shells is completely abolished. However, the mutant V289A is able to generate procapsids in vivo (Table 2), even though its in vitro assembly is slow.6 Because Val-289 is located at the base of the β1-turn and N terminus of helix 2, the volume of the tip the C-terminal HTH is altered with the substitution of a less bulky residue. (iii) The introduction of the additional negative charge on the surface might reduce the ability of scaffolding protein to interact with coat protein. Analysis of the residues at the turn in the HTH NMR structure shows that substitutions at or near the turn are unlikely to introduce a negative charge on the face of the helix where the main residues required for interaction with coat protein (Arg-293 and Lys-296) are located (49). Additionally, mutation of Ser-285 to aspartic acid (Table 2) or Lys-286 to glutamic acid (49) results in normal phage production, suggesting that introduction of a negative charge in this region does not have a significant detrimental effect. Thus, this last possibility seems unlikely to be the reason for the assembly defect of the V289D scaffolding protein. We favor the first hypothesis, which suggests that the coat protein of the scaffolding protein interaction domain must have very specific dimensions, shape, and relative helix orientation to function in PC assembly.
Residues Outside of the Hydrophobic Zipper Are Functional
Amino acid changes at the base of the HTH do not affect scaffolding protein assembly activity nearly as dramatically as changes within the hydrophobic core or turn, and the N272D variant actually shows improved HTH stability (Fig. 6a). The increased stability could be caused by the more efficient N-terminal helix capping by aspartic acid than by asparagine, leading to a more stable helix 1 (73). The rate of assembly of the full-length N272D protein is slower but it ultimately assembles nearly as well as WT scaffolding protein. An aspartic acid substitution at Ile-302, which is positioned at the opening of the HTH like Asn-272, is also fully functional in vivo (but was not analyzed in vitro). Thus, only the residues located within the HTH region significantly affect scaffolding protein function.
Because changing the bottom ends of the HTH helices does not greatly affect assembly, we hypothesized that the negative effect caused by substitution of amino acids in the core could be overcome with salt bridges between the end of the HTH plus a salt bridge in the center of the core. However, assembly activity is not recovered by replacement of one hydrophobic pair (Ile-276/Leu-299) with salt bridge network I276R:L299D and R303:N272D. This result is not very surprising because other studies have shown that salt bridges are usually weaker and less specific than hydrophobic interactions (74). Our observation that N272D increases the helical structure but decreases assembly activity suggests that the precise twist of the paired helices is important and that helix 2 has to be fixed in a certain orientation for proper binding to coat protein.
Studies on folding defects of mutant coat proteins (S223F and F353L) have shown that a suppressor of these mutations (T166I) appears to increase the binding of coat protein to scaffolding protein, thereby allowing proper PC assembly (75, 76). Moreover, folding defects of the coat protein mutants can be rescued by the presence of the GroE folding chaperone system (77–79). Only when these mutant coat proteins are properly folded does scaffolding protein bind them and proceed with assembly. These studies correlate with our conclusion that the scaffolding protein binding pocket of coat protein not only requires proper ionic interactions but also a specific shape.
Scaffolding/Coat Protein Interaction during Procapsid Assembly
Ionic interactions between scaffolding protein and coat protein have been identified as the major driving force for assembly of P22 PCs (42, 43, 67). These interactions are presumably rather weak to allow for easy dissociation upon PC shell maturation and may only require opposite charges interacting at specific distances. Scaffolding/coat protein interaction is mediated by the C-terminal HTH domain of scaffolding protein shown in Figs. 1 and 8. Residues Arg-293 and Lys-296, which are on the face of HTH helix 2, are the principal surface residues required for coat binding and procapsid assembly (49). The other charged residues on the surface of the C-terminal HTH domain modulate the scaffolding/coat protein interaction but even in aggregate are not essential (49). Interestingly most of the HTH zipper hydrophobic core substitutions studied here were found to have very strong effects on the binding of scaffolding protein to coat protein. We conclude from the analysis of these mutant scaffolding proteins that steric restrictions on the scaffolding protein-binding region are imposed by the coat protein, and these require a precisely folded scaffolding protein HTH domain and in so doing impart specificity on the interaction.
The scaffolding protein-binding site in monomeric coat protein of bacteriophage P22 (and all other such viruses) has not been identified. Structural studies of PCs with and without scaffolding protein have indicated scaffolding protein contact points near the strict and local 3-fold axes of the coat protein shell (the positions where three hexons or pentons meet) in these structures (25, 80, 81). According to current P22 coat protein models (25, 29, 82), this region likely corresponds largely to the N-terminal part of the protein. We have speculated from these models that negatively charged coat protein amino acids Asp-14, Glu-15, Glu-18, Glu-97, and/or possibly Asp-385 may be involved in the interaction with the positively charged Arg-293 and Lys-296 of the scaffolding protein HTH domain (49), and this idea fits well with the observation that this region undergoes a conformational change during PC maturation so that these amino acids are no longer at the interior surface (25, 29). However, in the absence of an atomic level resolution structure of the coat protein in PC shells, the structural details of the scaffolding protein binding site on coat protein will remain mysterious.
Acknowledgments
We thank Kelly Hughes for Salmonella strains and Dr. Marie Cantino for guidance and use of the University of Connecticut Electron Microscopy Center facility.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM076661 (to C. M. T.) and R01 AI074825 (to S. R. C.).
G. P. Padilla-Meier, unpublished data.
- HTH
- helix turn helix
- PC
- procapsids.
REFERENCES
- 1. Dokland T. (1999) Scaffolding proteins and their role in virus assembly. Cell Mol. Life Sci. 56, 580–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. King J., Casjens S. (1974) Catalytic head assembling protein in virus morphogenesis. Nature 251, 112–119 [DOI] [PubMed] [Google Scholar]
- 3. Prevelige P. E., Fane B. A. (2012) Building the machines. Scaffolding protein functions during bacteriophage morphogenesis. Adv. Exp. Med. Biol. 726, 325–350 [DOI] [PubMed] [Google Scholar]
- 4. Chang J. R., Spilman M. S., Rodenburg C. M., Dokland T. (2009) Functional domains of the bacteriophage P2 scaffolding protein. Identification of residues involved in assembly and protease activity. Virology 384, 144–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rishovd S., Marvik O. J., Jacobsen E., Lindqvist B. H. (1994) Bacteriophage P2 and P4 morphogenesis. Identification and characterization of the portal protein. Virology 200, 744–751 [DOI] [PubMed] [Google Scholar]
- 6. Black L. W., Showe M., Steven A. C. (1994) in Molecular Biology of Bacteriophage T4 (Karam J., ed) pp. 218–258, ASM Press, Washington, D. C [Google Scholar]
- 7. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 8. Medina E., Wieczorek D., Medina E. M., Yang Q., Feiss M., Catalano C. E. (2010) Assembly and maturation of the bacteriophage λ procapsid. GpC is the viral protease. J. Mol. Biol. 401, 813–830 [DOI] [PubMed] [Google Scholar]
- 9. Ray P., Murialdo H. (1975) The role of gene Nu3 in bacteriophage λ head morphogenesis. Virology 64, 247–263 [DOI] [PubMed] [Google Scholar]
- 10. Baines J., Weller S. (2003) in Viral Genome Packaging Machines (Catalano C. E., ed) pp. 135–150, Landes Bioscience, Georgetown, TX [Google Scholar]
- 11. Liu F. Y., Roizman B. (1991) The herpes simplex virus 1 gene encoding a protease also contains within its coding domain the gene encoding the more abundant substrate. J. Virol. 65, 5149–5156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gibson W. (2008) Structure and formation of the cytomegalovirus virion. Curr. Top. Microbiol. Immunol. 325, 187–204 [DOI] [PubMed] [Google Scholar]
- 13. Welch A. R., Woods A. S., McNally L. M., Cotter R. J., Gibson W. (1991) A herpesvirus maturational proteinase, assemblin. Identification of its gene, putative active site domain, and cleavage site. Proc. Natl. Acad. Sci. U.S.A. 88, 10792–10796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Casjens S., King J. (1974) P22 morphogenesis. I, Catalytic scaffolding protein in capsid assembly. J. Supramol. Struct. 2, 202–224 [DOI] [PubMed] [Google Scholar]
- 15. Cerritelli M. E., Studier F. W. (1996) Purification and characterization of T7 head-tail connectors expressed from the cloned gene. J. Mol. Biol. 258, 299–307 [DOI] [PubMed] [Google Scholar]
- 16. Bjornsti M. A., Reilly B. E., Anderson D. L. (1983) Morphogenesis of bacteriophage φ29 of Bacillus subtilis. Oriented and quantized in vitro packaging of DNA protein gp3. J. Virol. 45, 383–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dokland T., McKenna R., Ilag L. L., Bowman B. R., Incardona N. L., Fane B. A., Rossmann M. G. (1997) Structure of a viral procapsid with molecular scaffolding. Nature 389, 308–313 [DOI] [PubMed] [Google Scholar]
- 18. Huet A., Conway J. F., Letellier L., Boulanger P. (2010) In vitro assembly of the T = 13 procapsid of bacteriophage T5 with its scaffolding domain. J. Virol. 84, 9350–9358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cherwa J. E., Jr., Uchiyama A., Fane B. A. (2008) Scaffolding proteins altered in the ability to perform a conformational switch confer dominant lethal assembly defects. J. Virol. 82, 5774–5780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Conway J. F., Wikoff W. R., Cheng N., Duda R. L., Hendrix R. W., Johnson J. E., Steven A. C. (2001) Virus maturation involving large subunit rotations and local refolding. Science 292, 744–748 [DOI] [PubMed] [Google Scholar]
- 21. Morais M. C., Fisher M., Kanamaru S., Przybyla L., Burgner J., Fane B. A., Rossmann M. G. (2004) Conformational switching by the scaffolding protein D directs the assembly of bacteriophage φX174. Mol. Cell 15, 991–997 [DOI] [PubMed] [Google Scholar]
- 22. Suhanovsky M. M., Teschke C. M. (2011) Bacteriophage P22 capsid size determination. Roles for the coat protein telokin-like domain and the scaffolding protein amino terminus. Virology 417, 418–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Casjens S. (1979) Molecular organization of the bacteriophage P22 coat protein shell. J. Mol. Biol. 131, 1–14 [DOI] [PubMed] [Google Scholar]
- 24. Prasad B. V., Prevelige P. E., Marietta E., Chen R. O., Thomas D., King J., Chiu W. (1993) Three-dimensional transformation of capsids associated with genome packaging in a bacterial virus. J. Mol. Biol. 231, 65–74 [DOI] [PubMed] [Google Scholar]
- 25. Chen D. H., Baker M. L., Hryc C. F., DiMaio F., Jakana J., Wu W., Dougherty M., Haase-Pettingell C., Schmid M. F., Jiang W., Baker D., King J. A., Chiu W. (2011) Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc. Natl. Acad. Sci. U.S.A. 108, 1355–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Earnshaw W., Casjens S., Harrison S. (1976) Assembly of the head of bacteriophage P22, x-ray diffraction from heads, proheads, and related structures. J. Mol. Biol. 104, 387–410 [DOI] [PubMed] [Google Scholar]
- 27. Zhang Z., Greene B., Thuman-Commike P. A., Jakana J., Prevelige P. E., Jr., King J., Chiu W. (2000) Visualization of the maturation transition in bacteriophage P22 by electron cryomicroscopy. J. Mol. Biol. 297, 615–626 [DOI] [PubMed] [Google Scholar]
- 28. Jiang W., Li Z., Zhang Z., Baker M. L., Prevelige P. E., Jr., Chiu W. (2003) Coat protein fold and maturation transition of bacteriophage P22 seen at subnanometer resolutions. Nat. Struct. Biol. 10, 131–135 [DOI] [PubMed] [Google Scholar]
- 29. Parent K. N., Khayat R., Tu L. H., Suhanovsky M. M., Cortines J. R., Teschke C. M., Johnson J. E., Baker T. S. (2010) P22 coat protein structures reveal a novel mechanism for capsid maturation. Stability without auxiliary proteins or chemical cross-links. Structure 18, 390–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eppler K., Wyckoff E., Goates J., Parr R., Casjens S. (1991) Nucleotide sequence of the bacteriophage P22 genes required for DNA packaging. Virology 183, 519–538 [DOI] [PubMed] [Google Scholar]
- 31. Prevelige P. E., Jr., Thomas D., King J. (1988) Scaffolding protein regulates the polymerization of P22 coat subunits into icosahedral shells in vitro. J. Mol. Biol. 202, 743–757 [DOI] [PubMed] [Google Scholar]
- 32. Bazinet C., King J. (1988) Initiation of P22 procapsid assembly in vivo. J. Mol. Biol. 202, 77–86 [DOI] [PubMed] [Google Scholar]
- 33. Parent K. N., Zlotnick A., Teschke C. M. (2006) Quantitative analysis of multicomponent spherical virus assembly. Scaffolding protein contributes to the global stability of phage P22 procapsids. J. Mol. Biol. 359, 1097–1106 [DOI] [PubMed] [Google Scholar]
- 34. Parker M. H., Brouillette C. G., Prevelige P. E., Jr. (2001) Kinetic and calorimetric evidence for two distinct scaffolding protein binding populations within the bacteriophage P22 procapsid. Biochemistry 40, 8962–8970 [DOI] [PubMed] [Google Scholar]
- 35. Earnshaw W., King J. (1978) Structure of phage P22 coat protein aggregates formed in the absence of the scaffolding protein. J. Mol. Biol. 126, 721–747 [DOI] [PubMed] [Google Scholar]
- 36. King J., Lenk E. V., Botstein D. (1973) Mechanism of head assembly and DNA encapsulation in Salmonella phage P22. II. Morphogenetic pathway. J. Mol. Biol. 80, 697–731 [DOI] [PubMed] [Google Scholar]
- 37. Lenk E., Casjens S., Weeks J., King J. (1975) Intracellular visualization of precursor capsids in phage P22 mutant infected cells. Virology 68, 182–199 [DOI] [PubMed] [Google Scholar]
- 38. Prevelige P. E., Jr., Thomas D., King J., Towse S. A., Thomas G. J., Jr. (1990) Conformational states of the bacteriophage P22 capsid subunit in relation to self-assembly. Biochemistry 29, 5626–5633 [DOI] [PubMed] [Google Scholar]
- 39. Greene B., King J. (1996) Scaffolding mutants identifying domains required for P22 procapsid assembly and maturation. Virology 225, 82–96 [DOI] [PubMed] [Google Scholar]
- 40. Weigele P. R., Sampson L., Winn-Stapley D., Casjens S. R. (2005) Molecular genetics of bacteriophage P22 scaffolding protein's functional domains. J. Mol. Biol. 348, 831–844 [DOI] [PubMed] [Google Scholar]
- 41. Greene B., King J. (1999) In vitro unfolding/refolding of wild type phage P22 scaffolding protein reveals capsid-binding domain. J. Biol. Chem. 274, 16135–16140 [DOI] [PubMed] [Google Scholar]
- 42. Parent K. N., Doyle S. M., Anderson E., Teschke C. M. (2005) Electrostatic interactions govern both nucleation and elongation during phage P22 procapsid assembly. Virology 340, 33–45 [DOI] [PubMed] [Google Scholar]
- 43. Parker M. H., Prevelige P. E. (1998) Electrostatic interactions drive scaffolding/coat protein binding and procapsid maturation in bacteriophage P22. Virology 250, 337–349 [DOI] [PubMed] [Google Scholar]
- 44. Teschke C. M., King J. (1993) Folding of the phage P22 coat protein in vitro. Biochemistry 32, 10839–10847 [DOI] [PubMed] [Google Scholar]
- 45. Parker M. H., Stafford W. F., 3rd, Prevelige P. E., Jr. (1997) Bacteriophage P22 scaffolding protein forms oligomers in solution. J. Mol. Biol. 268, 655–665 [DOI] [PubMed] [Google Scholar]
- 46. Tuma R., Prevelige P. E., Jr., Thomas G. J., Jr. (1996) Structural transitions in the scaffolding and coat proteins of P22 virus during assembly and disassembly. Biochemistry 35, 4619–4627 [DOI] [PubMed] [Google Scholar]
- 47. Tuma R., Parker M. H., Weigele P., Sampson L., Sun Y., Krishna N. R., Casjens S., Thomas G. J., Jr., Prevelige P. E., Jr. (1998) A helical coat protein recognition domain of the bacteriophage P22 scaffolding protein. J. Mol. Biol. 281, 81–94 [DOI] [PubMed] [Google Scholar]
- 48. Parker M. H., Casjens S., Prevelige P. E., Jr. (1998) Functional domains of bacteriophage P22 scaffolding protein. J. Mol. Biol. 281, 69–79 [DOI] [PubMed] [Google Scholar]
- 49. Cortines J. R., Weigele P. R., Gilcrease E. B., Casjens S. R., Teschke C. M. (2011) Decoding bacteriophage P22 assembly. Identification of two charged residues in scaffolding protein responsible for coat protein interaction. Virology 421, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sun Y., Parker M. H., Weigele P., Casjens S., Prevelige P. E., Jr., Krishna N. R. (2000) Structure of the coat protein-binding domain of the scaffolding protein from a double-stranded DNA virus. J. Mol. Biol. 297, 1195–1202 [DOI] [PubMed] [Google Scholar]
- 51. Padilla-Meier G. P., Teschke C. M. (2011) Conformational changes in bacteriophage P22 scaffolding protein induced by interaction with coat protein. J. Mol. Biol. 410, 226–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Adams M. B., Brown H. R., Casjens S. (1985) Bacteriophage P22 tail protein gene expression. J. Virol. 53, 180–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Israel V. (1967) The production of inactive phage P22 particles following induction. Virology 33, 317–322 [DOI] [PubMed] [Google Scholar]
- 54. Schwarz J. J., Berget P. B. (1989) The isolation and sequence of missense and nonsense mutations in the cloned bacteriophage P22 tailspike protein gene. Genetics 121, 635–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Warming S., Costantino N., Court D. L., Jenkins N. A., Copeland N. G. (2005) Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Karlinsey J. E. (2007) λ-Red genetic engineering in Salmonella enterica serovar Typhimurium. Methods Enzymol. 421, 199–209 [DOI] [PubMed] [Google Scholar]
- 57. Parker M. H., Jablonsky M., Casjens S., Sampson L., Krishna N. R., Prevelige P. E., Jr. (1997) Cloning, purification, and preliminary characterization by circular dichroism and NMR of a carboxyl-terminal domain of the bacteriophage P22 scaffolding protein. Protein Sci. 6, 1583–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Botstein D., Waddell C. H., King J. (1973) Mechanism of head assembly and DNA encapsulation in Salmonella phage P22. I. Genes, proteins, structures, and DNA maturation. J. Mol. Biol. 80, 669–695 [DOI] [PubMed] [Google Scholar]
- 59. Winston F., Botstein D., Miller J. H. (1979) Characterization of amber and ochre suppressors in Salmonella typhimurium. J. Bacteriol. 137, 433–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Casjens S., Eppler K., Parr R., Poteete A. R. (1989) Nucleotide sequence of the bacteriophage P22 gene 19 to 3 region. Identification of a new gene required for lysis. Virology 171, 588–598 [DOI] [PubMed] [Google Scholar]
- 61. Fuller M. T., King J. (1981) Purification of the coat and scaffolding proteins from procapsids of bacteriophage P22. Virology 112, 529–547 [DOI] [PubMed] [Google Scholar]
- 62. Fuller M. T., King J. (1982) Assembly in vitro of bacteriophage P22 procapsids from purified coat and scaffolding subunits. J. Mol. Biol. 156, 633–665 [DOI] [PubMed] [Google Scholar]
- 63. Gope R., Serwer P. (1983) Bacteriophage P22 in vitro DNA packaging monitored by agarose gel electrophoresis. Rate of DNA entry into capsids. J. Virol. 47, 96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Teschke C. M. (1999) Aggregation and assembly of phage P22 temperature-sensitive coat protein mutants in vitro mimic the in vivo phenotype. Biochemistry 38, 2873–2881 [DOI] [PubMed] [Google Scholar]
- 65. Venkatachalam C. M. (1968) Stereochemical criteria for polypeptides and proteins. V. Conformation of a system of three linked peptide units. Biopolymers 6, 1425–1436 [DOI] [PubMed] [Google Scholar]
- 66. Prevelige P. E., Jr., Thomas D., King J. (1993) Nucleation and growth phases in the polymerization of coat and scaffolding subunits into icosahedral procapsid shells. Biophys. J. 64, 824–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zlotnick A., Suhanovsky M. M., Teschke C. M. (2012) The energetic contributions of scaffolding and coat proteins to the assembly of bacteriophage procapsids. Virology 428, 64–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Greene B., King J. (1994) Binding of scaffolding subunits within the P22 procapsid lattice. Virology 205, 188–197 [DOI] [PubMed] [Google Scholar]
- 69. Greene B., King J. (1999) Folding and stability of mutant scaffolding proteins defective in P22 capsid assembly. J. Biol. Chem. 274, 16141–16146 [DOI] [PubMed] [Google Scholar]
- 70. Greenfield N. J. (2006) Analysis of the kinetics of folding of proteins and peptides using circular dichroism. Nat. Protoc. 1, 2891–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Horovitz A., Matthews J. M., Fersht A. R. (1992) α-Helix stability in proteins. II. Factors that influence stability at an internal position. J. Mol. Biol. 227, 560–568 [DOI] [PubMed] [Google Scholar]
- 72. Luque I., Mayorga O. L., Freire E. (1996) Structure-based thermodynamic scale of α-helix propensities in amino acids. Biochemistry 35, 13681–13688 [DOI] [PubMed] [Google Scholar]
- 73. Serrano L., Fersht A. R. (1989) Capping and α-helix stability. Nature 342, 296–299 [DOI] [PubMed] [Google Scholar]
- 74. Hendsch Z. S., Tidor B. (1994) Do salt bridges stabilize proteins? A continuum electrostatic analysis. Protein Sci. 3, 211–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Parent K. N., Ranaghan M. J., Teschke C. M. (2004) A second-site suppressor of a folding defect functions via interactions with a chaperone network to improve folding and assembly in vivo. Mol. Microbiol. 54, 1036–1050 [DOI] [PubMed] [Google Scholar]
- 76. Parent K. N., Suhanovsky M. M., Teschke C. M. (2007) Phage P22 procapsids equilibrate with free coat protein subunits. J. Mol. Biol. 365, 513–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Aramli L. A., Teschke C. M. (1999) Single amino acid substitutions globally suppress the folding defects of temperature-sensitive folding mutants of phage P22 coat protein. J. Biol. Chem. 274, 22217–22224 [DOI] [PubMed] [Google Scholar]
- 78. Gordon C. L., King J. (1993) Temperature-sensitive mutations in the phage P22 coat protein which interfere with polypeptide chain folding. J. Biol. Chem. 268, 9358–9368 [PubMed] [Google Scholar]
- 79. Gordon C. L., Sather S. K., Casjens S., King J. (1994) Selective in vivo rescue by GroEL/ES of thermolabile folding intermediates to phage P22 structural proteins. J. Biol. Chem. 269, 27941–27951 [PubMed] [Google Scholar]
- 80. Thuman-Commike P. A., Greene B., Jakana J., Prasad B. V., King J., Prevelige P. E., Jr., Chiu W. (1996) Three-dimensional structure of scaffolding-containing phage P22 procapsids by electron cryomicroscopy. J. Mol. Biol. 260, 85–98 [DOI] [PubMed] [Google Scholar]
- 81. Thuman-Commike P. A., Greene B., Jakana J., McGough A., Prevelige P. E., Chiu W. (2000) Identification of additional coat-scaffolding interactions in a bacteriophage P22 mutant defective in maturation. J. Virol. 74, 3871–3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Teschke C. M., Parent K. N. (2010) “Let the phage do the work.” Using the phage P22 coat protein structures as a framework to understand its folding and assembly mutants. Virology 401, 119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]