

Background: Procaspase-3 is a critical protease in apoptosis.
Results: Procaspase-3 has less than 1/10,000,000 the activity of mature caspase-3 and does not detectably autoprocess. Small molecule and proteogenic fibrils promote procaspase-3 maturation through induced proximity to an active protease.
Conclusion: Fibrils enhance procaspase-3 maturation in vitro through colocalization with upstream proteases.
Significance: These studies demonstrate the importance of scaffolding and colocalization with active proteases for procaspase-3 processing and activation.
Keywords: Caspase, Drug Discovery, Enzyme Mechanisms, Kinetics, Protease, Fibrils, Procaspase, Zymogen
Abstract
Most proteases are expressed as inactive precursors, or zymogens, that become activated by limited proteolysis. We previously identified a small molecule, termed 1541, that dramatically promotes the maturation of the zymogen, procaspase-3, to its mature form, caspase-3. Surprisingly, compound 1541 self-assembles into nanofibrils, and localization of procaspase-3 to the fibrils promotes activation. Here, we interrogate the biochemical mechanism of procaspase-3 activation on 1541 fibrils in addition to proteogenic amyloid-β(1–40) fibrils. In contrast to previous reports, we find no evidence that procaspase-3 alone is capable of self-activation, consistent with its fate-determining role in executing apoptosis. In fact, mature caspase-3 is >107-fold more active than procaspase-3, making this proenzyme a remarkably inactive zymogen. However, we also show that fibril-induced colocalization of trace amounts of caspase-3 or other initiator proteases with procaspase-3 dramatically stimulates maturation of the proenzyme in vitro. Thus, similar to known cellular signaling complexes, these synthetic or natural fibrils can serve as platforms to concentrate procaspase-3 for trans-activation by upstream proteases.
Introduction
Proteases catalyze the irreversible post-translational modification of amide bond hydrolysis. Thus, cellular mechanisms exist to restrict spurious activation (1). For example, cellular inhibitors can often limit the activity of a mature protease. Moreover, most proteases are expressed as inactive precursors, known as zymogens, which require an external signal to generate the active enzyme (2, 3). Although the biological consequences of zymogen maturation are evident, important mechanistic details are not well understood (4).
Caspases are cysteine-class aspartyl-specific proteases that are expressed as inactive precursors, known as procaspases (5). To generate the mature enzyme, procaspases require cleavage after specific aspartic acid residues to remove an N-terminal prodomain and to form the large and small subunit, the latter processing event being critical for activation (Fig. 1A) (6, 7). In apoptosis, this maturation process proceeds via a signaling cascade (8). Upstream initiator procaspases-8, -9, and -10 are recruited to scaffolding complexes, such as the apoptosome or the death-inducing signaling complex, to generate active enzymes (9–13). Subsequently, the initiator caspases target downstream executioner procaspases-3 and -7 for activation (5, 10, 14–16). Together, these caspases can cleave >1000 downstream substrates that drive the apoptotic phenotype (17–19).
FIGURE 1.

General model for procaspase-3 activation. A, procaspase-3 variants used to explore potential activation mechanisms. Wild-type procaspase-3 is cleaved at Asp-9, Asp-28, and Asp-175. Cleavage in the intersubunit linker (Asp-175, red arrow) is essential to generate mature caspase-3. Cleavages in the prodomain (Asp-9 and Asp-28, black arrows) do not impact the in vitro activity of caspase-3. Note, caspases are expressed as constitutive dimers but illustrated here as monomers for simplicity. B, structure of compound 1541, which assembles into nanofibrils to activate procaspase-3. C, procaspase-3 activation may proceed through an initiation event that forms the first mature caspase-3 molecule, which can rapidly feedback to process additional procaspase-3 molecules in a propagation phase.
Significant efforts have been directed toward identifying small molecules that directly activate the procaspases for use as potential chemotherapeutic agents (20–22). Our laboratory previously discovered a small molecule, termed 1541, that promotes procaspase-3 activation (Fig. 1B). Remarkably, this small molecule spontaneously forms nanofibrils (4–5 nm thin and >1 μm long) that directly associate with procaspase-3 and promote activation (23). Natural amyloid-β (residues 1–40) fibrils also interact with procaspase-3 to increase its activity in vitro.
Importantly, such fibrous β-sheet structures have been correlated with caspase activation and neuronal cell death in many neurodegenerative diseases (24–30). Previous studies have even observed localization of active caspases to fibrillar structures in the cell (26, 31, 32). Although these reports are intriguing, evidence for direct procaspase activation on fibrils is limited. Furthermore, the mechanism by which procaspase maturation occurs upon localization to fibrils has not been previously described. Such characterization would offer a better understanding of the role of natural scaffolding mechanisms for procaspase maturation and may have significant implications in drug discovery of procaspase activators.
Here, we take advantage of the synthetic 1541 nanofibrils and amyloid-β fibrils to induce procaspase-3 activation and gain insight into the mechanism of zymogen maturation. Previous results hinted at an initiation and propagation phase to procaspase-3 activation (Fig. 1C) (22). We now show that initiation of procaspase-3 maturation is due to a trace amount of active caspase-3 or other active proteases. In contrast to previous reports (20–22, 33–35), we reveal that procaspase-3 has no detectable activity, and it does not autoprocess by either an intermolecular or intramolecular mechanism. Propagation of procaspase-3 activation is due to autocatalytic maturation of the proenzyme, whereby mature caspase-3 molecules that are formed rapidly feedback to cleave additional procaspase-3 molecules (Fig. 1C). Finally, both synthetic and natural fibrils accelerate this process by concentrating or colocalizing mature caspase-3 with its procaspase-3 substrate.
EXPERIMENTAL PROCEDURES
Materials
The mature caspase-3 antibody (9664) was purchased from Cell Signaling. Thermolysin and 1,1,1,3,3,3-hexafluoro-2-propanol were purchased from Sigma. Amyloid-β(1–40) was purchased from AnaSpec. Acetyl-Asp-Glu-Val-Asp-7-amino-4-trifluoromethylcoumarin (Ac-DEVD-afc)4 was purchased from SM Biochemicals LLC. Isopropyl β-d-thiogalactoside and acetyl-Asp-Glu-Val-Asp-chloromethyl ketone (Ac-DEVD-cmk) were purchased from Calbiochem. Benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone, acetyl-Ile-Glu-Thr-Asp-7-amino-4-trifluoromethylcoumarin (Ac-IETD-afc), and 7-amino-4-trifluoromethylcoumarin were purchased from Enzo Life Sciences. The pET23b and pET15b vectors were purchased from Novagen. Granzyme B was a generous gift from Dan Hostetter and Cheryl Tajon in the Craik laboratory at the University of California, San Francisco. Tobacco etch virus (TEV) protease was a generous gift from Charlie Morgan, and procaspase-3 (D175ENLYFQ) that has a TEV cleavage site inserted at the inter-subunit linker was a generous gift from Dan Gray at the Wells laboratory, University of California, San Francisco. All additional reagents were purchased from Sigma, unless otherwise noted.
Constructs
Full-length human procaspase-3 gene was cloned into pET23b, as described previously (22, 36). Truncated procaspase-8(217–479), lacking the death effector domains, was cloned into pET15b. Truncated procaspase-3 (29–277), lacking the prodomain, was cloned into pET23b. QuikChange mutagenesis (Stratagene) on the above constructs was used to generate the catalytically inactive procaspase-3 (C163A), the catalytically inactive, truncated procaspase-3 (29-277/C163A), the uncleavable procaspase-3 (D9A/D28A/D175A), procaspase-3 (T199A), procaspase-3 (S205A), and the catalytically inactive TEV-cleavable pro-3 (D175ENLYFQ/C163A).
Expression/Purification
For expression, all plasmids were transformed into the bacterial strain BL21 (DE3) pLysS. Wild-type, full-length procaspase-3 was expressed and purified as described previously (22, 36). Briefly, bacterial cells were grown at 37 °C to an A600 nm of ∼0.6, and subsequent overexpression was induced with 0.2 mm isopropyl β-d-thiogalactoside at 30 °C for 20 min. Cells were rapidly harvested and lysed. The clarified cell lysate was purified using a nickel-nitrilotriacetic acid affinity column, followed by anion-exchange chromatography. Purified procaspase-3 fractions were collected and stored at −80 °C.
Caspase-3 was overexpressed at 30 °C for 5 h from cells transformed with the pET23b containing full-length procaspase-3. Caspase-3 (T199A) and caspase-3 (S205A) were similarly overexpressed at 30 °C overnight from the respective full-length constructs. Caspase-8 was overexpressed at 12 °C overnight from cells transformed with the pET15b containing procaspase-8(217–479). All mature caspases were purified as described for procaspase-3.
All other constructs followed the same expression and purification protocols as wild-type procaspase-3 with only the overexpression time modified as follows: catalytically inactive procaspase-3 (C163A) for 15 h; catalytically inactive, truncated procaspase-3 (29-277/C163A) for 5 h; catalytically dead, TEV-cleavable procaspase-3 (D175ENLYFQ/C163A) for 5 h; and uncleavable procaspase-3 (D9A/D28A/D175A) for 2–8 h (exact conditions described in the text).
Synthesis of 1541
Synthesis, purification, and characterization of 1541 were performed as described previously (22).
Procaspase-3 Processing Assay (22)
Wild-type procaspase-3 was agitated in a caspase activity buffer (50 mm HEPES, pH 7.4, 50 mm KCl, 0.1 mm EDTA, 10 mm DTT, and 0.1% CHAPS) with 1541 or 2% DMSO alone for the indicated time intervals. Subsequent assays with procaspase-3 also included a 15-min preincubation with the irreversible inhibitor, Ac-DEVD-cmk, at the specified concentrations. Samples were quenched with LDS loading buffer and analyzed by SDS-PAGE. Protein bands were visualized by either silver stain analysis or Western blot.
Procaspase-3 Trans-activation Assays (33, 34)
Uncleavable procaspase-3 (D9A/D28A/D175A) or mature caspase-3 was agitated at 37 °C with the catalytically inactive procaspase-3 (C163A) in a caspase activity buffer (50 mm HEPES, pH 7.4, 50 mm KCl, 0.1 mm EDTA, 10 mm DTT, and 0.1% CHAPS) in the presence or absence of 1541. The irreversible inhibitor, Ac-DEVD-cmk, was added under the designated conditions to inhibit any cleaved caspase-3 impurity present in the uncleavable procaspase-3 expression. Samples were quenched with LDS loading buffer and analyzed by SDS-PAGE. Protein bands were visualized by either silver stain analysis or Western blot.
Procaspase-3 Self-activation Assays (22)
Wild-type procaspase-3 was preincubated for 15 min with DMSO alone or 1 nm Ac-DEVD-cmk in a caspase activity buffer (50 mm HEPES, pH 7.4, 50 mm KCl, 0.1 mm EDTA, 10 mm DTT, and 0.1% CHAPS). 1541 or DMSO was added to the corresponding samples and incubated at 37 °C. Granzyme B (2 nm) was added to wild-type procaspase-3 to determine the maximal activation levels possible. At the indicated time points, 50 μm Ac-DEVD-afc was added to each sample, and fluorescence intensity was monitored on a SpectraMax M5 (Molecular Devices) plate reader. Initial rates were plotted at each time point using Prism Version 5.0c.
Caspase Activity Assays
The activities of procaspase-3, procaspase-3 (D9A/D28A/D175A), caspase-3, caspase-3 (S205A), and caspase-3 (T199A) were measured as described previously (22, 37–40). Briefly, 5 μm procaspase-3, 0.1, 10, 20, and 50 μm procaspase-3 (D9A/D28A/D175A), 5 and 10 nm caspase-3, 5 and 10 nm caspase-3 (S205A), and 5 and 10 nm caspase-3 (T199A) were incubated in a caspase activity buffer (50 mm HEPES, pH 7.4, 50 mm KCl, 0.1 mm EDTA, 10 mm DTT, and 0.1% CHAPS). Processing of the fluorogenic substrate Ac-DEVD-afc was used to monitor activity. Km and Vmax values were determined from plots of initial rates versus substrate concentration in Prism Version 5.0c. Curves were fit using the standard Michaelis-Menten equation. Vmax values were transformed into kcat values using a linear plot of the fluorescence intensity of the 7-amino-4-trifluoromethylcoumarin standard versus concentration. Substoichiometric concentrations of Ac-DEVD-cmk were incorporated into subsequent assays to evaluate the impact of low levels of the cleaved caspase on procaspase activity. Notably, peptidic chloromethyl ketones have an ∼15-min half-life in the buffers containing reducing agent (41).
Covalent Modification of (Pro)Caspase-3
The extent of covalent modification of procaspase-3 and caspase-3 by Ac-DEVD-cmk was evaluated using an LCT-Premier LC/electrospray ionization-MS instrument (Waters) after 24 h of incubation in assay buffer (50 mm HEPES, pH 7.4, 50 mm KCl, 0.1 mm EDTA, and 10 mm DTT).
Calculation of Autocatalytic Activation Curves
To estimate autocatalytic conversion of procaspase-3 to active caspase-3, we fit our experimental data to the classic Michaelis-Menten equation: v = (Vmax·[S])/(Km + [S]).
Iterative application of this equation every minute of the reaction enabled us to estimate the instantaneous levels of procaspase-3 and caspase-3. Additionally, we evaluated how these factors would change in response to various catalytic efficiencies and different amounts of contaminating active caspase-3. The assumptions we have input for this model are as follows: kcat = 23.5 min−1 for mature caspase-3; kcat = 23.5 min−1·(10−7) for procaspase-3; Km = 140 μm for both caspase-3 and procaspase-3, and [S0] = 100 nm or 1 μm (procaspase-3). Initial levels of caspase-3 were set at 0 or 0.2% of [S0].
Cosedimentation Assays with 1541 (23, 42)
20 μl of 0.5 mm 1541 or DMSO alone was added to 1 ml of 0.128, 0.064, 0.032, 0.016, 0.008, 0.004, 0.002, 0.001, and 0 mg/ml procaspase-3, caspase-3, caspase-8, or TEV protease in a caspase assay buffer (20 mm HEPES, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 1 mm DTT, 1.5% sucrose, and 0.1% CHAPS). 10 μl of 0.5 mm 1541 was added to 500 μl of 0.016, 0.008, 0.004, 0.002, 0.001, and 0 mg/ml granzyme B in a caspase assay buffer (50 mm HEPES, pH 7.4, 50 mm KCl, 0.1 mm EDTA, 1 mm DTT, and 0.1% CHAPS). Thermolysin was dissolved in 2.5 m NaCl and 10 mm CaCl2 to 1.28, 0.64, 0.32, 0.16, 0.08, 0.04, 0.02, 0.01, and 0 mg/ml, and diluted 10× into a caspase assay buffer (20 mm HEPES, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 1 mm DTT, 1.5% sucrose, and 0.1% CHAPS) to a final volume of 1 ml. 20 μl of 0.5 mm 1541 or DMSO was next added to the thermolysin mixtures. All samples were vortexed and immediately centrifuged at 16,200 × g for 15 min. The supernatant was aspirated, and 100 μl assay buffer was added to the pellet in each tube. Samples were diluted with 4× LDS sample buffer (Invitrogen) and analyzed by SDS-PAGE. Bands were visualized by Coomassie (Bio-Rad) stain, imaged on a LI-COR Odyssey Infrared Imaging System, and quantified by ImageJ. Granzyme B samples were normalized for direct comparison with other enzymes.
Rate of Procaspase-3 Processing by Upstream Proteases
20 μl of 0.5 mm 1541 or DMSO alone was added to 1 ml of 200 nm procaspase-3 (29-277/C163A) in a caspase activity buffer (20 mm HEPES, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 1 mm DTT, 1.5% sucrose, and 0.1% CHAPS). 2 nm caspase-8, 2 nm caspase-3, 20 nm TEV, or 200 nm TEV was subsequently added. Aliquots from each sample were taken at the indicated time points, quenched with LDS loading buffer, and analyzed by SDS-PAGE. Protein bands were visualized by silver stain and quantified by ImageJ.
Similarly, 20 μl of 0.5 mm 1541 or DMSO was added to 1 ml of 200 nm procaspase-3 (29-277/C163A) in a caspase activity buffer (50 mm HEPES, pH 7.4, 50 mm KCl, 0.1 mm EDTA, 1 mm DTT, and 0.1% CHAPS), 2 nm granzyme B was added, and processing was monitored as described above.
Finally, procaspase-3 (29-277/C163A) susceptibility to processing by thermolysin was determined, as described previously (43). Briefly, thermolysin was dissolved in 2.5 m NaCl and 10 mm CaCl2 to 0.5 or 0.05 mg/ml. 100 μl of 10 μm procaspase-3 (29-277/C163A) was added to 860 μl of caspase buffer. 20 μl of 2.5 mm 1541 or DMSO was subsequently added, followed by 20 μl of the specified thermolysin stocks. Processing was monitored over time as described above except using Sypro Ruby protein gel stain (Invitrogen).
Catalytic Efficiency of Procaspase-3 Cleavage (44, 45)
50 μl of 2 μm procaspase-3 (29-277/C163A) followed by 10 μl of 0.5 mm 1541 or DMSO were added to 390 μl of buffer (20 mm HEPES, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 1 mm DTT, 1.5% Sucrose, and 0.1% CHAPS) in 12 separate 1.5-ml Eppendorf tubes. 50 μl of a dilution series of an upstream protease was added last to corresponding samples to initiate the reaction. We agitated the samples at 37 °C and quenched the reactions with LDS loading buffer at the indicated time points. We subsequently analyzed the samples by SDS-PAGE followed by silver stain to visualize the bands. We quantified the disappearance of the substrate with increasing enzyme concentrations and established the amount of an upstream protease corresponding to 50% cleavage (E½). Assuming pseudo-first order kinetics, the following equation gives an approximate catalytic efficiency (kcat/Km): kcat/Km = ln(2)/(E½·t).
Amyloid-β(1–40) Preparation
Aβ(1–40) fibrils were formed as described previously (23, 46). Briefly, 250 μl of a caspase activity buffer (20 mm HEPES, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 1.5% sucrose, 10 mm DTT, 0.1% CHAPS) was added to the lyophilized solid to 50 μm. The peptide was agitated at 37 °C for 4–6 h. Fibril formation was confirmed by an increase in fluorescence intensity upon the addition of 15 μl of sample to 250 μl 5 μm thioflavin T (Sigma).
Procaspase-3 Processing with Aβ(1–40)
250 nm procaspase-3 was agitated in a caspase activity buffer (20 mm HEPES, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 1.5% sucrose, 10 mm DTT, 0.1% CHAPS) with 20 μm Aβ(1–40) fibrils (4 h preincubation) or buffer alone for the indicated time intervals. Processing was monitored by SDS-PAGE as described above.
Trans-activation of Procaspase-3 with Aβ(1–40)
5 nm caspase-3 was agitated with 200 nm procaspase-3 (29-277/C163A) in a caspase activity buffer (20 mm HEPES, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 1.5% sucrose, 10 mm DTT, 0.1% CHAPS) with 20 μm Aβ(1–40) fibrils (4 h of preincubation) or buffer alone for the indicated time intervals. Processing was monitored by SDS-PAGE as described above.
Cosedimentation of Caspases with Aβ(1–40)
50 μm Aβ(1–40) peptide or buffer alone was incubated at 37 °C for 6 h. 50-μl of 1.0 mg/ml procaspase-3, caspase-3, caspase-8, caspase-9, and caspase-6 was added to 450 μl of 20 μm Aβ(1–40) fibrils (6 h of preincubation). The samples were vortexed briefly and centrifuged at 20,817 × g for 20 min. The supernatant was aspirated, and 100 μl of assay buffer was added to the pellet in each tube. The amount of enzyme in each pellet was assessed as described for 1541 samples.
RESULTS
1541 Nanofibrils Enhance Autocatalytic Maturation of Procaspase-3
Previously, we reported that 1541 nanofibrils induce procaspase-3 activation to form mature caspase-3 (23). The addition of 25 μm 1541 to 100 nm wild-type procaspase-3 results in a burst of activity after a 2-h incubation, using cleavage of Ac-DEVD-afc as a reporter substrate (Fig. 2A) (22). This procaspase-3 activation curve is similar to the classic S-shaped curve obtained for auto-activation of other zymogens, such as trypsinogen (47, 48). Based on our data, we suspected an autocatalytic activation mechanism for procaspase-3, where mature caspase-3 rapidly processes its zymogen substrate, procaspase-3, in a second order reaction (Fig. 1C). This model predicts an initially accelerating activity curve that saturates as the procaspase-3 substrate is depleted to give the observed S-shaped curve.
FIGURE 2.

Mature caspase-3 drives activation of procaspase-3 in the presence of 1541. A, activation was monitored for 100 nm procaspase-3 alone (dark circles), 100 nm procaspase-3 with granzyme B (open circles), and 100 nm procaspase-3 with 25 μm 1541 (purple circles). The tetrapeptide substrate, Ac-DEVD-afc, was added at the indicated time points, and initial rates were plotted as a function of time. B, activities were monitored in the presence of 1 nm caspase-3, procaspase-3 (dark squares), procaspase-3 with granzyme B (open squares), and procaspase-3 with 25 μm 1541 (green squares). C, enzyme activity was measured in the presence of 1 nm Ac-DEVD-cmk, procaspase-3 (dark diamonds), procaspase-3 with granzyme B (open diamonds), and procaspase-3 with 25 μm 1541 (blue diamonds).
To confirm the role of active caspase-3 in accelerating wild-type procaspase-3 activation, we explored how changing the mature caspase-3 concentration affects the kinetics of wild-type procaspase-3 activation. The rate of procaspase-3 activation upon inclusion of even a 1% stoichiometric amount of active caspase-3 drastically shortens the lag period by 2.5-fold to ∼40 min (Fig. 2B). In contrast, addition of a 1% equivalent of an irreversible active site inhibitor, Ac-DEVD-cmk, to wild-type procaspase-3 delayed activation to ∼5 h (Fig. 2C). Notably, Ac-DEVD-cmk preferentially labels and inactivates mature caspase-3 relative to procaspase-3 (Fig. 3). These data highlight the importance of mature caspase-3 in the procaspase-3 activation process and support an autocatalytic activation mechanism.
FIGURE 3.

Irreversible inhibitor of mature caspase-3. A, structure of Ac-DEVD-cmk. B, 5 or 500 μm Ac-DEVD-cmk was added to 5 μm caspase-3 or 5 μm wild-type procaspase-3. After a 24-h incubation at 37 °C, covalent modification was evaluated by mass spectrometry.
1541 Fibrils Enhance Procaspase-3 Processing by Several Upstream Proteases
The results above demonstrate that the fibrils plus trace amounts of active caspase-3 increase procaspase-3 activation kinetics. To measure the extent to which the nanofibrils enhance the natural zymogen maturation process, we further quantified the magnitude of the acceleration. We first characterized the apparent increase in caspase-3 catalytic efficiency in the presence of 1541 fibrils. The catalytically inactive procaspase-3 (C163A) lacking its 28-amino acid prodomain (29-277) was used as the substrate (Fig. 1A). This variant permits evaluation of the processing rate at the single critical activating cut site between the large and small subunit without the confounding effects of procaspase-3 autocatalytic activation (Fig. 1C). In a preliminary competition assay with Ac-DEVD-afc, we found that the Km value for cleaving this zymogen substrate is greater than 100 μm (data not shown). Thus, we kept the proenzyme concentration at 200 nm, which is well below its Km value. Using these experimental conditions, we found caspase-3 to show an ∼17-fold increase in catalytic efficiency (kcat/Km) for cleaving this zymogen variant in the presence of 1541 (Fig. 4A and Table 1).
FIGURE 4.

Effective change in the catalytic efficiency of upstream proteases due to 1541 fibrils. A, dilution series of caspase-3 was added to the truncated, inactive procaspase-3 (29-277/C163A) in the absence (DMSO) or presence of 10 μm 1541. At 60 min, samples were quenched with LDS loading buffer, analyzed by SDS-PAGE, silver-stained, and band intensities quantified. B, processing of the inactive procaspase-3 (29–277/C163A) by a dilution series of mature caspase-8 with and without 10 μm 1541 was assessed at 20 min. C, processing of the inactive procaspase-3 by a dilution series of granzyme B with and without 10 μm 1541 was evaluated at 90 min. Replicate gels are not shown for clarity. The asterisks indicate roughly 50% cleavage of procaspase-3.
TABLE 1.
1541 fibrils increase the catalytic efficiency of upstream proteases against the inactive procaspase-3 (C163A) substrate
| Proteasea | 1541 | Time | kcat/Km | Ratiob |
|---|---|---|---|---|
| μm | min | m−1 s−1 | ||
| Caspase-3 | 0 | 60 | 2.8 × 103 | |
| Caspase-3 | 10 | 60 | 4.8 × 104 | 17 |
| Caspase-8 | 0 | 20 | 2.7 × 104 | |
| Caspase-8 | 10 | 20 | 7.5 × 105 | 28 |
| Granzyme B | 0 | 90 | 8.3 × 104 | |
| Granzyme B | 10 | 90 | 4.1 × 105 | 5 |
a Substrate = inactive procaspase-3 (29–277/C163A).
b Ratio = (catalytic efficiency with 1541)/(catalytic efficiency without 1541).
We next evaluated whether this increase in activity is generalizable to other known upstream proteases, such as caspase-8 and granzyme B. Interestingly, the catalytic efficiency for caspase-8 cleaving the zymogen was enhanced 28-fold in the presence of 1541 compared with the absence (Fig. 4B and Table 1). For granzyme B, there was only a 5-fold enhancement in the presence compared with absence of 1541 (Fig. 4C and Table 1). These results reinforce the importance of an active upstream protease in promoting procaspase-3 activation and further quantify the impact of 1541 nanofibrils on procaspase-3 maturation by active proteases, including caspase-3.
Fibrils Concentrate Procaspase-3 with Mature Proteases to Enhance Processing
1541 nanofibrils increase procaspase-3 processing by caspase-3 as well as other upstream proteases. This enhancement in procaspase-3 maturation could result from three possible mechanistic effects as follows: an increase of the intrinsic activity of the upstream proteases, increased susceptibility of procaspase-3 to processing, or increased proximity of procaspase-3 to these active proteases.
We first evaluated whether 1541 altered the catalytic activity of some known activating proteases, such as caspase-3, caspase-8, and granzyme B, against their preferred tetrapeptide substrates, Ac-DEVD-afc, Ac-IETD-afc, and Ac-IETD-afc, respectively. No change in the catalytic efficiencies was observed upon the addition of fibrils (Fig. 5, A–C). These data show that the fibrils do not directly impact the intrinsic activities of these activating proteases. The unaltered activities further suggest that the fibrils do not globally alter the conformation of the mature enzymes.
FIGURE 5.
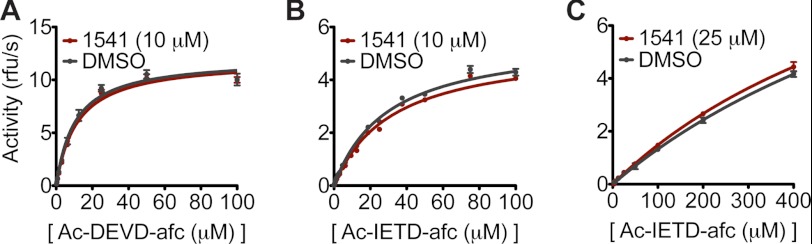
Activities of caspase-3, caspase-8, and granzyme B against tetrapeptide substrates are similar with or without 1541. A, activity of caspase-3 (5 nm) against Ac-DEVD-afc was measured in the presence and absence of 10 μm 1541. B, activity of capsase-8 (20 nm) against Ac-IETD-afc was monitored with and without 10 μm 1541. Notably, inhibition of caspase-3 and caspase-8 was observed at higher concentrations of 1541 (>30 μm); however, compound concentration remained below this value to focus on activation alone. C, activity of granzyme B against Ac-IETD-afc was measured with and without 25 μm 1541.
Although 1541 does not directly impact the mature enzyme conformation or activity, it is possible that the nanofibrils alter the conformation of the precursors to make the cleavage site more accessible to proteolysis. Pulse proteolysis with the broadly specific protease, thermolysin, has previously been used to assess conformational changes and protein stability (49). 1541 fibrils did not affect the rate of processing of the inactive procaspase-3 (C163A) by thermolysin (Fig. 6A). These data suggest that the fibrils do not alter the conformation of procaspase-3 to make it generally more susceptible to proteolysis.
FIGURE 6.

Rate of procaspase-3 cleavage by an active protease depends on its catalytic efficiency and the extent of binding to fibrils. A, 0.001 mg/ml (upper panel) and 0.01 mg/ml (lower panel) thermolysin were added to 1 μm procaspase-3 (29-277/C163A) in the presence of 50 μm 1541 or DMSO alone. Samples were quenched at the indicated time points and analyzed by SDS-PAGE. B, 10 μm 1541 was added to a dilution series of procaspase-3, caspase-3, caspase-8, TEV protease, thermolysin, and granzyme B, followed by centrifugation at 16,100 × g for 15 min. The pellet was analyzed by SDS-PAGE followed by Coomassie staining to determine the amount of enzyme that bound to 1541 nanofibrils. C, 2 nm caspase-3, caspase-8, or granzyme B was added to 200 nm truncated, dead procaspase-3 (29-277/C163A) in the presence of 10 μm 1541 or DMSO. Samples were collected at the indicated time points and quenched with LDS loading buffer. After analysis by SDS-PAGE and silver stain, percent cleavage of the procaspase was determined by quantifying band intensities. D, TEV cleavage site was engineered into the inactive procaspase-3, rendering it susceptible to proteolysis and activation by only TEV protease. 200 nm of the inactive TEV-cleavable procaspase-3 (D175ENLYFQ/C163A) was incubated with 10 μm 1541 or DMSO alone in the presence of 20 or 200 nm TEV protease. Processing was monitored as described above. Replicate gels are not shown for clarity.
Finally, we explored if the fibrils promoted activation through colocalization or concentration of procaspase-3 with the activating proteases. We previously showed a direct association between procaspase-3 and the 1541 nanofibrils using transmission electron microscopy and cosedimentation studies (23). Here, we evaluated if the upstream proteases can also associate directly with the fibrils using the cosedimentation assays. As seen in Fig. 6B, caspase-3 binds and saturates the fibrils at roughly the same concentration as procaspase-3. Caspase-8 even shows slightly better binding and saturation behavior (Fig. 6B) consistent with the greater enhancement in catalytic efficiency seen in Fig. 4B. Granzyme B interacts less with the fibrils and shows the smallest change in activity in the presence of the fibrils. These studies support the notion that colocalization of an active protease with the procaspase-3 substrate is critically important for activation to occur.
This colocalization mechanism was further established by measuring the rate of processing of the truncated, inactive procaspase-3 (29-277/C163A) by low concentrations of caspase-3, caspase-8, and granzyme B (Fig. 6C). As expected, very little cleavage of the inactive zymogen was observed by caspase-3 or caspase-8 in the absence of 1541. However, upon the inclusion of 1541, the half-lives (t½) were reduced from undetectable levels to 220 and 8 min by caspase-3 and caspase-8, respectively. The relative processing rates for caspase-3 and caspase-8 in the presence of 1541 are consistent with the relative catalytic efficiencies of the respective enzymes.
In contrast, granzyme B does bind but not well to the fibrils, which provides an important distinction in comparison with caspase-3 and caspase-8. 1541 nanofibrils only slightly enhance granzyme B processing of procaspase-3, reducing the t½ from 77 to 21 min. Although granzyme B alone cleaves procaspase-3 much more rapidly than caspase-8, granzyme B processes procaspase-3 roughly 3-fold slower than caspase-8 in the presence of 1541. This is also reflected by the much smaller change in catalytic efficiency seen for granzyme B compared with caspase-3 and caspase-8 against the procaspase-3 substrate in the presence of the fibrils (Table 1). Both observations are consistent with colocalization of the upstream protease with its procaspase-3 substrate being especially important for maturation.
We subsequently explored the ability of 1541 nanofibrils to impact procaspase-3 cleavage by noncognate proteases, such as tobacco etch virus (TEV) protease and thermolysin to evaluate if procaspase-3 was more susceptible to proteolysis in general. 1541 nanofibrils do not significantly affect the rate of procaspase-3 processing by thermolysin (Fig. 6A). To enable TEV to cleave procaspase-3, we replaced the caspase cleavage site in the inter-subunit linker with the recognition sequence for TEV protease (D175ENLYFQ). Previous experiments showed this procaspase-3 mutant could be cleaved by TEV (7). However, 1541 did not enhance the rate of cleavage of this variant by TEV (Fig. 6D). Correspondingly, both TEV and thermolysin do not interact with the 1541 fibrils to a significant degree (Fig. 6B). Thus, those proteases that can bind and colocalize to the fibrils showed dramatic enhancement in processing of procaspase-3. Those that cannot bind to the fibrils showed no enhancement in the rate at which they cleaved procaspase-3.
Decreased Catalytic Efficiency of Resistance Mutants
In previous studies (22), we identified point mutations in procaspase-3 (S205A or T199A) that reduced activation by 1541 but could still be cleaved by granzyme B to generate an active enzyme. We wished to test if the resistance to activation of these variants could result from either decreased interaction with the fibrils or from diminished catalytic efficiency relative to the wild-type enzyme.
Kinetic studies (Table 2) show that the catalytic efficiency for cleaving the preferred substrate Ac-DEVD-afc for caspase-3 (S205A) and caspase-3 (T199A) is indeed reduced 8- and 7-fold, respectively. This is mostly due to a significant decrease in the kcat for both mutants. Similar results were shown for Ac-IETD-afc, a tetrapeptide substrate that mimics the cleavage site in the inter-subunit linker of procaspase-3. The catalytic efficiency against this substrate was reduced by 4- and 14-fold for the S205A and T199A variants, respectively. These reductions in kcat/Km offset the apparent 17-fold increase in activity that results from colocalization on the fibrils. Furthermore, the procaspase-3-resistant mutants are susceptible to enhanced processing by wild-type caspase-3 in the presence of 1541, which would indicate that the mutant proenzymes still interact with the fibrils (data not shown). Thus, the reduction in catalytic efficiency for these variant enzymes, largely explains why these mutations lead to greater resistance to 1541-stimulated activation.
TABLE 2.
Decreased catalytic efficiency of caspase-3 resistance mutations tested on two synthetic peptide substrates
| Construct | Km | kcat | kcat/Km | Ratioa |
|---|---|---|---|---|
| μm | s−1 | m−1s−1 | ||
| Ac-DEVD-afc | ||||
| Wild-type | 12.0 ± 0.7 | 9.11 ± 0.15 | 7.6 × 105 | 1.0 |
| T199A | 16.2 ± 0.9 | 1.46 ± 0.02 | 9.1 × 104 | 0.14 |
| S205A | 5.9 ± 0.3 | 0.63 ± 0.01 | 1.1 × 105 | 0.12 |
| Ac-IETD-afc | ||||
| Wild-type | 140 ± 10 | 2.25 ± 0.04 | 1.6 × 104 | 1.0 |
| T199A | 100 ± 10 | 0.12 ± 0.01 | 1.2 × 103 | 0.07 |
| S205A | 56 ± 3 | 0.25 ± 0.01 | 4.4 × 103 | 0.27 |
a Ratio = (catalytic efficiency of mutant)/(catalytic efficiency of wild type).
Procaspase-3 Does Not Self-activate by an Intermolecular or Intramolecular Mechanism but Requires an Active Protease to Initiate Processing
The above results elucidated the importance of trace levels of mature caspase-3 in rapidly activating procaspase-3 and described how the fibrils act to enhance this process. However, a detailed mechanistic understanding of the initial event that leads to the first caspase-3 molecules was still lacking. This initiating event could originate from one of two sources as follows: procaspase-3 could cleave itself by either a cis- or trans-mechanism to generate an initial population of the mature enzyme (Fig. 7A) or a trace amount of mature caspase-3 initially present in the procaspase-3 preparations could cleave procaspase-3 to generate more mature caspase-3 (Fig. 7B).
FIGURE 7.

Two possible models for initiation of procaspase-3 activation. A, initial event derives from a procaspase-3 molecule either cutting itself in cis (intramolecular) or cutting another procaspase-3 molecule in trans (intermolecular) upon interaction with the fibrils. B, initial event involves a trace amount of active caspase-3 that cleaves procaspase-3 upon colocalization to the nanofibrils. In either model, propagation is greatly accelerated by the accumulation of active caspase-3. Note that procaspase-3 molecules are only illustrated on one face of 1541 nanofibrils for simplicity; however, previous studies show that they coat the full surface (23).
Concurrent with the burst in activation observed in Fig. 2A, wild-type procaspase-3 processing to the large and small subunit of active caspase-3 in the presence of 1541 nanofibrils occurs after a lag period of roughly 2 h (Fig. 8A) (22). Either proposed model for the initiation of procaspase-3 maturation is consistent with this lag or delay (Fig. 7). Thus, we first assessed if concentrating procaspase-3 on the fibrils is sufficient to promote an initial trans-activation event, where one proenzyme molecule, termed “the initiator,” cleaves another proenzyme molecule, termed “the substrate” (Fig. 7A). The catalytically inactive mutant of procaspase-3 (C163A) served as the substrate, because it can be cleaved but cannot initiate. The catalytically competent, yet uncleavable procaspase-3 served as the initiator; here, the critical aspartic acid residues recognized by the protease are mutated to alanines (D9A/D28A/D175A, Fig. 1A) (34). 1541 or vehicle alone was added to equimolar enzyme mixtures of the initiator and the substrate procaspase-3 (250 nm). At the indicated time points, samples were quenched with LDS loading buffer, analyzed by SDS-PAGE, and visualized by silver stain (Fig. 8B). No processing of the substrate variant (C163A) was observed in the presence or absence of 1541. Furthermore, even a 4-fold stoichiometric excess of the uncleavable initiator procaspase-3 did not promote processing of the proenzyme (data not shown).
FIGURE 8.

Enhanced susceptibility of procaspase-3 to processing by mature caspase-3 in the presence of 1541 fibrils. A, activation as a function of time for 250 nm wild-type procaspase-3 in the absence (−) or presence (+) of 10 μm 1541. Procaspase-3 is processed at three sites (Asp-9, Asp-28, and Asp-175), which leads to multiple bands by SDS-PAGE. The final cleavage products are residues 29–175 (large subunit) and 176–277 (small subunit). B, uncleavable procaspase-3 (D9A/D28A/D175A, 250 nm) was incubated with a catalytically inactive procaspase-3 (C163A, 250 nm) with (+) or without (−) 10 μm 1541. C, higher concentration of uncleavable procaspase-3 (5 μm) was added to 5 μm dead procaspase-3 in the presence of 5% Ac-DEVD-cmk (250 nm). Either 50 μm 1541 or DMSO alone was added to the samples. Processing was monitored by Western blot with an antibody specific for the C terminus of the large subunit of cleaved caspase-3 (Cell Signaling , catalog no. 9664). D, 250 nm Ac-DEVD-cmk, an irreversible inhibitor that selectively labels mature caspase-3 under the assay conditions, was added to 250 nm wild-type procaspase-3. 1541 or DMSO alone was subsequently added. Processing was monitored by silver stain analysis. E, self-activation of a higher concentration of wild-type procaspase-3 (5 μm) in the presence of 20% (1 μm) Ac-DEVD-cmk was monitored by Western blot. F, processing was examined for the inactive procaspase-3 (C163A, 250 nm) upon addition of mature caspase-3 (5 nm).
To detect any trace cleavage of the inactive procaspase-3 (5 μm) by the uncleavable proenzyme (5 μm), we further analyzed processing at extended times using more sensitive Western blots. We added sub-stoichiometric concentrations of Ac-DEVD-cmk to ensure no feedback activation by mature caspase-3. Still, no detectable change in the amount of procaspase-3 or its cleavage products was observed with an antibody that preferentially recognizes the large subunit of mature caspase-3 (Fig. 8C). This indicates that 1541 fibrils do not promote trans-activation of procaspase-3 by another proenzyme molecule. Moreover, trans-activation of procaspase-3 alone is not detectable at physiologically relevant concentrations of the proenzyme (∼100 nm) or even at much higher concentrations (50, 51).
Nonetheless, we noted that incubation of wild-type procaspase-3 at high concentrations (>1 μm) could generate the large and small subunit of the mature enzyme, even in the absence of 1541 (Fig. 9). These results in addition to previous reports would suggest that the proenzyme can auto-activate (20, 21, 33–35); however, the above proteolytic susceptibility assay shows that intermolecular processing between two procaspase-3 molecules is restricted. Therefore, we assessed if intramolecular processing of procaspase-3 can generate a trace amount of mature caspase-3, which can then rapidly feedback to promote autocatalytic maturation of the proenzyme (Fig. 7B).
FIGURE 9.
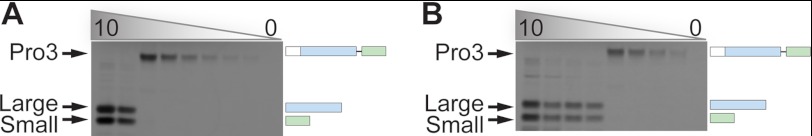
Wild-type procaspase-3 activation. Dilution series of wild-type procaspase-3 (starting at 10 μm) was incubated at 37 °C for 4 h (A) or 24 h (B). The reactions were quenched with LDS loading buffer, analyzed by SDS-PAGE, and visualized by Coomassie staining. Note the delay or lag in processing of the 2.5 and 1.25 μm procaspase-3 samples between the 4- and 24-h time points.
To evaluate this mechanism, we needed to isolate wild-type procaspase-3 activity alone and to eliminate the contribution of newly generated caspase-3 to the activation process. We used the covalent inhibitor, Ac-DEVD-cmk, to preferentially label and inactivate any mature caspase-3 while preserving procaspase-3 (Fig. 3A). At stoichiometric concentrations (5 μm), the inhibitor covalently modifies mature caspase-3 (5 μm) to >97% by mass spectrometry, yet labels <4.0% of wild-type procaspase-3 (5 μm) over the same time course (Fig. 3B). Even with 500 μm inhibitor added to 5 μm procaspase-3 (100-fold excess), we see only ∼70% single-site modification with some nonspecific covalent modification of surface-exposed cysteine residues. Thus, although procaspase-3 can bind and react with Ac-DEVD-cmk, it is much slower and less specific than labeling of the active site of mature caspase-3.
We next added stoichiometric amounts of Ac-DEVD-cmk (250 nm) to wild-type procaspase-3 (250 nm) with and without 1541 under our standard assay conditions. No discernible processing to the large and small subunit of the mature caspase occurred for up to 8 h, as monitored by silver stain (Fig. 8D). At higher concentrations of wild-type procaspase-3 (5 μm) with sub-stoichiometric amounts of Ac-DEVD-cmk (1 μm), processing over time was still not observed with or without 1541 by Western blot analysis (Fig. 8E). Thus, the proenzyme alone appears to be incapable of intramolecular auto-activation. These results in conjunction with the lack of trans-processing argue strongly against a model whereby procaspase-3 activation occurs due to auto-proteolysis (Fig. 7A).
Thus, the data indicate that trace amounts of active caspase-3 are present in standard procaspase-3 preparations that facilitate procaspase-3 activation (Fig. 7B). Indeed, Western blot analysis clearly shows the existence of trace amounts of mature caspase-3 in our standard preparations of procaspase-3 before the start of our activation assays (Fig. 8, C and E).
This begs the following question. Can trace levels of caspase-3 support activation of procaspase-3 in the presence of our fibrils? A sub-stoichiometric amount of active caspase-3 (5%) was added to the inactive procaspase-3 (C163A) and found to markedly increase proenzyme processing in the presence of the nanofibrils (Fig. 8F). Note that 1541 does not alter the cleavage products generated from procaspase-3 activation. SDS-PAGE and mass spectrometry analyses both show the same processed products of procaspase-3 irrespective of the presence of the fibrils (data not shown). We also demonstrated above that the activity of mature caspase-3 against a tetrapeptide substrate (Ac-DEVD-afc) is unaffected by 10 μm 1541 (Fig. 5A). This indicates that the nanofibrils do not further stimulate the activity of mature caspase-3 but rather promote enhanced cleavage of procaspase-3 by the active enzyme when concentrated on the nanofibrils.
Procaspase-3 Is an Extremely Inactive Zymogen
Given the fact that we were unable to see any evidence for processing driven in cis or in trans by the proenzyme form, we were motivated to compare the basal activities of procaspase-3 and caspase-3. Using the standard tetrapeptide substrate, Ac-DEVD-afc, we found that mature caspase-3 has a catalytic efficiency (kcat/Km) of 7.6 × 105 m−1 s−1 and a Km of 12.0 μm, in agreement with previous reports (Table 3) (52–54).
TABLE 3.
Contaminants of mature caspase-3 present with procaspase-3 dominates activity measurements
| Construct | I/Ea | Km | kcat | kcat/Km |
|---|---|---|---|---|
| μm | s−1 | m−1 s−1 | ||
| Caspase-3 | 0 | 12.0 ± 0.7 | 9.11 ± 0.15 | 7.6 × 105 |
| Caspase-3 | 0.05 | 13.3 ± 0.6 | 9.34 ± 0.14 | 7.0 × 105 |
| Pro-3 (D3A)b | 0 | 11.2 ± 0.4 | 0.0031 ± (1.0 × 10−4) | 2.8 × 102 |
| Pro-3 (D3A)b,c | 0.01 | 370 ± 30 | 0.00098 ± (1.0 × 10−4) | <3.0 |
| Pro-3 (D3A)b,c | 0.05 | 890 ± 190 | 0.00069 ± (1.2 × 10−4) | <1.0 |
| Pro-3 (WT) | 0 | 9.8 ± 0.7 | 0.00048 ± (1 × 10−5) | 4.9 × 101 |
| Pro-3 (WT) | 0.01 | No activity observed | ||
| Pro-3 (D3A)d | 0 | 9.8 ± 0.6 | 0.00033 ± (1 × 10−5) | 3.4 × 101 |
| Pro-3 (D3A)d | 0.01 | No activity observed |
a I/E = [Ac-DEVD-cmk]/[enzyme].
b Expression time = 8 h.
c Saturation was not reached.
d Expression time = 2.5 h. Pro-3 (D3A) = uncleavable procaspase-3 (D9A/D28A/D75A). Pro-3 (WT) = wild-type procaspase-3.
We found that the catalytic efficiency of our standard preparation of wild-type procaspase-3 (4.9 × 101 m−1 s−1) is roughly 15,000-fold less than the mature enzyme (Table 3). However, the Km value measured (9.8 μm) is suspiciously similar to the cleaved caspase-3 (12.0 μm), and trace amounts of caspase-3 are present, as shown in Fig. 8E. These data prompted us to determine whether the procaspase-3 activity that we observed was dominated by a caspase-3 contaminant. Indeed, the addition of 1% stoichiometric equivalent of Ac-DEVD-cmk inhibitor to 5 μm procaspase-3 reduced the activity against Ac-DEVD-afc to undetectable levels. Control studies show that the addition of 5% of Ac-DEVD-cmk to caspase-3 minimally impacted its catalytic efficiency, showing that sub-stoichiometric concentrations of the inhibitor did not unexpectedly trigger gross adverse effects on enzyme activity (Table 3).
To quantify the amount of caspase-3 contaminant in our wild-type procaspase-3 preparation, we took 5 μm of the proenzyme and added a dilution series of Ac-DEVD-cmk. After a 1-h incubation with the inhibitor, 100 μm Ac-DEVD-afc was added to each sample. Activity was monitored, and initial rates were plotted versus inhibitor concentration. The initial plateau of the activity was determined to be the concentration of contaminating caspase-3, which we estimate at 10 nm active caspase-3 or 0.2% of the total amount of proenzyme (Fig. 10A).
FIGURE 10.

Multiple mature caspase-3 products identified by active site titration with Ac-DEVD-cmk. An active site titration with Ac-DEVD-cmk was performed against 5 μm wild-type procaspase-3, which was expressed for 20 min (A), 5 μm uncleavable procaspase-3 (D9A/D28A/D175A), which was expressed for 8 h (B), or 10 μm uncleavable procaspase-3, which was expressed for 2.5 h (C). The titration was plotted on a log scale. The asterisk indicates stoichiometric concentrations of Ac-DEVD-cmk relative to enzyme concentrations. The initial drop in activity for all three procaspase preparations occurred at sub-stoichiometric concentrations. Note the distinct levels of maximum activity for each proenzyme batch. This indicates that increased expression times lead to increased levels of contaminating active enzyme. Also note the second species detected in the uncleavable procaspase-3 expressed for 8 h. Based on the kinetic measurements described in the text, this second species weakly interacts with Ac-DEVD-cmk and is most likely an alternate cleavage product or a hemi-cleaved dimer.
As an orthogonal test of the intrinsic activity of the full-length procaspase-3, we assayed the activity of the uncleavable procaspase-3 (D9A/D28A/D175A), reasoning that it should be incapable of autocatalytic maturation (34). Consistent with previous reports, we found a catalytic efficiency roughly 3000-fold lower for the triple mutant (2.8 × 102 m−1 s−1) relative to the mature enzyme (Table 3) (52–54). However, the Km value for the uncleavable procaspase-3 (11.2 μm) was again suspiciously similar to caspase-3. Indeed, we found a drastic decrease in the catalytic efficiency upon the addition of 1% Ac-DEVD-cmk to 3.0 m−1 s−1, due to both an increase in Km and decrease in kcat. These kinetic values were reduced further to 1.0 m−1 s−1 upon the inclusion of 5% Ac-DEVD-cmk (Table 3). Because such low concentrations of Ac-DEVD-cmk do not covalently modify procaspase-3 (Fig. 3), there appears to be at least two contaminant populations present in the uncleavable procaspase-3 preparations. The initial drop in activity by 90-fold suggests a population of mature caspase-3 that binds Ac-DEVD-cmk well, similar to the fully mature caspase-3.
A second population of cleaved caspase-3 appears to be present that does not bind tightly to the inhibitor, as shown by the subsequent smaller 3-fold drop in activity (Table 3). This population, which is resistant to inhibition by Ac-DEVD-cmk and retains diminished activity, appears to also be present in recombinant mature caspase-3 but not the inactive procaspase-3 (C163A) (data not shown). This indicates that the second contaminant is also a cleavage product of full-length proenzyme, likely due to an alternate cleavage site in the intersubunit linker or a hemi-cleaved dimer generated during expression in Escherichia coli (Fig. 11) (55–57). Indeed, shorter expression times (2.5 versus 8 h) resulted in roughly 80-fold lower catalytic activity for the uncleavable procaspase-3 (D9A/D28A/D175A) (Table 3). No activity was detectable for this protein upon the addition of 1% Ac-DEVD-cmk.
FIGURE 11.

Wild-type procaspase-3 and uncleavable procaspase-3 (D9A/D28A/D175A) have very small levels of mature caspase-3 in the preparations. A, after ion-exchange chromatography on wild-type procaspase-3, fractions were collected. Aliquots of each fraction were analyzed by SDS-PAGE followed by Coomassie stain. B, similar procedure was performed on the uncleavable procaspase-3 (D9A/D28A/D175A). C, uncleavable procaspase-3 from batches expressed for 8 h (red) and 2.5 h (blue) was run on a gel filtration column. No differences or impurities were detected by gel filtration. D, 15 μl of 5 μm samples of the uncleavable procaspase-3 (D9A/D28A/D175A) from different batches expressed for different times were analyzed by SDS-PAGE followed by Western blot with an antibody that recognizes the C terminus of the cleaved caspase-3 large subunit (Cell Signaling, catalog no. 9664). Because a large excess of the full-length procaspase-3 is loaded relative to the trace caspase-3 contaminants in each sample, the antibody nonspecifically recognizes the full-length band as well.
We next estimated the amount of caspase-3 contaminant in 10 μm uncleavable procaspase-3 (D9A/D28A/D175A), expressed for 2.5 h. Again, by dosing in Ac-DEVD-cmk to determine where the activity plateaued, we estimate a 40 nm contamination of caspase-3 (0.4%). Using a similar approach, we observed at least two distinct contaminants in the batch of uncleavable procaspase-3 expressed for 8 h, consistent with the kinetic observations described (Fig. 10, B and C).
High Zymogenicity of Procaspase-3
The ratio of the activity of a mature enzyme to the activity of its precursor is referred to as a zymogenicity value. Zymogenicity describes the extent to which an enzyme is trapped as its inactive precursor, with larger values signifying a more restrained proenzyme. Previous measurements for the zymogenicity of procaspase-3 have been estimated at >10,000 (39, 52, 58). However, we felt this value should be revisited given the clear presence of active caspase-3 in procaspase-3 preparations, which we could eliminate using Ac-DEVD-cmk. Our experiments indicate procaspase-3 is restrained to an even greater extent with a zymogenicity >107, essentially the detection limit of the activity assay (Table 4).
TABLE 4.
Summary of zymogenicity values
This zymogenicity value was determined by comparing the maximum concentration of procaspase-3 (50 μm), where we observed no activity, with the limit of detection in our catalytic efficiency assays against Ac-DEVD-afc. Because we do not observe any activity for the proenzyme, our assessment of procaspase-3 zymogenicity includes the assumption that the Km value is similar for both the mature enzyme and its precursor. This approximation is a lower limit because our results above suggest that the Km value for procaspase-3 against Ac-DEVD-afc is actually much greater than caspase-3. We next evaluated the limit of detection by diluting caspase-3 below a concentration (5 pm) where no activity could be observed against Ac-DEVD-afc. This is a conservative estimate because dilute enzymes can associate with the surface of assay plates. Even with the conservative approximations, the ratio of these two values gives us a zymogenicity estimate of >107.
Amyloid-β Fibrils and 1541 Nanofibrils Activate Procaspase-3 by a Similar Mechanism
In previous studies, we showed that the proteogenic amyloid-β(1–40) fibrils also promoted procaspase-3 activation (23). Here, we sought to determine whether the activation mechanism by amyloid-β fibrils is analogous to that of 1541.
First, we wished to determine whether there was direct interaction between the fibrils, procaspase-3, and caspase-3. Indeed, procaspase-3, caspase-3, as well as caspase-8 can bind and cosediment with amyloid-β(1–40) fibrils (Fig. 12A and data not shown). Next, we sought to evaluate if this association results in increased maturation of the wild-type precursor to a large and small subunit. The amyloid-β fibrils stimulated processing of procaspase-3 as a function of time; however, 1541 acts ∼2–4-fold faster (Fig. 12B). The incubation period before robust procaspase-3 cleavage is more variable for the amyloid-β fibrils than for 1541 nanofibrils, depending on the exact preparation of the fibrils (data not shown). Transmission electron microscopy studies have shown that amyloid-β(1–40) can also form small oligomers as well as longer fibrils (59, 60), whereas 1541 fibrils are longer and more regular (23). The heterogeneity in the amyloid-β(1–40) fibrils may account for the differences seen in procaspase-3 activation kinetics.
FIGURE 12.

Amyloid-β(1–40) fibrils bind and enhance procaspase-3 activation. A, 50 μm amyloid-β(1–40) was incubated at 37 °C for 6 h to form fibrils. In 500 μl of buffer, 20 μm Aβ(1–40) fibrils and 0.1 mg/ml procaspase-3 or caspase-3 was added, incubated at room temperature for 5 min, and centrifuged at 20,800 × g for 20 min. The pellets were analyzed for the amounts of the respective enzymes. B, after a 4-h incubation at 37 °C, amyloid-β(1–40) or buffer alone was added to 250 nm wild-type procaspase-3. Processing was evaluated at the indicated time points. C, processing of 250 nm wild-type procaspase-3 with or without amyloid-β(1–40) fibrils was evaluated in the presence of 50 nm Ac-DEVD-cmk. D, similar preparation of amyloid-β(1–40) or buffer alone was incubated with 200 nm truncated inactive procaspase-3 (29-277/C163A) and 5 nm caspase-3. Processing was again evaluated at the indicated time points.
Similar to 1541, the activation of wild-type procaspase-3 on the amyloid-β fibrils is blocked by addition of sub-stoichiometric concentrations of Ac-DEVD-cmk, suggesting a need for an initiator protease (Fig. 12C). This is consistent with trace amounts of mature caspase-3 being necessary to drive proenzyme maturation. Furthermore, the amyloid-β(1–40) fibrils also enhance the rate of procaspase-3 (29-277/C163A) processing by exogenous caspase-3 (Fig. 12D). The variability in the rates of activation makes it difficult to evaluate a precise change in the catalytic efficiency of caspase-3 against the zymogen precursor.
Although caspase-8 can bind the amyloid-β(1–40) fibrils, we do not observe a significant change in procaspase-3 processing, in contrast to the 28-fold enhancement seen with 1541 fibrils (Fig. 4B). A number of factors may contribute to the difference in rate enhancement, including the orientation of caspase-8 on the amyloid-β fibrils or its ability to interact with amyloid-β oligomers versus fibrils. The data show amyloid-β(1–40) can enhance processing by promoting colocalization of active caspase-3 and procaspase-3, albeit more slowly than for 1541.
DISCUSSION
Procaspase-3 Is Activated by Initiator Proteases on Synthetic and Natural Scaffolds
Our studies demonstrate that scaffolding, such as synthetic or natural fibrils, colocalizes procaspase-3 with caspase-3 to promote explosive activation in vitro. A collection of experiments, including cosedimentation studies with 1541 and amyloid-β(1–40) fibrils, mutational studies on procaspase-3, and thorough biochemical characterization of procaspase-3 activity, show that procaspase-3 activation does not happen to any extent by procaspase-3 alone (Fig. 7A). Instead, proenzyme maturation requires a small amount of an activated caspase to bind and colocalize with the procaspase-3 on an ordered fibrillar scaffold (Fig. 7B).
Interestingly, procaspase-3 activation by upstream proteases on 1541 nanofibrils exhibits some protease specificity. For example, the 1541 nanofibrils show the greatest enhancement and binding for caspase-8, followed by caspase-3 and granzyme-B. We show that this colocalization of procaspase-3 with upstream proteases results in an effective change in the catalytic efficiency of the mature enzymes for processing the proenzyme, but it does not change the intrinsic activity of the activating protease to cleave soluble synthetic substrates. However, 1541 nanofibrils do not enhance proteolysis by some noncognate proteases like thermolysin or TEV protease, even when presented to procaspase-3 variants containing consensus TEV protease cleavage sites in place of the natural caspase cleavage site.
The specificity of this process seems to also be at the level of the chemical composition of the fibril. Previous and unpublished studies show fibril-forming variants of 1541 can be specific for activation of procaspases-3, -6, and -7 (22, 23).5 We show here that procaspase-3 can be activated on amyloid-β(1–40) fibrils by a similar mechanism as on 1541 fibrils. Additional studies to explore the influence of different fibrillar structures on the catalytic efficiency of various upstream proteases against procaspase-3 will be instructive in identifying biologically relevant activation platforms.
Because the caspases initiate fate-determining transformations in the cells, it is not surprising that their activity would be highly restricted in the absence of a signaling event (9). Procaspases are classically recruited and activated on scaffolding complexes, such as the apoptosome, the inflammasome, and the death-inducing signaling complex (5, 8–10, 14, 15, 61). The induced proximity model that describes clustering and activation of procaspase-8 and procaspase-9 upon localization to such platforms provides a similar model for procaspase-3 activation (34, 35, 58, 62). Our data further suggests that procaspase-3 recruitment alone is not sufficient for auto-activation, but induced proximity with an active caspase is critical for maturation of the proenzyme on fibrils.
Previous confocal microscopy and immunoprecipitation studies have illustrated a direct interaction between mature caspases and specific intracellular fibrillar structures. For example, caspase-9 and caspase-3 appear to colocalize with cytokeratin 18 in epithelial cells upon the addition of an apoptotic inducer (31, 32). In neurodegenerative disorders, such as Huntington disease, caspases can also localize to aggregates or fibrils associated with disease progression (26). Also, the death effector domain motifs in the prodomain of caspase-8 can assemble into filaments in cells to promote apoptosis (63, 64). Although localization has been demonstrated previously, our results show that fibrils can serve as a direct platform for procaspase recruitment and maturation in vitro. Furthermore, we detail the specific mechanism that results in procaspase activation upon recruitment to the fibrils in vitro. However, additional studies are necessary to understand if these mechanisms apply to the cellular activities of 1541 (22), amyloid-β fibrils, or other intracellular fibrillar structures (28, 65).
Previous studies demonstrate that enzymes can retain activity upon fibril formation or fibril association. Zymogens in the blood coagulation cascade are also well known to be activated on fibers. For example, tissue plasminogen activator is stimulated by fibrinogen and more dramatically by fibrin (66). Remarkably, amyloid-β fibrils also enable activation of tissue plasminogen activator (67). Furthermore, distinct aggregation states and cleavage products of the amyloid-β precursor protein have distinct impact on the tissue plasminogen activator catalytic efficiency against its substrate plasminogen. This observation has implicated amyloid-β fibrils in the pathogenesis of hereditary cerebral hemorrhage with amyloidosis-Dutch type.
Lag in Procaspase-3 Activation Is Due to a Second-order Reaction Rate
Our results show that procaspase-3 lacks any detectable activity and cannot auto-process. Furthermore, 1541 nanofibrils form immediately upon addition to buffer (23). Both procaspase-3 and mature caspase-3 can also associate rapidly with the fibrils. Thus, the lag observed for procaspase-3 activation in the presence of 1541 nanofibrils is attributable to the rate of reaction of procaspase-3 cleavage by caspase-3. A distinct delay or lag in activation is also observed for wild-type procaspase-3 activation alone at high concentrations of the proenzyme (Fig. 9). The 1541 fibrils act to enhance the natural activation process through concentration or colocalization of procaspase-3 with mature caspase-3.
A significant lag in activation would be predicted to exist even in the presence of a trace amount of mature enzyme. We further confirmed this observation through calculation of the activation curves for procaspase-3 using the Michaelis-Menten equation, taking into account an autocatalytic maturation of 100 nm procaspase-3 upon the inclusion of a trace amount of caspase-3 (0.2 nm). These initial parameters were based on our typical assay conditions, which contain 100 nm procaspase-3. We also assumed a Km of 140 μm, which represents the Km value measured for caspase-3 against the Ac-IETD-afc substrate. With these parameters, we calculated an S-shaped activation curve, including the lag, which closely matched our experimental data (data not shown). Thus, the activity of mature caspase-3 alone in our population of wild-type procaspase-3 is sufficient to explain the activation curves observed experimentally. Similar activation curves have been demonstrated for autocatalytic activation of trypsinogen by a low level of trypsin (47, 48).
Importance of Zymogenicity in the Mechanism of Caspase Activation
Numerous structural and biochemical studies have delineated the unique mechanisms for maintaining proteases as inactive zymogens (3). Such mechanisms include an N-terminal domain that may occlude substrate from docking, a conformational change that may alter or disorder substrate binding or catalytic residues, and oligomerization or interaction with an activating partner protein or small molecule (1). In almost all cases, a proteolytic processing event is critical for activation of the proenzyme by another protease.
Interestingly, different proteases are locked in this inactive or zymogen state to variable extents (58, 68). Table 4 summarizes the zymogenicity values for some well characterized proteases. For example, tissue plasminogen activator exhibits a zymogenicity between 2 and 10, which reflects a relatively small difference between the activity of the cleaved form and its full-length precursor (69–72). In stark contrast, trypsinogen and chymotrypsinogen are much more inactive, which is revealed by zymogenicities on the order of 104 to 106 (73, 74). Although the precursors for trypsin and chymotrypsin display nominal activity in vitro, their cellular regulation is due to processing from upstream proteases. Other proteases have intermediate values such as factor XIIa, which has a zymogenicity value of 4000 (75). The tremendous range in zymogenicity values likely reflects on the in vivo function and activation of the proteases (76).
Our results suggest that procaspase-3 has a much greater zymogenicity than other proteases and is at least 3 orders of magnitude less active than previous reports, based upon the activity of the uncleavable mutant (D9A/D28A/D175A) (39, 58). Key to this assessment was the unexpected discovery that active caspase-3 is present in preparations of the “uncleavable” procaspase-3 (D9A/D28A/D175A). The active product was probably generated from noncaspase proteolysis during expression in E. coli. When the trace contaminants were inactivated using sub-stoichiometric amounts of the active site inhibitor, Ac-DEVD-cmk, we detected no activity. Based on the limit of detection of the assay, we estimate the activity for procaspase-3 to be 1/10,000,00 that of mature caspase-3.
The extreme zymogenicity of procaspase-3 is not surprising given the fact that caspase-3 executes the final and fate-determining stages of apoptosis. Moreover, this high zymogenicity barrier probably accounts for the lack of any evidence for procaspase-3 auto-processing. Precursors that exhibit such high zymogenicities have typically required harsh conditions in vitro to observe any activity. Studies with trypsinogen and chymotrypsinogen used high enzyme concentrations in the micromolar to millimolar range, low pH conditions, high ionic strength buffers, and long time courses on the orders of days to detect any processing by these precursors (34, 48, 74, 77, 78).
Challenges for Small Molecule Discovery
Our results strongly suggest that auto-activation of procaspase-3 is extremely restricted or nonexistent under typical cellular enzyme concentrations, ∼100 nm, as well as at higher proenzyme concentrations. Activation almost certainly requires at least trace amounts of active caspase or an initiator protease to trigger processing. Based on the zymogenicity approximation here and modeling activation curves using the Michaelis-Menten equation, we estimate a small molecule activator would need to stimulate the activity of the proenzyme over 1000-fold to generate 0.2% fractional activity to promote autocatalytic maturation of the entire procaspase-3 population within a 24-h period. Even increasing the enzyme concentration 10-fold, to super-physiological levels (1 μm), would only decrease this barrier to ∼100-fold.
The high energetic zymogenicity barrier indicates a challenging endeavor for a small molecule to directly bind and activate procaspase-3. From our results, procaspase-3 maturation is critically dependent on the presence of active caspase. Others have reported small molecule activators of procaspase-3. In the case of PAC-1, this small molecule is proposed to function by removing an inhibitory Zn2+ ion (33). Clark and co-workers report another compound identified using a DOCK screen (21). This small molecule activator is proposed to bind to an allosteric site on procaspase-3 to promote activation. Additional studies will be important to elucidate if these compounds work by a direct activation of the proenzyme or possibly act on trace contaminants of mature caspase-3 to initiate the process.
The above considerations do not mean that one cannot activate a protease precursor with a small molecule. In fact, an endogenous small molecule, inositol hexaphosphate, is known to bind an allosteric site on the cysteine protease domain of MARTXVc toxin to promote activation (80). It would be interesting to explore the zymogenicity of the cysteine protease domain. Perhaps enzymes with low zymogenicity values would be better candidates for small molecule activator discovery.
Our results indicate that increasing the activity of the mature caspase-3 or another upstream protease may also promote activation of the procaspase-3. Previous studies have demonstrated conformational flexibility of the mature caspases (81, 82). As little as a 2-fold change in the catalytic efficiency of caspase-3 would be enough to promote autocatalytic maturation of the proenzyme with a 0.2% contaminant. Our studies also suggest that facilitating an ordered oligomerization or colocalization of procaspase-3 with the mature caspase-3 would also stimulate activation. The latter two approaches, however, depend on the cellular concentrations of both pro- and mature enzymes and are not direct activators of the proenzyme per se. Importantly, successful protein engineering approaches for procaspase activation have been described for both these strategies (7, 35, 79, 83–86).
Acknowledgments
We thank O. Julien for support and for critically reviewing the manuscript. We thank D. Hostetter and C. Tajon from the Craik laboratory at the University of California, San Francisco, for providing granzyme B. We thank the Wells laboratory and the Small Molecule Discovery Center (University of California, San Francisco) for useful suggestions.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 CA136779, Grant F32 CA119641-03 from NCI (to D. W. W.), and Grant F32 AI077177 (to N. J. A.). This work was also supported by an Achievement Rewards for College Scientists (ARCS) Foundation Award (to J. A. Z.), a Scleroderma Research Foundation Evnin-Wright Fellowship (to J. A. Z.), and the Multiple Myeloma Translational Initiative (MMTI) at UCSF.
J. A. Zorn, D. W. Wolan, and J. A. Wells, unpublished results.
- afc
- amino-4-trifluoromethylcoumarin
- Aβ(1–40)
- amyloid-β(1–40)
- cmk
- chloromethyl ketone
- TEV
- tobacco etch virus
- LDS
- lithium dodecyl sulfate.
REFERENCES
- 1. Turk B. (2006) Targeting proteases. Successes, failures, and future prospects. Nat. Rev. Drug Discov. 5, 785–799 [DOI] [PubMed] [Google Scholar]
- 2. Khan A. R., James M. N. (1998) Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 7, 815–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stroud R. M., Kossiakoff A. A., Chambers J. L. (1977) Mechanisms of zymogen activation. Annu. Rev. Biophys. Bioeng. 6, 177–193 [DOI] [PubMed] [Google Scholar]
- 4. Shen A. (2010) Allosteric regulation of protease activity by small molecules. Mol. Biosyst. 6, 1431–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Donepudi M., Grütter M. G. (2002) Structure and zymogen activation of caspases. Biophys. Chem. 101, 145–153 [DOI] [PubMed] [Google Scholar]
- 6. Stennicke H. R., Salvesen G. S. (2000) Caspases. Controlling intracellular signals by protease zymogen activation. Biochim. Biophys. Acta 1477, 299–306 [DOI] [PubMed] [Google Scholar]
- 7. Gray D. C., Mahrus S., Wells J. A. (2010) Activation of specific apoptotic caspases with an engineered small molecule-activated protease. Cell 142, 637–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Salvesen G. S., Riedl S. J. (2008) Caspase mechanisms. Adv. Exp. Med. Biol. 615, 13–23 [DOI] [PubMed] [Google Scholar]
- 9. Li J., Yuan J. (2008) Caspases in apoptosis and beyond. Oncogene 27, 6194–6206 [DOI] [PubMed] [Google Scholar]
- 10. Yuan S., Yu X., Asara J. M., Heuser J. E., Ludtke S. J., Akey C. W. (2011) The Holo-apoptosome. Activation of procaspase-9 and interactions with caspase-3. Structure 19, 1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Renatus M., Stennicke H. R., Scott F. L., Liddington R. C., Salvesen G. S. (2001) Dimer formation drives the activation of the cell death protease caspase 9. Proc. Natl. Acad. Sci. U.S.A. 98, 14250–14255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donepudi M., Mac Sweeney A., Briand C., Grütter M. G. (2003) Insights into the regulatory mechanism for caspase-8 activation. Mol. Cell 11, 543–549 [DOI] [PubMed] [Google Scholar]
- 13. Yang X., Chang H. Y., Baltimore D. (1998) Autoproteolytic activation of pro-caspases by oligomerization. Mol. Cell 1, 319–325 [DOI] [PubMed] [Google Scholar]
- 14. Twiddy D., Cohen G. M., Macfarlane M., Cain K. (2006) Caspase-7 is directly activated by the approximately 700-kDa apoptosome complex and is released as a stable XIAP-caspase-7 approximately 200-kDa complex. J. Biol. Chem. 281, 3876–3888 [DOI] [PubMed] [Google Scholar]
- 15. Yin Q., Park H. H., Chung J. Y., Lin S. C., Lo Y. C., da Graca L. S., Jiang X., Wu H. (2006) Caspase-9 holoenzyme is a specific and optimal procaspase-3 processing machine. Mol. Cell 22, 259–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Muzio M., Salvesen G. S., Dixit V. M. (1997) FLICE induced apoptosis in a cell-free system. Cleavage of caspase zymogens. J. Biol. Chem. 272, 2952–2956 [DOI] [PubMed] [Google Scholar]
- 17. Mahrus S., Trinidad J. C., Barkan D. T., Sali A., Burlingame A. L., Wells J. A. (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 134, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dix M. M., Simon G. M., Cravatt B. F. (2008) Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell 134, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lüthi A. U., Martin S. J. (2007) The CASBAH. A searchable database of caspase substrates. Cell Death Differ. 14, 641–650 [DOI] [PubMed] [Google Scholar]
- 20. Putt K. S., Chen G. W., Pearson J. M., Sandhorst J. S., Hoagland M. S., Kwon J. T., Hwang S. K., Jin H., Churchwell M. I., Cho M. H., Doerge D. R., Helferich W. G., Hergenrother P. J. (2006) Small molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy. Nat. Chem. Biol. 2, 543–550 [DOI] [PubMed] [Google Scholar]
- 21. Schipper J. L., MacKenzie S. H., Sharma A., Clark A. C. (2011) A bifunctional allosteric site in the dimer interface of procaspase-3. Biophys. Chem. 159, 100–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wolan D. W., Zorn J. A., Gray D. C., Wells J. A. (2009) Small molecule activators of a proenzyme. Science 326, 853–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zorn J. A., Wille H., Wolan D. W., Wells J. A. (2011) Self-assembling small molecules form nanofibrils that bind procaspase-3 to promote activation. J. Am. Chem. Soc. 133, 19630–19633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nikolaev A., McLaughlin T., O'Leary D. D., Tessier-Lavigne M. (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457, 981–989 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25. Wellington C. L., Ellerby L. M., Gutekunst C. A., Rogers D., Warby S., Graham R. K., Loubser O., van Raamsdonk J., Singaraja R., Yang Y. Z., Gafni J., Bredesen D., Hersch S. M., Leavitt B. R., Roy S., Nicholson D. W., Hayden M. R. (2002) Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington disease. J. Neurosci. 22, 7862–7872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hermel E., Gafni J., Propp S. S., Leavitt B. R., Wellington C. L., Young J. E., Hackam A. S., Logvinova A. V., Peel A. L., Chen S. F., Hook V., Singaraja R., Krajewski S., Goldsmith P. C., Ellerby H. M., Hayden M. R., Bredesen D. E., Ellerby L. M. (2004) Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington disease. Cell Death Differ. 11, 424–438 [DOI] [PubMed] [Google Scholar]
- 27. Wellington C. L., Hayden M. R. (2000) Caspases and neurodegeneration. On the cutting edge of new therapeutic approaches. Clin. Genet. 57, 1–10 [DOI] [PubMed] [Google Scholar]
- 28. Umeda T., Tomiyama T., Sakama N., Tanaka S., Lambert M. P., Klein W. L., Mori H. (2011) Intraneuronal amyloid β oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J. Neurosci. Res. 89, 1031–1042 [DOI] [PubMed] [Google Scholar]
- 29. Frost B., Diamond M. I. (2010) Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 11, 155–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Graham R. K., Ehrnhoefer D. E., Hayden M. R. (2011) Caspase-6 and neurodegeneration. Trends Neurosci. 34, 646–656 [DOI] [PubMed] [Google Scholar]
- 31. Lee J. C., Schickling O., Stegh A. H., Oshima R. G., Dinsdale D., Cohen G. M., Peter M. E. (2002) DEDD regulates degradation of intermediate filaments during apoptosis. J. Cell Biol. 158, 1051–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dinsdale D., Lee J. C., Dewson G., Cohen G. M., Peter M. E. (2004) Intermediate filaments control the intracellular distribution of caspases during apoptosis. Am. J. Pathol. 164, 395–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peterson Q. P., Goode D. R., West D. C., Ramsey K. N., Lee J. J., Hergenrother P. J. (2009) PAC-1 activates procaspase-3 in vitro through relief of zinc-mediated inhibition. J. Mol. Biol. 388, 144–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roy S., Bayly C. I., Gareau Y., Houtzager V. M., Kargman S., Keen S. L., Rowland K., Seiden I. M., Thornberry N. A., Nicholson D. W. (2001) Maintenance of caspase-3 proenzyme dormancy by an intrinsic “safety catch” regulatory tripeptide. Proc. Natl. Acad. Sci. U.S.A. 98, 6132–6137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Colussi P. A., Harvey N. L., Shearwin-Whyatt L. M., Kumar S. (1998) Conversion of procaspase-3 to an autoactivating caspase by fusion to the caspase-2 prodomain. J. Biol. Chem. 273, 26566–26570 [DOI] [PubMed] [Google Scholar]
- 36. Denault J. B., Salvesen G. S. (2003) Expression, purification, and characterization of caspases. Curr. Protoc. Protein Sci. Chapter 21, Unit 21.13 [DOI] [PubMed] [Google Scholar]
- 37. Stennicke H. R., Salvesen G. S. (1997) Biochemical characteristics of caspases-3, -6, -7, and -8. J. Biol. Chem. 272, 25719–25723 [DOI] [PubMed] [Google Scholar]
- 38. Stennicke H. R., Salvesen G. S. (1998) Properties of the caspases. Biochim. Biophys. Acta 1387, 17–31 [DOI] [PubMed] [Google Scholar]
- 39. Stennicke H. R., Salvesen G. S. (1999) Catalytic properties of the caspases. Cell Death Differ. 6, 1054–1059 [DOI] [PubMed] [Google Scholar]
- 40. Pop C., Timmer J., Sperandio S., Salvesen G. S. (2006) The apoptosome activates caspase-9 by dimerization. Mol. Cell 22, 269–275 [DOI] [PubMed] [Google Scholar]
- 41. Graybill T. L., Dolle R. E., Helaszek C. T., Ator M. A., Strasters J. (1995) Synthesis and evaluation of diacylhydrazines as inhibitors of the interleukin-1β-converting enzyme (ICE). Bioorg. Med. Chem. Lett. 5, 1197–1202 [Google Scholar]
- 42. Coan K. E., Shoichet B. K. (2008) Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 130, 9606–9612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Park C., Marqusee S. (2005) Pulse proteolysis. A simple method for quantitative determination of protein stability and ligand binding. Nat. Methods 2, 207–212 [DOI] [PubMed] [Google Scholar]
- 44. Timmer J. C., Zhu W., Pop C., Regan T., Snipas S. J., Eroshkin A. M., Riedl S. J., Salvesen G. S. (2009) Structural and kinetic determinants of protease substrates. Nat. Struct. Mol. Biol. 16, 1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pop C., Salvesen G. S., Scott F. L. (2008) Caspase assays. Identifying caspase activity and substrates in vitro and in vivo. Methods Enzymol. 446, 351–367 [DOI] [PubMed] [Google Scholar]
- 46. Smith T. J., Stains C. I., Meyer S. C., Ghosh I. (2006) Inhibition of β-amyloid fibrillization by directed evolution of a β-sheet presenting miniature protein. J. Am. Chem. Soc. 128, 14456–14457 [DOI] [PubMed] [Google Scholar]
- 47. Wu J. W., Wu Y., Wang Z. X. (2001) Kinetic analysis of a simplified scheme of autocatalytic zymogen activation. Eur. J. Biochem. 268, 1547–1553 [PubMed] [Google Scholar]
- 48. Kassell B., Kay J. (1973) Zymogens of proteolytic enzymes. Science 180, 1022–1027 [DOI] [PubMed] [Google Scholar]
- 49. Park C., Marqusee S. (2006) Quantitative determination of protein stability and ligand binding by pulse proteolysis. Curr. Protoc. Protein Sci. Chapter 20, Unit 20.11 [DOI] [PubMed] [Google Scholar]
- 50. Pop C., Chen Y. R., Smith B., Bose K., Bobay B., Tripathy A., Franzen S., Clark A. C. (2001) Removal of the prodomain does not affect the conformation of the procaspase-3 dimer. Biochemistry 40, 14224–14235 [DOI] [PubMed] [Google Scholar]
- 51. Svingen P. A., Loegering D., Rodriquez J., Meng X. W., Mesner P. W., Jr., Holbeck S., Monks A., Krajewski S., Scudiero D. A., Sausville E. A., Reed J. C., Lazebnik Y. A., Kaufmann S. H. (2004) Components of the cell death machine and drug sensitivity of the National Cancer Institute Cell Line Panel. Clin. Cancer Res. 10, 6807–6820 [DOI] [PubMed] [Google Scholar]
- 52. Pop C., Feeney B., Tripathy A., Clark A. C. (2003) Mutations in the procaspase-3 dimer interface affect the activity of the zymogen. Biochemistry 42, 12311–12320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ganesan R., Mittl P. R., Jelakovic S., Grütter M. G. (2006) Extended substrate recognition in caspase-3 revealed by high resolution x-ray structure analysis. J. Mol. Biol. 359, 1378–1388 [DOI] [PubMed] [Google Scholar]
- 54. Karki P., Lee J., Shin S. Y., Cho B., Park I. S. (2005) Kinetic comparison of procaspase-3 and caspase-3. Arch. Biochem. Biophys. 442, 125–132 [DOI] [PubMed] [Google Scholar]
- 55. Zhou Q., Salvesen G. S. (1997) Activation of procaspase-7 by serine proteases includes a noncanonical specificity. Biochem. J. 324, 361–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Denault J. B., Békés M., Scott F. L., Sexton K. M., Bogyo M., Salvesen G. S. (2006) Engineered hybrid dimers. Tracking the activation pathway of caspase-7. Mol. Cell 23, 523–533 [DOI] [PubMed] [Google Scholar]
- 57. Berger A. B., Witte M. D., Denault J. B., Sadaghiani A. M., Sexton K. M., Salvesen G. S., Bogyo M. (2006) Identification of early intermediates of caspase activation using selective inhibitors and activity-based probes. Mol. Cell 23, 509–521 [DOI] [PubMed] [Google Scholar]
- 58. Salvesen G. S., Dixit V. M. (1999) Caspase activation. The induced-proximity model. Proc. Natl. Acad. Sci. U.S.A. 96, 10964–10967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee J., Culyba E. K., Powers E. T., Kelly J. W. (2011) Amyloid-β forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 7, 602–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stine W. B., Jungbauer L., Yu C., LaDu M. J. (2011) Preparing synthetic Aβ in different aggregation states. Methods Mol. Biol. 670, 13–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schroder K., Tschopp J. (2010) The inflammasomes. Cell 140, 821–832 [DOI] [PubMed] [Google Scholar]
- 62. Srinivasula S. M., Ahmad M., Fernandes-Alnemri T., Alnemri E. S. (1998) Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol. Cell 1, 949–957 [DOI] [PubMed] [Google Scholar]
- 63. Yuan R. T., Young S., Liang J., Schmid M. C., Mielgo A., Stupack D. G. (2012) Caspase-8 isoform 6 promotes death effector filament formation independent of microtubules. Apoptosis 17, 229–235 [DOI] [PubMed] [Google Scholar]
- 64. Siegel R. M., Martin D. A., Zheng L., Ng S. Y., Bertin J., Cohen J., Lenardo M. J. (1998) Death-effector filaments. Novel cytoplasmic structures that recruit caspases and trigger apoptosis. J. Cell Biol. 141, 1243–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bucciantini M., Calloni G., Chiti F., Formigli L., Nosi D., Dobson C. M., Stefani M. (2004) Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J. Biol. Chem. 279, 31374–31382 [DOI] [PubMed] [Google Scholar]
- 66. Kranenburg O., Bouma B., Kroon-Batenburg L. M., Reijerkerk A., Wu Y. P., Voest E. E., Gebbink M. F. (2002) Tissue-type plasminogen activator is a multiligand cross-β structure receptor. Curr. Biol. 12, 1833–1839 [DOI] [PubMed] [Google Scholar]
- 67. Kingston I. B., Castro M. J., Anderson S. (1995) In vitro stimulation of tissue-type plasminogen activator by Alzheimer amyloid β-peptide analogues. Nat. Med. 1, 138–142 [DOI] [PubMed] [Google Scholar]
- 68. Tachias K., Madison E. L. (1996) Converting tissue-type plasminogen activator into a zymogen. J. Biol. Chem. 271, 28749–28752 [DOI] [PubMed] [Google Scholar]
- 69. Boose J. A., Kuismanen E., Gerard R., Sambrook J., Gething M. J. (1989) The single-chain form of tissue-type plasminogen activator has catalytic activity. Studies with a mutant enzyme that lacks the cleavage site. Biochemistry 28, 635–643 [DOI] [PubMed] [Google Scholar]
- 70. Tate K. M., Higgins D. L., Holmes W. E., Winkler M. E., Heyneker H. L., Vehar G. A. (1987) Functional role of proteolytic cleavage at arginine 275 of human tissue plasminogen activator as assessed by site-directed mutagenesis. Biochemistry 26, 338–343 [DOI] [PubMed] [Google Scholar]
- 71. Madison E. L., Kobe A., Gething M. J., Sambrook J. F., Goldsmith E. J. (1993) Converting tissue plasminogen activator to a zymogen. A regulatory triad of Asp-His-Ser. Science 262, 419–421 [DOI] [PubMed] [Google Scholar]
- 72. Madison E. L., Sambrook J. E. (1993) Probing structure-function relationships of tissue-type plasminogen activator by oligonucleotide-mediated site-specific mutagenesis. Methods Enzymol. 223, 249–271 [DOI] [PubMed] [Google Scholar]
- 73. Robinson N. C., Neurath H., Walsh K. A. (1973) The relation of the -amino group of trypsin to enzyme function and zymogen activation. Biochemistry 12, 420–426 [DOI] [PubMed] [Google Scholar]
- 74. Gertler A., Walsh K. A., Neurath H. (1974) Catalysis by chymotrypsinogen. Demonstration of an acyl-zymogen intermediate. Biochemistry 13, 1302–1310 [DOI] [PubMed] [Google Scholar]
- 75. Silverberg M., Kaplan A. P. (1982) Enzymatic activities of activated and zymogen forms of human Hageman factor (factor XII). Blood 60, 64–70 [PubMed] [Google Scholar]
- 76. Neurath H., Walsh K. A. (1976) Role of proteolytic enzymes in biological regulation (a review). Proc. Natl. Acad. Sci. U.S.A. 73, 3825–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lonsdale-Eccles J. D., Neurath H., Walsh K. A. (1978) Probes of the mechanism of zymogen catalysis. Biochemistry 17, 2805–2809 [DOI] [PubMed] [Google Scholar]
- 78. Kay J., Kassell B. (1971) The autoactivation of trypsinogen. J. Biol. Chem. 246, 6661–6665 [PubMed] [Google Scholar]
- 79. Muzio M., Stockwell B. R., Stennicke H. R., Salvesen G. S., Dixit V. M. (1998) An induced proximity model for caspase-8 activation. J. Biol. Chem. 273, 2926–2930 [DOI] [PubMed] [Google Scholar]
- 80. Lupardus P. J., Shen A., Bogyo M., Garcia K. C. (2008) Small molecule-induced allosteric activation of the Vibrio cholerae RTX cysteine protease domain. Science 322, 265–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hardy J. A., Lam J., Nguyen J. T., O'Brien T., Wells J. A. (2004) Discovery of an allosteric site in the caspases. Proc. Natl. Acad. Sci. U.S.A. 101, 12461–12466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Scheer J. M., Romanowski M. J., Wells J. A. (2006) A common allosteric site and mechanism in caspases. Proc. Natl. Acad. Sci. U.S.A. 103, 7595–7600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. MacCorkle R. A., Freeman K. W., Spencer D. M. (1998) Synthetic activation of caspases. Artificial death switches. Proc. Natl. Acad. Sci. U.S.A. 95, 3655–3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tse E., Rabbitts T. H. (2000) Intracellular antibody caspase-mediated cell killing. An approach for application in cancer therapy. Proc. Natl. Acad. Sci. U.S.A. 97, 12266–12271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Oberst A., Pop C., Tremblay A. G., Blais V., Denault J. B., Salvesen G. S., Green D. R.(2010) Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J. Biol. Chem. 285, 16632–16642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chang D. W., Xing Z., Capacio V. L., Peter M. E., Yang X. (2003) Interdimer processing mechanism of procaspase-8 activation. EMBO J. 22, 4132–4142 [DOI] [PMC free article] [PubMed] [Google Scholar]