

Background: Plant NTRC contains a flavin, a disulfide group, and an extra thioredoxin module.
Results: The flavin cofactor in NTRC is oxidized following two intramolecular reactions that are altered by peroxiredoxin.
Conclusion: In comparison with canonical NTRs, NTRC shows additional conformational dynamics affected by peroxiredoxin.
Significance: NTRC is involved in the response to oxidative stress and in maintaining the redox homeostasis of plastids.
Keywords: Electron Transfer, Peroxiredoxin, Protein Dynamics, Redox Regulation, Thioredoxin, NTRC, Chloroplast NADPH-dependent Thioredoxin Reductase C
Abstract
NADPH-dependent thioredoxin reductases (NTRs) contain a flavin cofactor and a disulfide as redox-active groups. The catalytic mechanism of standard NTR involves a large conformational change between two configurations. Oxygenic photosynthetic organisms possess a plastid-localized NTR, called NTRC, with a thioredoxin module fused at the C terminus. NTRC is an efficient reductant of 2-Cys peroxiredoxins (2-Cys Prxs) and thus is involved in the protection against oxidative stress, among other functions. Although the mechanism of electron transfer of canonical NTRs is well established, it is not yet known in NTRC. By employing stopped-flow spectroscopy, we have carried out a comparative kinetic study of the electron transfer reactions involving NTRC, the truncated NTR module of NTRC, and NTRB, a canonical plant NTR. Whereas the three NTRs maintain the conformational change associated with the reductive cycle of catalysis, NTRC intramolecular electron transfer to the thioredoxin module presents two kinetic components (kET of ∼2 and 0.1 s−1), indicating the occurrence of additional dynamic motions. Moreover, the dynamic features associated with the electron transfer to the thioredoxin module are altered in the presence of 2-Cys Prx. NTRC shows structural constraints that may locate the thioredoxin module in positions with different efficiencies for electron transfer, the presence of 2-Cys Prx shifting the conformational equilibrium of the thioredoxin module to a specific position, which is not the most efficient.
Introduction
Flavoproteins, acting as redox enzymes, play different roles in many cellular processes by transferring electrons to a protein substrate or another electron acceptor, as quinones or dithiols (1–3). NADPH-dependent thioredoxin reductases (NTRs),2 in particular, are flavo-disulfide enzymes that act as key modulators determining the redox state of their thiol-containing thioredoxin (Trx) substrates (1, 4, 5). Trxs are small proteins (∼12 kDa) containing two redox-active cysteine residues (6, 7), which act as redox regulators involved in the defense against oxidative stress by modifying the redox state of disulfide groups of numerous proteins. Trxs are as well able to deliver reducing equivalents to peroxiredoxins (Prxs), and they are therefore involved in the mechanism of defense against oxidative stress (5–8).
In plants, as in other organisms, the NTR/Trx system plays an important role in redox regulation, although plants are unique in having a rather complex family of genes encoding Trxs (5, 6, 9). Plant cells cope with oxidative stress by using a number of antioxidant systems. In particular, one of the plant systems that control intracellular hydrogen peroxide levels is based on the activity of Prxs, thiol-based peroxidases that are reduced by specific NTR/Trx systems (10–12) and, in the case of some plant Prxs, also by the glutaredoxin system (13). Plant genomes contain numerous genes coding for Prxs, with cytosolic, plastidial, and even nuclear localization. Remarkably, the chloroplast is the plant organelle with the highest content of Prxs forms. The Arabidopsis chloroplast is equipped with four Prx: two almost identical typical 2-Cys Prxs, stated A and B, the atypical Prx Q, and a type II Prx, stated Prx IIE (10, 11).
The Arabidopsis genome contains two genes (NTRA and NTRB) encoding canonical NTRs. NTRB is more abundant in the cytosol, whereas NTRA is more abundant in the mitochondria (14). As canonical NTRs, both NTRA and NTRB are NADPH-dependent thioredoxin reductases containing a bound FAD cofactor and an active two-Cys motif (5). The structure of Arabidopsis NTRB has been solved at 2.5 Å resolution, revealing a dimeric enzyme structurally similar to that of the previously solved Escherichia coli protein (4, 15, 16). Thus, each NTR monomer presents two domains, one containing the FAD cofactor and the other the active 2-Cys group together with the NADPH-binding site (15). The catalytic mechanism of NTRs has been intensively studied in the prokaryotic enzymes (4, 17–21), and the structural similarities suggest a similar enzymatic mechanism in plant NTRs. Thus, the reduction cycle involves large interdomain conformational movements, the protein shifting between two conformers (4, 16, 18, 19, 21). Interconversion of conformers enables FAD reduction by NADPH and then FAD oxidation by the redox-active disulfide at the other domain of the enzyme. Fast kinetic studies have established that the large interdomain rotation allowing FAD reduction by NADPH is probably the rate-limiting step of catalysis (4, 16, 18).
In plant chloroplasts, Trxs were considered to be exclusively reduced by a ferredoxin-dependent thioredoxin reductase (FTR), so that these organelles were thought to have unique properties regarding redox regulation (5). This view of the system of redox regulation in chloroplasts changed after the discovery of a plastid-localized NTR, termed NTRC, which contains a joint Trx module at the C terminus connected by an ∼20-amino acid flexible linker (22). NTRC is exclusive of oxygenic photosynthetic organisms: plants, algae, and some, not all, cyanobacteria (22, 23). NTRC is able to conjugate both NTR and Trx activities to efficiently reduce 2-Cys Prxs (24–26). By doing so, NTRC was proposed to perform an antioxidant function. Indeed, it was shown that a peroxide-sensitive cyclase involved in the chlorophyll biosynthesis pathway was protected by the NTRC/2-Cys Prx system in Arabidopsis (27). More recently, it has been established that NTRC functions not only as antioxidant but also in redox regulation of starch synthesis (28) or aromatic amino acids and auxin synthesis (29). Moreover, the localization of NTRC in both chloroplasts and non-green plastids has been established (30), and thus, it may be considered an important component of redox regulation in plants.
A reaction mechanism was proposed for NTRC in its reaction with 2-Cys Prx (31), according to which the catalytic unit is a homodimer, the transfer of electrons taking place from the NTR module of one subunit to the Trx module of the other subunit. Although NTRC can form oligomeric states, the dimeric species has been proposed as the functional unit (31, 32). There are two additional examples of hybrid proteins with NTR and Trx modules, both found in bacteria. The first one was identified in Mycobacterium leprae (33). This bacterial protein and NTRC are organized in the same way, and there is very little difference in the linker region (22, 33). The second one is the protein AhpF from Salmonella typhimurium (34), which is formed by an NTR domain at the C terminus, and a double Trx fold at the N terminus containing only a single disulfide active center.
Despite the good knowledge of the dynamics and mechanism of electron transfer of canonical NTRs, nothing is yet known of these processes in NTRC. Thus, a pertinent question is whether the conformational change mechanism described for the canonical E. coli NTR is also occurring in the bimodular NTRC. With the objective of addressing this question, we have carried out a kinetic study of the electron transfer (ET) properties of rice NTRC in a comparative way with both the truncated NTR-like module (NTRCM) and the canonical plant NTRB.
EXPERIMENTAL PROCEDURES
Expression and purification of recombinant wild-type NTRB from wheat, NTRC, and 2-Cys Prx from rice, as well as the Trxs x and f1 from Arabidopsis, were carried out as described previously (22, 24). The truncated NTR domain (NTRCM) of NTRC was obtained by expressing an N-terminal His-tagged polypeptide, including residues 37–359 of the full-length NTRC, thus excluding the putative transit peptide and the Trx domain, as described previously (22). The double C140S/C143S (NTRCCys-1) and C377S/C380S (NTRCCys-2) NTRC mutants, lacking the 2-Cys active group in the NTR or Trx modules, respectively, were generated by site-directed mutagenesis (35). These mutant versions of NTRC eluted as dimers in gel filtration experiments but showed altered tendency to aggregate as compared with the wild-type enzyme (35).
In all cases, proteins were produced as N-terminal His-tagged forms. Purification was achieved in one chromatographic step by FPLC using 5-ml HiTrap chelating HP columns (GE Healthcare), loaded with nickel and prepared as described by the manufacturer. The recombinant proteins were eluted with a 0.004–0.5 m imidazole gradient in 20 mm phosphate buffer, pH 7.5, supplemented with 0.5 m NaCl and 10% glycerol. Protein fractions were dialyzed against 5 mm phosphate buffer, pH 7.5, supplemented with 5 mm imidazole, 50 mm NaCl, and 0.02% (w/v) Triton X-100, concentrated by ultrafiltration, and stored at −80 °C until its use. NTR concentrations were determined spectrophotometrically using absorption coefficients of 12.2 (NTRB) and 15.2 (NTRC and NTRCM) mm−1 cm−1 at 456 nm, calculated using the method described in Mayhew and Massey (36).
NADPH titrations were performed in a diode array spectrometer Hewlett Packard 8452A in 100 mm phosphate buffer, pH 7.5, under anaerobic conditions by previous sample bubbling with argon. Fixed amounts of the nucleotide were added to estimate the NADPH/NTR ratio needed to accomplish full protein reduction. Experiments of NTRs reduction by 5-deazariboflavin (dRf) were performed under anaerobiosis by illuminating solutions of NTRs, containing 1 μm dRf and 5 mm EDTA, with successive pulses of white light (37).
Stopped-flow experiments were carried out in 20 mm phosphate buffer, pH 7.5, at 10 °C under anaerobic conditions, using a μSFM-20 device fitted with a TC-50/10 cuvette and coupled to a MOS-450 spectrophotometer (Bio-Logic, France) (38). All the experiments were performed in the presence of 20 mm glucose and 5 units ml−1 of both glucose oxidase and catalase, to eliminate oxygen traces. NTR reduction by either NADPH or NADH was followed under pseudo-first order conditions, at 456 and 540–580 nm, by mixing 200-μl solutions of oxidized NTRs 5–8 μm with small volumes of concentrated nucleotide solutions (0.3–3 mm) to obtain final NTR/NAD(P)H ratios ≪1. To observe intramolecular ET reactions, similar experiments were carried out by mixing 200-μl solutions of oxidized NTRs 15–30 μm with small volumes of concentrated NADPH solutions (0.09–0.3 mm) to obtain final NTR/NADPH ratios of ≥1. In some experiments, different amounts of 2-Cys Prx or the Trxs x and f1 were also added to the concentrated NADPH solutions. In all cases, the observed rate constants (kobs) were calculated by fitting the kinetic traces to mono- or multiexponential processes by using the Bio-Kine32 software package from Bio-Logic. Estimated errors in the determined values were ±5%. Kinetic analyses to estimate minimal values for the association constants and the apparent electron transfer rate constants in NAD(P)H concentration dependence experiments were carried out according to the two-step reaction mechanisms previously proposed (39). Estimated errors in the calculated values were ±10%.
RESULTS
NTR Reductive Reactions
With the aim of establishing the pathway of ET during NTRC calalysis, and to determine the role of the NTR module of the enzyme, NTRC, NTRB, and NTRCM samples were subjected to one- and two-electron reduction under steady-state anaerobic conditions either with NADPH (two-electron donor) (Fig. 1) or the dRf/EDTA system (one-electron donor) (37). Whereas in the case of NADPH reduction of either NTRB or NTRCM a 2:1 NADPH/NTR stoichiometry was enough to accomplish complete protein reduction, a 3:1 stoichiometry was required for the NADPH/NTRC reaction, in agreement with the presence of an additional 2-Cys active group in NTRC. The main features of the absorption spectra of the oxidized and NADPH-reduced species were very similar in the three NTRs (Fig. 1) and almost identical for NTRB and NTRCM (data not shown). However, reduction of NTRC, NTRB, or NTRCM by dRf was a very slow process (illumination times ≥30 min) and led to the observation of absorption spectra with features not corresponding to typical reduced flavoproteins (data not shown) (2, 3), suggesting alterations either in the flavin cofactor or its environment. Thus, as described previously for the proteins from other organisms (4, 19, 40), NTRC is strictly a two-electron acceptor protein, as the other plant NTRs.
FIGURE 1.

Representative spectra for the progressive reduction of 20 μm NTRB (upper panel) and NTRC (lower panel) samples by adding repeated fixed amounts (1 μm) of NADPH; top spectra, initial spectra without NADPH; bottom spectra, plus 45 μm (upper panel) or 65 μm (lower panel) NADPH. Other experimental conditions were as described under “Experimental Procedures.”
Plant NTRs can be reduced by both NADPH and NADH, although with a clear preference for the former, as measured by stopped-flow spectrophotometry (Fig. 2, upper panel). NADPH-dependent reduction under pseudo-first order conditions, with an excess of nucleotide with respect to NTR, occurred in three phases (Fig. 2), either with NTRC, NTRB, or NTRCM. The first and faster step was basically complete within the dead time of the instrument (kobs ≥250 s−1), making it not possible to analyze the effect of NADPH concentration on this phase. This fast phase can be monitored either as a small component of a fast absorbance decrease at 456 nm or better as a fast initial absorbance increase at 540–580 nm (Fig. 2, upper panel inset). The fast phase accounted roughly for about 10–15% of the change observed at 456 nm, as deduced from the incomplete amplitude at this wavelength for NTR reduction, taking into account the enzyme concentration present in the assay. The next kinetic phase represented ≈70% of the total absorbance decrease at 456 nm, showing a hyperbolic dependence of kobs on NADPH concentration, its value increasing until reaching a saturation plateau at relatively low concentrations of the nucleotide (Fig. 2, lower panel). Equivalent observed rate constants for this intermediate phase were obtained at 540 or 580 nm from the disappearance of the initial increase arising from the first phase (Fig. 2, upper panel inset). Finally, a slower phase was observed, either with NTRC, NTRB, or NTRCM, as an additional decrease in absorbance at 456 nm (Fig. 2, upper panel). This phase represented <25% of the total changes at this wavelength, and the observed rate constants were independent of NADPH concentration (data not shown), with an apparent value (k3′) of ≈2.2 s−1 (Table 1).
FIGURE 2.

Upper panel, transient kinetics of NTRC reduction by NAD(P)H were observed by stopped flow at 456 nm; kinetic traces could be adjusted to multiexponential fits, as indicated under “Results.” NTRC- and NAD(P)H-concentrated samples were mixed to obtain final concentrations of 7.5 μm NTRC and 27 μm NADPH or 270 μm NADH. Upper panel inset, NTRC reduction by NADPH measured at 540 nm. Lower panel, dependence on NADPH concentration of the observed rate constants (kobs) of the intermediate phase for NTRC reduction. Lower panel inset, dependence on NADH concentration of kobs for NTRC reduction. Experiments were carried out in 20 mm phosphate buffer, pH 7.5, at 10 °C under anaerobic conditions. NTRC reduction was followed by mixing 200-μl solutions of NTRC 8 μm with small volumes of concentrated nucleotide solutions (0.3–3 mm). Continuous lines represent theoretical fits according to the reaction mechanisms previously proposed (39). Other experimental conditions were as described under “Experimental Procedures.”
TABLE 1.
Kinetic parameters for plant NTRs electron transfer reactions as determined by stopped-flow spectroscopy
| Reaction | KAa | k2′b | k3′c | kF1′d | kF2′e |
|---|---|---|---|---|---|
| m−1 | s−1 | s−1 | s−1 | s−1 | |
| NADPH ⇒ NTRB | 9.7 × 105 | 16.0 | 2.3 | ||
| NADPH ⇒ NTRC | 2.8 × 105 | 26.0 | 2.2 | 1.90 | 0.13 |
| NADPH ⇒ NTRCM | 2.5 × 105 | 29.0 | 2.5 | ||
| NADH ⇒ NTRB | 1.8 × 104 | 12.8 | 1.2 | ||
| NADH ⇒ NTRC | 1.6 × 104 | 13.3 | 1.2 | ||
| NADH ⇒ NTRCM | 0.7 × 104 | 18.2 | 1.1 |
a KA is the association rate constant obtained from the concentration dependence of the observed intermediate phase of reduction.
b k2′ is the first-order apparent electron transfer rate constant for the NTR domain rearrangement step, estimated by extrapolating the kobs of the intermediate phase to infinite NAD(P)H concentration.
c k3′ is the first-order rate constant for the NTR-NAD(P)+ rearrangement step, estimated by the concentration-independent kobs of the third phase.
d kFl′ is the first-order electron transfer rate constants for the first phase of NTRC intramolecular ET.
e kF2′ is the first-order electron transfer rate constants for the second phase of NTRC intramolecular ET.
Prior stopped-flow experiments with other NTRs have reported the very rapid formation of a NADPH/flavin charge-transfer complex (kobs ≈950 s−1), with absorbance bands in the 500–800-nm region that fit with the absorbance changes associated with the fast initial phase described here (4, 18, 41). The second phase has been reported to involve the decay of the NADPH-flavin charge-transfer complex and the reduction of the flavin cofactor, probably followed by electron equilibration between the FAD and the 2-Cys group, a process limited by a large conformational change involving interdomain rotation (4, 18). From the NADPH concentration dependence of kobs shown in Fig. 2 (lower panel), it is possible to estimate minimal values for both the NADPH/enzyme association constant (KA) and an apparent electron transfer rate constant (k2′) that corresponds to the reduction/rearrangement process (18, 39). These values are included in Table 1 for the three proteins studied here. Thus, for all NTRs, comparable values both for KA (≈3–10 × 105 m−1; KD ≈3–1 μm) and k2′ (≈16–29 s−1) have been estimated. Finally, the third phase has been proposed to represent further NADP+/flavin interactions at a rate limited either by NADP+ release and/or the structural rearrangement from the conformer unable to be reduced by NADPH to the nucleotide-reducible form (18). Very similar k3′ values (≈2 s−1) can be obtained for this process with NTRC, NTRB, or NTRCM. Thus, the kinetic behavior observed here for the three plant enzymes analyzed indicates that plant NTRs, including NTRC, show a reductive mechanism similar to that previously described for NTR from E. coli.
Plant NTRs also react, although much less efficiently, with NADH (Fig. 2), showing multiexponential kinetics equivalent to those previously observed with NADPH, from which minimal values for both the KA (≈1–2 × 104 m−1; KD ≈100–50 μm) and the apparent electron transfer rate constants that reflect the different rearrangement steps (k2′ ≈13–18 s−1; k3′ ≈1.2 s−1) can be also estimated (Table 1).
NTRC Intramolecular Electron Transfer Kinetics
Additional stopped-flow experiments were carried out by mixing NTRs with NADPH at an NTR/NADPH stoichiometry ≥1:1, with the aim of observing (in the case of NTRC) further intramolecular ET without interferences arising from additional re-reductions by the excess of NADPH (Fig. 3, upper panel). At a short time scale (< 0.5 s), each NTR, either NTRC, NTRB, or NTRCM, showed a similar decrease of absorbance at 456 nm, which agrees with the fast initial reduction by NADPH previously described (Figs. 2 and 3). At longer time scales, a slower reoxidation process was observed with NTRC, revealed by a recovery of the absorbance values at 456 nm (Fig. 3, upper panel) and the monitoring of protein spectra at different times after the NADPH addition (data not shown). This reoxidation reaction was shown to be a complex process that could be deconvoluted in three kinetic phases (Fig. 3, upper panel). The faster step, which partially overlaps with the reduction by NADPH, accounted for ≈40% of the total change observed at 456 nm, with kobs being independent of NADPH concentration, thus suggesting the occurrence of an intramolecular reaction with an ET rate (kF1′) ≈2 s−1 (Fig. 3, lower panel, and Table 1). The next step represented again ≈40% of the total absorbance increase, and the kobs values were also independent of NADPH concentration, with an ET rate (kF2′) ≈0.1 s−1 (Fig. 3, lower panel, and Table 1). Additionally, a very much slower step was observed as a final recovery of the absorbance at 456 nm (Fig. 3, upper panel). This phase represented ≈20% of the changes at this wavelength, with kobs <0.01 s−1, and it was also apparently independent of NADPH concentration (data not shown); this phase has not been further studied as it can be considered as irrelevant in catalysis, and it may be better explained as residual oxidation reactions with unknown protein internal electron acceptors (see below).
FIGURE 3.

Upper panel, transient kinetics for NTRC intramolecular electron transfer, after reduction by NADPH, as observed by stopped flow at 456 nm in the absence or presence of 2-Cys Prx or Trx x; kinetic traces of reoxidation could be adjusted to multiexponential fits, as indicated under “Results.” NTRC and NADPH concentrated samples were mixed to obtain final concentrations of 15 μm NTRC and 15 μm NADPH, in the absence or the presence of 50 μm 2-Cys Prx or Trx x, as indicated. For comparative purposes, a similar experiment was carried out with NTRCM. Lower panel, dependence on NADPH concentration of the kobs for the two reoxidation phases. NTRC reactions were followed by mixing 200-μl solutions of NTRC 15 μm with small volumes of concentrated NADPH solutions (0.09–0.3 mm). Other experimental conditions were as described in Fig. 2.
Similar stopped-flow experiments, carried out by mixing both NTRB and NTRCM with NADPH at an NTR/NADPH ≥1:1 stoichiometry, did not show any of the two initial reoxidation phases observed with the NTRC enzyme, but only the very slow one, as shown in Fig. 3 (upper panel) for NTRCM. This is in agreement with the assignment of these two faster phases to the intramolecular ET toward the Trx module of NTRC, which is absent in NTRB and NTRCM. Thus, from a comparison of the kinetic behavior of the complete NTRC bimodular enzyme with that of the truncated NTRCM form and canonical NTRB, it can be concluded that the previously NADPH-reduced NTR module of NTRC was further oxidized following two intramolecular reaction processes with different efficiency. The possibility that these two reactions correspond to different conformational states of NTRC will be discussed below.
The previous analysis of the interaction between NTRC and 2-Cys Prx allowed us to propose that NTRC conjugates both NTR and Trx activities to efficiently reduce 2-Cys Prx (24, 25, 31), and thus, it could be considered that the NTRC pathway acts in parallel to the FTR/Trxs pathway in chloroplast redox regulation. However, an additional possibility to be taken into account is that the NTR module of NTRC is able to reduce other Trxs than its own Trx module. Here, we have addressed this possibility by analyzing the interaction of NTRC with two representative chloroplastic Trxs, the f1-type and the x-type. Stopped-flow experiments showed that the kinetic behavior described above for NTRC was not affected in the presence of 50 μm of either Trx f1 (data not shown) or Trx x (Fig. 3, upper panel). However, the addition of 2-Cys Prx did exert a remarkable effect on the kinetics of NTRC intramolecular ET (Fig. 3, upper panel). Thus, in the presence of 2-Cys Prx, the first faster phase of reoxidation was absent, whereas the total absorbance change remained unaltered or even slightly increased. This amplitude conservation was a consequence of the increase in the amplitude of the second phase up to 80% of the total observed change (Fig. 3, upper panel), without affecting the kobs values and being independent on 2-Cys Prx concentration (data not shown).
As compared with canonical NTRs (as NTRB), NTRC contains an additional redox-active site (the extra 2-Cys group in the Trx module), and thus also an additional ET pathway via internal dithiol-disulfide exchange. It has been previously suggested that this internal reaction proceeds from the NTR module dithiol of one monomer to the additional Trx module disulfide of the other, within a dimeric enzyme functional unit (5, 31). This possibility was addressed here following the kinetic behavior of the NTRC double mutants C140S/C143S (NTRCCys-1) and C377S/C380S (NTRCCys-2) lacking, respectively the 2-Cys groups at the NTR and Trx modules by site-directed mutagenesis replacement of Cys by Ser.
Both NTRCCys-1 and NTRCCys-2 mutants were efficiently reduced by NADPH, as observed by stopped-flow experiments (Fig. 4) and steady-state monitoring of the absorption spectra (data not shown), showing multiexponential kinetics and kobs values similar to those observed with NTRC, NTRB, and NTRCM. However, at longer time scales, no fast reoxidation reactions were observed in any case (Fig. 4, traces A and B), thus suggesting again that the two initial oxidation processes detected in the entire NTRC protein can be assigned to intramolecular ET involving the two 2-Cys redox-active groups. In the case of the NTRCCys-1 mutant, no reoxidation reaction was observed at all, as expected for an enzyme lacking the primary electron acceptor of the flavin group, whereas the NTRCCys-2 mutant showed just the very slow kinetics of the third phase described above (kobs <0.01 s−1).
FIGURE 4.
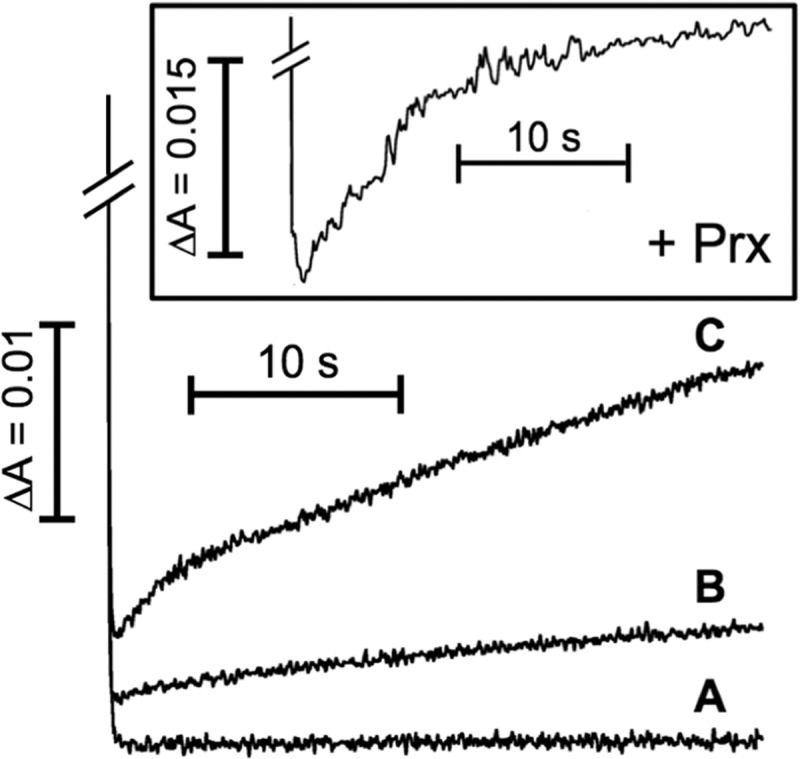
Transient kinetics for the reoxidation reactions in NTRCCys-1 (A) or NTRCCys-2 (B) samples and in NTRCCys-1/NTRCCys-2 1:1 mixtures (C). Inset, reoxidation reaction in NTRCCys-1/NTRCCys-2 1:1 mixtures in the presence of 50 μm 2-Cys Prx. Intramolecular electron transfer in the NTRC mutants, after reduction by NADPH, was followed by stopped flow at 456 nm. NTRC mutants and NADPH concentrated samples were mixed to obtain final concentrations of 20 μm protein and 20 μm NADPH (A and B) or 30 μm protein and 30 μm NADPH (C, and inset). Other experimental conditions were as described in Fig. 3.
It has been suggested that in NTRC, the ET exchange involving the two 2-Cys redox-active groups occurs between the NTR and Trx modules from different monomers in the dimeric enzyme (31). This proposal has been confirmed here by stopped-flow kinetic experiments with 1:1 NTRCCys-1/NTRCCys-2 mixtures. Whereas internal ET inside the same monomer of NTRC would result in totally inactive mutant mixtures, a process involving inter-monomeric exchange would determine the formation of a 25% of active NTRCCys-1/NTRCCys-2 dimers, in which ET from the active 2-Cys group in the NTR module of NTRCCys-2 to the active Trx module of NTRCCys-1 is feasible (31), and this is indeed what it was observed with NTRCCys-1/NTRCCys-2 mixtures. Although both the small amplitude of the signal arising from just 25% of active NTRCCys-1/NTRCCys-2 dimers and the interference of the absorbance decay due to reduction by NADPH hindered the observation of the two faster phases of intramolecular ET, they were still clearly visible (Fig. 4, trace C). Finally, a similar result to that described above for the NTRC wild-type enzyme was observed when 2-Cys Prx was added to 1:1 NTRCCys-1/NTRCCys-2 mixture experiments, 2-Cys Prx affecting the behavior of the active complexes by eliminating the first fast phase, and concurrently increasing the amplitude of the second phase (Fig. 4, inset).
DISCUSSION
The solved structures of canonical NTRs show that the protein is formed by two different domains of which one binds the FAD cofactor, and the other contains the NADPH-binding site and the 2-Cys group (4, 15, 16). The catalytic mechanism of canonical NTRs, as described for the E. coli enzyme, implicates the existence of two protein conformers and a large interdomain rotation to allow NADPH and the 2-Cys group to be alternatively close and react with the FAD cofactor (5, 16). The different reaction steps involved in the catalytic cycle have been characterized by kinetic measurements and structural analyses of both wild-type and mutant NTR enzymes (4, 18–21). Thus, in the E. coli protein, from the multiexponential kinetics for NTR reduction by NADPH, it is deduced that a charge transfer NADPH-enzyme complex is formed after the initial NADPH binding (4, 18, 41). Further flavin reduction and electron equilibration with the 2-Cys group are limited by the rate of interconversion between the two conformers, whereas a third slower kinetic component has been assigned to the formation of a reduced flavin/NADP+ charge transfer complex, maybe reflecting the structural rearrangement back to the nucleotide-reducible NTR form (16, 18, 19).
Besides plant NTRC, there are two additional examples found in bacteria of hybrid proteins with NTR and Trx modules. The protein from M. leprae (33) has been described to catalyze free Trx reduction, although with low efficiency, by following the canonical conformational change mechanism. However, no intramolecular disulfide ET exchange was detected in the Mycobacterium fusion enzyme (33). In the case of the homodimeric AhpF enzyme from S. typhimurium (42), a Trx module mediates the reduction of a 2-Cys-type Prx via intra-subunit ET (34). Thus, the mechanism of the Trx module reduction can vary from one type of fusion enzyme to the other.
Steady-state NTRB and NTRCM titration with NADPH led to a nucleotide/enzyme stoichiometry of 2 for full protein reduction. This is in agreement with the fact that canonical NTRs can exist as E (oxidized), EH2, and EH4 forms, and thus two NADPH molecules are required to completely reduce the protein (1, 4). NTRC, however, requires three nucleotide molecules for full reduction, as a consequence of the presence of an extra 2-Cys active group. Our data clearly indicate that NTRC is a reductase for its own Trx module and that electrons have to be transferred from NADPH to the internal Trx domain via intramolecular ET.
Kinetic data for NTRC, NTRB, or NTRCM reduction show a multiexponential behavior similar to that previously described in canonical NTRs. The values reported here for the different catalytic steps, i.e. the NADPH/NTR KA, the interconversion rate for the structural rearrangement of the FAD and NADPH domains (k2′), and the slower kinetic component (k3′), are in the range of the values previously described for the E. coli enzyme (18), considering that our experiments have been carried out at 10 °C to slow down the rates for a better detection. Thus, it is demonstrated that the presence of the extra Trx module in the bimodular NTRC enzyme does not restrict or affect the conformational motions involved in the reductive reactions. In addition, it is interesting to note that, although NTRs preferentially use NADPH as electron donor, qualitatively similar results were observed when using NADH with NTRC, NTRB, or NTRCM, although the values for the NADH/NTR KA are ≈25–50 times lower as compared with NADPH, and also a slightly lower value for the rearrangement constant k2′ was calculated. It has been reported previously that the use of different electron donor systems is sufficient to cause small alterations in the observed rate for the conformational change in NTR (17). However, the similar kinetic constants values observed here with NADPH/NADH are not only in agreement with the assignment of the first order kinetic reduction steps to conformational changes but also that these motions are relatively independent of the nucleotide donor used.
The final acceptor of canonical NTRs is a Trx. Structural analyses have shown that the interdomain movement in E. coli NTR also exposes the reduced disulfide group to be accessible to the Trx partner. In addition, the observed experimental ET rates of Trx reduction are equivalent (≈30–80 s−1) to the rate of interconversion between the two conformers (≈50 s−1) (17, 40). Consequently, according to the “conformational gating” proposal, the ET rate determined for Trx reduction is assumed to reflect this interconversion process between conformers, which thus is thought to be the turnover limiting step (16). In NTRC, however, the “Trx substrate” is already bound to the NTR module, and thus intramolecular ET processes are expected. The structure of NTRC is not yet solved, but a model structure, using the homologous Arabidopsis NTRB and E. coli Trx as templates, is available (5). The model shows that the NTR module is bound to the Trx module through an ∼20-residue flexible linker at the C-terminal part of the NTR domain containing the NADPH-binding site. In this model, the Trx domain was located close to the NTR domain active site of the second subunit of the NTRC dimer, thus suggesting an inter-subunit ET. However, the possibility of a monomeric active enzyme, able to approach the Trx module and the active 2-Cys in the NTR module through large conformational movements, was not discarded (5).
The experimental results here presented clearly detected intramolecular reactions in NTRC that are absent in NTRCM and involve the 2-Cys groups. The reduction of the Trx module occurs according to two first order kinetic components. Two possible explanations could account for these two reoxidation processes. As a first proposal, it would be possible that the two kinetic phases reflect the ET from the 2-Cys group in the NTR module to both the Trx domain from the same monomer or the other monomer in the dimeric enzyme. According to this possibility, the two kinetic phases would be assigned to the ET to Trx modules located at different distances from the NTR module active site: one closer (intermonomeric ET) than the other (intramonomeric ET), but in any case in fixed positions. However, this hypothesis can be ruled out when considering the results obtained with the NTRC C140S/C143S and C377S/C380S double mutants lacking the 2-Cys groups in the NTR and Trx modules, respectively. Thus, in NTRCCys-1/NTRCCys-2 1:1 mixtures, both kinetic phases are still visible in a situation in which only the intermonomeric ET, but not the intramonomeric one, is possible (31).
An alternative explanation for the two phases of intramolecular ET in NTRC is based on the existence of an equilibrium between two different structural conformations of the Trx module relative to the NTR module active site (Fig. 5). These two orientations would establish different distances between the two 2-Cys active sites, which results in the 1 order of magnitude difference in the observed intramolecular ET rates (1.90 versus 0.13 s−1); these rate constants could reflect either true ET rates or rate-limiting structural rearrangements, as already described in the reductive cycle of canonical NTRs. In this sense, it is interesting to note the coincidence of the values (≈2 s−1) for the slower step in NTRC reduction by NADPH and the faster reoxidation phase. However, that this coincidence reflects the same structural rearrangement process cannot be unequivocally demonstrated. Remarkably, the rate constant values observed here are at least 10 times lower that those reported for the E. coli NTR/Trx system (4, 17, 40), and a low catalytic activity has been also reported for the fusion enzyme from Mycobacterium (33). Thus, it seems that the intermodular-binding linker in NTRC fixes the Trx domain in nonoptimal configurations related to free Trx, and thus the fusion enzyme shows restricted conformations that limit the kinetic efficiency. In this sense, the results obtained in the presence of 2-Cys Prx, the physiological substrate, are of great interest. Whereas the presence of Trxs did not affect the NTRC dynamic motions, the binding of 2-Cys Prx shifted the equilibrium of the Trx module orientations in a way that favors a specific but less efficient conformation for ET (Fig. 5). Therefore, our results support the occurrence of additional dynamic motions in NTRC that are absent in NTRB and NTRCM, and are thus imposed by the extra Trx module, but are also influenced by the presence of the 2-Cys Prx substrate. These dynamics motions involving the NTR and Trx modules are finally the limiting step in the NTRC enzyme turnover, in strong contrast with the canonical enzymes, in which the NTR interdomain rotation is the limiting step (16). In addition, as stated before, the results observed with the NTRC double mutants, missing the 2-Cys group either in the NTR or the Trx modules, agree with the hypothesis of a catalytic cross-reaction between subunits, as previously suggested from steady-state experiments (Fig. 5) (31).
FIGURE 5.

Proposed model for the reaction mechanism of NTRC. In NTRC dimers, the minimal catalytic unit, electron transfer from NTR to Trx modules occurs via inter-subunit pathways. In the absence of 2-Cys Prx (upper panel), the equilibrium between different structural conformations of the Trx domain related to the NTR module in each monomer (indicated as dashed gray arrows) would establish different electron transfer rates. 2-Cys Prx shifts the equilibrium restricting the Trx module to a specific configuration (lower panel). Each monomer in NTRC dimers is colored in dark or light gray, respectively; white arrows indicate electron transfer pathways.
As a final conclusion, our experimental results show not only that plant NTRB and NTRC maintain the conformational changes previously associated with the process of catalysis in canonical NTRs but also unveil additional dynamic motions and restricted configurations in NTRC, associated with the ET to the extra Trx module, that are altered in the presence of 2-Cys Prx.
Acknowledgments
We thank Pilar León and María-Belén Pascual for technical assistance.
This work was supported by Andalusian Government European Regional Development Fund-cofinanced Grants BIO-022 and CVI-4528 (to J. A. N.) and European Regional Development Fund-cofinanced Grant BIO2010-15430 from Ministry of Science and Innovation and Andalusian Government Grant BIO-182 and CVI-5919 (to F. J. C.).
- NTR
- NADPH-dependent thioredoxin reductase
- dRf
- 5-deazariboflavin
- ET
- electron transfer
- NTRB
- NADPH-dependent thioredoxin reductase B
- NTRC
- chloroplast NADPH-dependent thioredoxin reductase
- NTRCM
- truncated NTR domain of NTRC
- NTRCCys-1 and NTRCCys-2
- NTRC mutants lacking the 2-cysteines group in the NTR or thioredoxin modules, respectively, by replacement to serines
- Trx
- thioredoxin
- Prx
- peroxiredoxin
- k2′ and k3′
- apparent first-order electron transfer rate constants for the second and third phase of NTRs reduction by NAD(P)H
- kF1′ and kF2′
- first-order electron transfer rate constants for the first and second phase of NTRC intramolecular ET.
REFERENCES
- 1. Williams C. H., Jr. (1992) in Chemistry and Biochemistry of Flavoenzymes (Müller F., ed) Vol. 3, pp. 121–211, CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 2. Massey V. (2000) The chemical and biological versatility of riboflavin. Biochem. Soc. Trans. 28, 283–296 [PubMed] [Google Scholar]
- 3. van Berkel W. J. (2008) in Wiley Encyclopedia of Chemical Biology, pp. 1–11, John Wiley & Sons, Inc., New York [Google Scholar]
- 4. Williams C. H., Jr., (1995) Mechanism and structure of thioredoxin reductase from Escherichia coli. FASEB J. 9, 1267–1276 [DOI] [PubMed] [Google Scholar]
- 5. Jacquot J. P., Eklund H., Rouhier N., Schürmann P. (2009) Structural and evolutionary aspects of thioredoxin reductases in photosynthetic organisms. Trends Plant Sci. 14, 336–343 [DOI] [PubMed] [Google Scholar]
- 6. Gelhaye E., Rouhier N., Navrot N., Jacquot J. P. (2005) The plant thioredoxin system. Cell. Mol. Life Sci. 62, 24–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collet J. F., Messens J. (2010) Structure, function, and mechanism of thioredoxin proteins. Antioxid. Redox. Signal. 13, 1205–1216 [DOI] [PubMed] [Google Scholar]
- 8. Meyer Y., Buchanan B. B., Vignols F., Reichheld J. P. (2009) Thioredoxins and glutaredoxins. Unifying elements in redox biology. Annu. Rev. Genet. 43, 335–367 [DOI] [PubMed] [Google Scholar]
- 9. Meyer Y., Reichheld J. P., Vignols F. (2005) Thioredoxins in Arabidopsis and other plants. Photosynth. Res. 86, 419–433 [DOI] [PubMed] [Google Scholar]
- 10. Bhatt I., Tripathi B. N. (2011) Plant peroxiredoxins. Catalytic mechanisms, functional significance and future perspectives. Biotechnol. Adv. 29, 850–859 [DOI] [PubMed] [Google Scholar]
- 11. Dietz K. J. (2003) Plant peroxiredoxins. Annu. Rev. Plant Biol. 54, 93–107 [DOI] [PubMed] [Google Scholar]
- 12. Wood Z. A., Schröder E., Robin Harris J., Poole L. B. (2003) Structure, mechanism, and regulation of peroxiredoxins. Trends Biochem. Sci. 28, 32–40 [DOI] [PubMed] [Google Scholar]
- 13. Rouhier N., Lemaire S. D., Jacquot J. P. (2008) The role of glutathione in photosynthetic organisms. Emerging functions for glutaredoxins and glutathionylation. Annu. Rev. Plant Biol. 59, 143–166 [DOI] [PubMed] [Google Scholar]
- 14. Reichheld J. P., Meyer E., Khafif M., Bonnard G., Meyer Y. (2005) AtNTRB is the major mitochondrial thioredoxin reductase in Arabidopsis thaliana. FEBS Lett. 579, 337–342 [DOI] [PubMed] [Google Scholar]
- 15. Dai S., Saarinen M., Ramaswamy S., Meyer Y., Jacquot J. P., Eklund H. (1996) Crystal structure of Arabidopsis thaliana NADPH-dependent thioredoxin reductase at 2.5 Å resolution. J. Mol. Biol. 264, 1044–1057 [DOI] [PubMed] [Google Scholar]
- 16. Lennon B. W., Williams C. H., Jr., Ludwig M. L. (2000) Twists in catalysis. Alternating conformations of Escherichia coli thioredoxin reductase. Science 289, 1190–1194 [DOI] [PubMed] [Google Scholar]
- 17. Lennon B. W., Williams C. H., Jr. (1995) Effect of pyridine nucleotide on the oxidative half-reaction of Escherichia coli thioredoxin reductase. Biochemistry 34, 3670–3677 [DOI] [PubMed] [Google Scholar]
- 18. Lennon B. W., Williams C. H., Jr. (1997) Reductive half-reaction of thioredoxin reductase from Escherichia coli. Biochemistry 36, 9464–9477 [DOI] [PubMed] [Google Scholar]
- 19. Veine D. M., Ohnishi K., Williams C. H., Jr. (1998) Thioredoxin reductase from Escherichia coli. Evidence of restriction to a single conformation upon formation of a cross-link between engineered cysteines. Protein Sci. 7, 369–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Veine D. M., Mulrooney S. B., Wang P. F., Williams C. H., Jr. (1998) Formation and properties of mixed disulfides between thioredoxin reductase from Escherichia coli and thioredoxin. Evidence that cysteine 138 functions to initiate dithiol-disulfide interchange and to accept the reducing equivalent from reduced flavin. Protein Sci. 7, 1441–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Negri A., Rodríguez-Larrea D., Marco E., Jiménez-Ruiz A., Sánchez-Ruiz J. M., Gago F. (2010) Protein-protein interactions at an enzyme-substrate interface. Characterization of transient reaction intermediates throughout a full catalytic cycle of Escherichia coli thioredoxin reductase. Proteins 78, 36–51 [DOI] [PubMed] [Google Scholar]
- 22. Serrato A. J., Pérez-Ruiz J. M., Spínola M. C., Cejudo F. J. (2004) A novel NADPH thioredoxin reductase, localized in the chloroplast, which deficiency causes hypersensitivity to abiotic stress in Arabidopsis thaliana. J. Biol. Chem. 279, 43821–43827 [DOI] [PubMed] [Google Scholar]
- 23. Pascual M. B., Mata-Cabana A., Florencio F. J., Lindahl M., Cejudo F. J. (2010) Overoxidation of 2-Cys peroxiredoxin in prokaryotes. Cyanobacterial 2-Cys peroxiredoxins sensitive to oxidative stress. J. Biol. Chem. 285, 34485–34492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pérez-Ruiz J. M., Spínola M. C., Kirchsteiger K., Moreno J., Sahrawy M., Cejudo F. J. (2006) Rice NTRC is a high efficiency redox system for chloroplast protection against oxidative damage. Plant Cell 18, 2356–2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moon J. C., Jang H. H., Chae H. B., Lee J. R., Lee S. Y., Jung Y. J., Shin M. R., Lim H. S., Chung W. S., Yun D. J., Lee K. O., Lee S. Y. (2006) The C-type Arabidopsis thioredoxin reductase ANTR-C acts as an electron donor to 2-Cys peroxiredoxins in chloroplasts. Biochem. Biophys. Res. Commun. 348, 478–484 [DOI] [PubMed] [Google Scholar]
- 26. Alkhalfioui F., Renard M., Montrichard F. (2007) Unique properties of NADP-thioredoxin reductase C in legumes. J. Exp. Bot. 58, 969–978 [DOI] [PubMed] [Google Scholar]
- 27. Stenbaek A., Hansson A., Wulff R. P., Hansson M., Dietz K. J., Jensen P. E. (2008) NADPH-dependent thioredoxin reductase and 2-Cys peroxiredoxins are needed for the protection of Mg-protoporphyrin monomethyl ester cyclase. FEBS Lett. 582, 2773–2778 [DOI] [PubMed] [Google Scholar]
- 28. Michalska J., Zauber H., Buchanan B. B., Cejudo F. J., Geigenberger P. (2009) NTRC links built-in thioredoxin to light and sucrose in regulating starch synthesis in chloroplasts and amyloplasts. Proc. Natl. Acad. Sci. U.S.A. 106, 9908–9913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lepistö A., Kangasjärvi S., Luomala E. M., Brader G., Sipari N., Keränen M., Keinänen M., Rintamäki E. (2009) Chloroplast NADPH-thioredoxin reductase interacts with photoperiodic development in Arabidopsis. Plant Physiol. 149, 1261–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kirchsteiger K., Ferrández J., Pascual M. B., González M., Cejudo F. J. (2012) NADPH thioredoxin reductase C is localized in plastids of photosynthetic and nonphotosynthetic tissues and is involved in lateral root formation in Arabidopsis. Plant Cell 24, 1534–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pérez-Ruiz J. M., Cejudo F. J. (2009) A proposed reaction mechanism for rice NADPH thioredoxin reductase C, an enzyme with protein disulfide reductase activity. FEBS Lett. 583, 1399–1402 [DOI] [PubMed] [Google Scholar]
- 32. Wulff R. P., Lundqvist J., Rutsdottir G., Hansson A., Stenbaek A., Elmlund D., Elmlund H., Jensen P. E., Hansson M. (2011) The activity of barley NADPH-dependent thioredoxin reductase C is independent of the oligomeric state of the protein. Tetrameric structure determined by cryo-electron microscopy. Biochemistry 50, 3713–3723 [DOI] [PubMed] [Google Scholar]
- 33. Wang P. F., Marcinkeviciene J., Williams C. H., Jr., Blanchard J. S. (1998) Thioredoxin reductase-thioredoxin fusion enzyme from Mycobacterium leprae. Comparison with the separately expressed thioredoxin reductase. Biochemistry 37, 16378–16389 [DOI] [PubMed] [Google Scholar]
- 34. Reynolds C. M., Poole L. B. (2001) Activity of one of two engineered heterodimers of AhpF, the NADH/peroxiredoxin oxidoreductase from Salmonella typhimurium, reveals intrasubunit electron transfer between domains. Biochemistry 40, 3912–3919 [DOI] [PubMed] [Google Scholar]
- 35. Pérez-Ruiz J. M., González M., Spínola M. C., Sandalio L. M., Cejudo F. J. (2009) The quaternary structure of NADPH thioredoxin reductase C is redox-sensitive. Mol. Plant 2, 457–467 [DOI] [PubMed] [Google Scholar]
- 36. Mayhew S. G., Massey V. (1969) Purification and characterization of flavodoxin from Peptostreptococcus elsdenii. J. Biol. Chem. 244, 794–802 [PubMed] [Google Scholar]
- 37. Tollin G. (1995) Use of flavin photochemistry to probe intraprotein and interprotein electron transfer mechanisms. J. Bioenerg. Biomembr. 27, 303–309 [DOI] [PubMed] [Google Scholar]
- 38. Hervás M., López-Maury L., León P., Sánchez-Riego A. M., Florencio F. J., Navarro J. A. (2012) ArsH from the cyanobacterium Synechocystis sp. PCC 6803 is an efficient NADPH-dependent quinone reductase. Biochemistry 51, 1178–1187 [DOI] [PubMed] [Google Scholar]
- 39. Meyer T. E., Zhao Z. G., Cusanovich M. A., Tollin G. (1993) Transient kinetics of electron transfer from a variety of c-type cytochromes to plastocyanin. Biochemistry 32, 4552–4559 [DOI] [PubMed] [Google Scholar]
- 40. Navarro J. A., Gleason F. K., Cusanovich M. A., Fuchs J. A., Meyer T. E., Tollin G. (1991) Kinetics of electron transfer from thioredoxin reductase to thioredoxin. Biochemistry 30, 2192–2195 [DOI] [PubMed] [Google Scholar]
- 41. Massey V., Matthews R. G., Foust G. P., Howell L. G., Williams C. H., Zanetti G., Ronchi S. (1970) in Pyridine Nucleotide-dependent Dehydrogenases (Sund H., ed) pp. 393–411, Springer-Verlag, Berlin [Google Scholar]
- 42. Poole L. B., Reynolds C. M., Wood Z. A., Karplus P. A., Ellis H. R., Li Calzi M. (2000) AhpF and other NADH/peroxiredoxin oxidoreductases, homologues of low Mr thioredoxin reductase. Eur. J. Biochem. 267, 6126–6133 [DOI] [PubMed] [Google Scholar]