

Abstract
We have recently reported the discovery of numerous new compounds that are selective inhibitors of all of the subtypes of the adenosine receptor family via a pharmacophore database searching and screening strategy. During the course of this work we made the unexpected discovery of a potent A2B receptor antagonist, 4-methyl-7-methoxyquinazolyl-2-(2′-amino-4′-imidazolinone) (38, CMB 6446), which showed selectivity for this receptor and functioned as an antagonist, with a binding Ki value of 112 nM. We explored the effects of both substituent- and ring-structural variations on the receptor affinity in this series of derivatives, which were found to be mostly non-selective adenosine receptor ligands with Ki values in the micromolar range. Since no enhancement of A2B receptor affinity of 38 was achieved, the previously reported pharmacophore-based searching strategy yielded the most potent and selective structurally-related hit in the database originally searched.
Introduction
The adenosine receptors (ARs) are G protein-coupled receptors consisting of four subtypes, A1, A2A, A2B, and A3.1 Until recently,2 no selective antagonists of the A2B subtype were known, among either xanthines or non-xanthines. It has been difficult to characterize the pharmacological actions of this AR subtype using only non-selective agonists and antagonists.
Our approach to the identification of novel AR antagonists has been unified pharmacophore-based screening.3 During this study we discovered one compound that showed significant binding to all AR subtypes at 10 micromolar concentration (38, CMB 6446), but also showed some selectivity for the A2BAR. In order to determine whether this compound that bound to the A2BAR was already optimized for potency among the members of this chemical series present in the database originally searched, we conducted substructure-based searching and AR assays on other substructure ‘hits’. These other ‘hits’ provided a wide range of substituents on the quinazoline ring. These experiments would give us information regarding the overall quality of the pharmacophore-based queries and structure–activity relationship data. We synthesized five additional compounds for the purpose of testing the importance on one of the ring nitrogens for adenosine receptor recognition.
Few A2BAR subtype-selective antagonists are known, but the biological interest in this subtype is growing. The alkylxanthine theophylline is a weak, non-selective AR antagonist, used therapeutically for the treatment of asthma. The use of theophylline has been associated with unpleasant side effects, such as insomnia and diuresis. The development of a theophylline-like drug with reduced side effects is desirable. The mechanism of action of theophylline in asthma has been controversial for decades,4 and only recently did antagonism of the A2BAR subtype become a likely mechanism.5,6 The A2BAR is expressed in some mast cells, such as canine mastocytoma cells and human HMC-1 cells, in which it is responsible for triggering acute Ca2+ mobilization. Thus, a novel nonpurine A2BAR antagonist, to complement the recently reported A2BAR selective xanthines,2 would be useful as a probe of the relationship of this receptor subtype to disease states.
Results
To further understand the structure–activity relationships of the most potent A2B antagonist discovered (38, CMB 6446) in our previously reported pharmacophore searching efforts,3 and to explore structural modifications that might lead to enhanced selectivity we set about the iterative collection, synthesis and assay of structurally related compounds. In order to facilitate this process we used 2-guanidinylquinazoline substructure searches in ISISBase to search a large available database of compounds (~100,000 from Express-Pick™, available from ChemBridge corporation) the same database that was originally used for the pharmacophore based queries (CNS-Set™). Compounds that matched the 2-guanidinylquinazoline-substructure query from either database were selected for assay.
In addition to 38, the additional quinazoline derivatives, 3–53, were tested for affinity in radioligand binding assays at ARs as shown in Tables 1–5. Compounds 3–48 were obtained from the database, and the synthetic procedures for compounds not previously reported are provided. Scheme 1 shows an overview of the chemistry that was used in the synthesis of these compounds. Substituted anilines of general structure I were subjected to acetone in the presence of iodine to give substituted 1,2-dihydro-2,2,4-trimethylquinoline derivatives of general structure II.9 Derivatives of this type were treated with cyanoguanidine to give substituted 4-methylquinazolyl-2-guanidines (III),10 and then further derivatized to give compounds of the general structure IV.11 Alternately compounds of general structure III could be selectively mono- or di-alkylated or acylated to give derivatives of the general structure V.12
Table 1.
Affinities of quinazoline derivatives in radioligand binding assays at A1, A2A, and A2B adenosine receptorsa–c
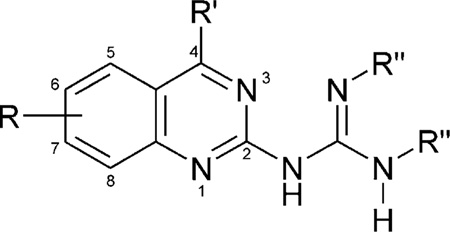 | |||||||
|---|---|---|---|---|---|---|---|
| Compd | R | R′ | R″ | rA1a | rA2Ab | hA2Bc | rA1/hA2B |
| 3 | — | CH3 | H, H | 3.77 ± 0.84 | 5.05 ± 0.62 | 36 ± 7% (10−5) | <1 |
| 4 | — | CH3 | CH3CH2CO, H | 2.13 ± 0.31 | 4.28 ± 1.15 | 39 ± 0% (10−5) | <1 |
| 5 | — | CH3 | C6H5CO, H | 0.988 ± 0.171 | 0.296 ± 0.063 | 35 ± 2% (10−5) | <1 |
| 6 | — | CH3 | CH3CO,CH3CO | 57% (10−4) | <10% (10−5) | ||
| 7 | — | OH | H, H | 36 ± 4% (10−4) | 46.0 ± 13.0 | <10% (10−5) | |
| 8 | 5-OCH3 | CH3 | H, H | 5.16 ± 1.32 | 8.41 ± 2.66 | 10 ± 4% (10−5) | <1 |
| 9 | 5-OCH3 | CH3 | CH3CO,CH3CO | 23.2 ± 8.7 | <10%(10−4) | ||
| 10 | 6-CH3 | CH3 | H, H | 1.22 ± 0.14 | 2.94 ± 1.22 | 39 ± 5% (10−5) | <1 |
| 11 | 6-CH3 | CH3 | CH3CH2CO, H | 7.33 ± 2.20 | 9.22 ± 1.59 | 27 ± 9% (10−5) | |
| 12 | 6-CH3 | OCH3 | H, H | 43 | 8.41 ± 2.72 | 50 ± 1% (10−5) | — |
| 13 | 6-OCH3 | CH3 | CH3,H | 12.2 ± 2.9 | 1.58 ± 0.30 | 53 ± 2% (10−5) | |
| 14 | 6-OCH3 | CH3 | CH3CO, H | <10% (10−5) | 50 ± 1% (10−5) | >1 | |
| 15 | 6-OCH3 | CH3 | 2:CH3CH2CO | 11.7 ± 3.1 | 34 ± 2% (10−5) | 1.97 ± 0.09 | 5.9 |
| 16 | 6-OCH3 | CH3 | C6H5CO, H | <10% (10−5) | 34 ± 4% (10−5) | >1 | |
| 17 | 6-OCH2CH3 | CH3 | CH3CO,CH3CO | 9.54 ± 3.41 | 17.2 ± 8.5 | <10% (10−5) | |
| 18 | 6-CH3,7-CH3 | CH3 | H, H | 2.30 ± 0.37 | 6.8 ± 1.8 | 3.44 ± 0.18 | 0.67 |
| 19 | 6-CH3,7-CH3 | CH3 | CH3CO, H | 2.49 ± 0.43 | 3.90 ± 1.76 | 41 ± 2% (10−5) | <1 |
| 20 | 6-CH3,7-CH3 | CH3 | 2: CH3CH2CO, | 38 ± 3% (10−4) | <10% (10−5) | ||
| 21 | 6-CH3,8-CH3 | CH3 | H, H | 2.01 ± 0.03 | 2.2 ± 0.5 | 45 ± 6% (10−5) | <1 |
| 22 | 7-CH3 | CH3 | CH3CH2CO, H | 10.1 ± 5.2 | 22.5 ± 8.1 | 32 ± 7% (10−5) | |
| 23 | 7-CH3 | CH3 | CH3CO,CH3CO | 9.22 ± 2.77 | 21.4 ± 5.7 | <10% (10−5) | |
| 24 | 7-CH3 | CH2CH3 | H, H | 0.359 ± 0.095 | 0.551 ± 0.102 | 1.36 ± 0.31 | 0.26 |
| 25 | 7-OCH3 | CH3 | H, H | 1.62 ± 0.13 | 2.14 ± 0.56 | 0.215 ± 1.040 | 7.5 |
| 26 | 7-OCH3 | CH3 | C6H5CO, H | 14.2 ± 3.3 | 12.4 ± 7.0 | <10% (10−5) | |
| 27 | 7-OCH3 | CH3 | CH3CO,CH3CO | 47 ± 5% (10−4) | <10% (10−5) | ||
| 28 | 7-OCH2CH3 | CH3 | CH3CO, H | 21.5 ± 10.2 | 32 | <10% (10−5) | |
| 29 | 8-CH3 | CH3 | H, H | 3.96 ± 1.22 | 1.87 ± 0.96 | 47 ± 1% (10−5) | <1 |
| 30 | 8-OCH3 | CH3 | H, H | 0.413 ± 0.087 | 0.258 ± 0.069 | 0.592 ± 0.120 | 0.70 |
| 31 | 8-OCH2CH3 | CH3 | H, H | 1.33 ± 0.61 | 0.624 ± 0.250 | 1.78 ± 0.03 | 0.75 |
Displacement of specific [3H]R-PIA binding in rat brain membranes, expressed as Ki ± SEM in µM (n = 3–5), or as a percentage of specific binding displaced at the indicated concentration (M).
Displacement of specific [3H]CGS 21680 binding in rat striatal membranes. Expressed a Ki ± SEM in µM (n = 3–6), or as a percentage of specific binding displaced at the indicated concentration (M).
Displacement of specific [3H]ZM241,385 binding at human A2B receptors expressed in HEK cells, in membranes, expressed as Ki ± SEM in µM (n = 3–4), or as a percentage of specific binding displaced at the indicated concentration (M).
Table 5.
Affinities of 4-methylquinoline derivatives in radioligand binding assays A1, A2A, and A2B receptorsa,b,c
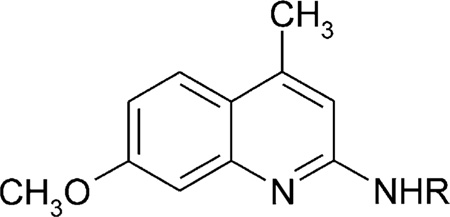 | ||||
|---|---|---|---|---|
| Compd | R | rA1a | rA2Ab | hA2Bc |
| 49 | COCH3 | 6.30 ± 1.13 | 45.7 ± 4.6 | <10% (10−5) |
| 50 | COOCH3 | 21.1 ± 7.3 | <10% (10−4) | <10% (10−5) |
| 52 | C(NH)NH2 | 19.8 ± 2.6 | 17.2 ± 1.9 | <10% (10−5) |
| 53 | C(NH)NH-CO2C(CH3)3 | 8.75 ± 2.77 | 5.06 ± 1.69 | <10% (10−5) |
| 51 | C(NH)N (CO2C(CH3)3)2 | <10% (10−5) | 40.7 ± 10.1 | <10% (10−5) |
Displacement of specific [3H]R-PIA binding in rat brain membranes, expressed as Ki ± SEM in µM (n = 3–5), or as a percentage of specific binding displaced at the indicated concentration (M).
Displacement of specific [3H]CGS 21680 binding in rat striatal membranes. Expressed a Ki ± SEM in µM (n = 3–6), or as a percentage of specific binding displaced at the indicated concentration (M).
Displacement of specific [3H]ZM241,385 binding at human A2B receptors expressed in HEK cells, in membranes, expressed as Ki ± SEM in µM (n = 3–4), or as a percentage of specific binding displaced at the indicated concentration (M).
Scheme 1.

General synthetic method for analogues in the quinazoline series. R = alkyl, alkoxy. Reagents: (i) iodine/acetone, reflux; (ii) HCl gas; (iii) cyanoguanidine; (iv) ethylbromoa cetate/DMF or glycine/water reflux; (v) acyl anhydride/DMSO.
We also prepared a series of new substituted quinoline derivatives (see Scheme 2). Compounds 49–53 were prepared in order to test the requirement for the N3 nitrogen in AR binding. These substituted 4-methylquinoline derivatives were prepared from 2-amino-4-methyl-7-methoxyquinoline (Scheme 2). Compounds 49 and 50 were synthesized with typical acylation methods. Compound 51 was prepared using di-Boc-protected triflylguanidine,20 and the deprotection of one or both of the Boc groups gave a mixture of compounds 52 (30%) and 53 (57%).
Scheme 2.

The synthesis of the quinoline derivatives. Reagents: (i) acetic anhydride, TEA, cat. DMAP in CH2Cl2; (ii) methylchl oroformate, TEA, CH2Cl2; (iii) di-Boc-N-triflylguanidine, TEA, CH2Cl2; (iv) 5% TFA/CH2Cl2.
The potency of test compounds at the human A2BAR was evaluated in binding assays and in a functional assay. As an initial indication of selectivity, the binding was compared at rat A1 and A2AARs. There appeared to be a great freedom of substitution of both the quinazoline ring system and the pendant guanidino group, as judged from retention of binding at these three ARs.
Compound 38, the previously-identified lead, was 10.6- and 21.4-fold selective for human A2B versus rat A1 and A2AARs, with a Ki value of 112 nM. At the human A3AR a Ki value of 1.85 ± 0.62 µM was determined,8 thus selectivity with respect to this subtype was 16.5-fold. A related derivative, 37, was slightly less selective for the A2AAR. The N3 was found to be necessary for recognition by the A2BAR, since 3-deaza analogues did not bind at that subtype (cf., 25 and it deaza analogue 51). Nevertheless, analogues lacking the N3 retained affinity for A1 and A2AARs. In addition to binding effects, the functional effect of 38 in inhibiting the A2BAR-mediated rise in intracellular calcium in HEK-A2B cells was examined. At 100 nM, 38 inhibited calcium mobilization stimulated by NECA (100 nM) by roughly 70%.
Discussion
There is evidence that both the A2BAR and A3AR may play a role in asthma.5,13–15 The A3AR mediates the degranulation of rat RBL mast-like cells and is present in high density in human blood eosinophils. The availability of antagonists selective for the A2BAR shall provide an opportunity to explore the importance of these two AR subtypes in asthma and other inflammatory diseases.
Xanthine derivatives displaying selectivity for the A2B subtype in binding assays were recently reported.2,6 Several such xanthines were found to have antagonistic effects in functional assays.2,16 In addition to xanthine antagonists of ARs, there is need to identify non-xanthine structures. We initially set out to use information derived from known A3AR antagonists both to identify new leads for this receptor family and to explore the pharmacophore relationships within the receptor family. Further, we also wished to explore the utility of pharmacophore database queries for the discovery of new leads.3 An unexpected added benefit of our approach was the discovery of new selective leads for related AR subtypes, A1, A2A, and A2B. This ability to find new structural series from pharmacophore information for an existing bioactive structural series is advantageous in the development of new drug leads.
Pharmacophore-based screening of a diverse chemical library has identified 4-methylquinazoline derivatives as AR antagonists. Analogues of 4-substituted quinazolines have been selected via 2D substructure queries or synthesized, then tested in receptor binding, and found to be generally non-selective. These derivatives distantly resemble isoquinolines reported as A3AR antagonists.19 Most of the structural variation occurred at the 2-position, with guanidino and substituted guanidino groups. Simple functional group substitution of the phenyl ring was included. Nearly all of the compounds bound to A1, A2A, and A2BARs in the micromolar range. Dramatic flexibility of substitution of the 2-guanidino group was possible with retention of affinity. Bulky substituents (such as dihydropyrimidines 39–41 and benzocycloheptanones 42–48) did not prevent the binding to ARs. However the di-Boc derivative 51 did not bind appreciably at any of the ARs examined.
One imidazolinone derivative, 38, was roughly an order of magnitude selective for human A2B versus rat A1 and A2AARs. Further binding and functional studies within the same species will be required to rigorously establish the true selectivity of this compound. Interestingly this compound was selected in the initial pharmacophore-based searching.3 The full characterization of this compound will require comparative binding studies within the same species. This derivative functionally antagonized the Ca2+ response mediated by the recombinant human A2BAR expressed in HEK293 cells.
In addition to 38, potent binding to the human A2BAR (Ki < 1 µM) was noted for the unsubstituted guanidine derivatives 25 and 30 and for the other unsubstituted imidazolinone derivatives 33 and 37. Compound 25, which displayed a low degree of selectivity for the A2BAR, was similar to 38 in the position of substitution of the quinazoline, that is, a 7-methyl group replaced the 7-methoxy group. Other analogues substituted at the 7-position were not A2B-selective. The N-benzoyl derivative 5 and the unsubstituted guanidine 24 were potent at A1 and A2AARs and less potent at the A2BAR. The N-propionyl derivative 15 was more potent at the human A2BAR than at rat A1 and A2AARs. The quinazoline N3 was found to be necessary for recognition by the A2BAR but not by A1 and A2AARs.
The lack of high affinity binding to ARs or distinct SAR patterns within the series suggests that there may be novel mode of binding. Further experiments will be needed to define the competitive nature of the antagonism by these quinazoline derivatives.
Thus, at least in this case, the pharmacophore-based query selected the most potent and selective compound in this structural series present in the entire database (of 9600 compounds) that was originally searched. The search of a larger database (~100,000 compounds) using 2D sub-structure queries based on 38 did not identify more potent or selective compounds within this particular series. Further experiments will need to be performed to determine if the results obtained from these types of queries against other targets may also yield analogous results. Refinement of searching methods will be of increasing value to researchers in medicinal chemistry with the recent advent of substructure- and similarity-based compound acquisition via a web browser interface and the availability of larger numbers of diverse compounds from combinatorial and parallel synthesis. Further experiments of this type with purine receptors, as well as application of the search strategies that we have been developing, will be the topic of future publications.
Experimental
Chemistry
All proton NMR spectra were acquired on a Bruker AC-300 spectrometer using DMSO-d6 as the solvent, unless otherwise indicated, and reported as δ from TMS as the internal standard. Other than compounds 49–53 (see detailed experimental and procedures below) the compounds used in this study were obtained from the ChemBridge Corporation collection, described at http://www.hit2lead.com. General procedures and experimental details are provided below. Following the procedures described and substituting the corresponding substituted anilines (Formula I, Scheme 1) for p-methoxyaniline, substituted quinolines (Formula II) were obtained.8 Compounds 3,17 (10, 13, 14, 16, 17),12 (18, 25),18 (32, 33, 35, 36, 37, 38)11 may be prepared according to the literature procedures. The combustion analytical data were obtained on new compounds from NuMega Resonance Labs (San Diego, CA, USA).
Preparation of substituted quinazolines
4-Methyl-6-methoxyquinazolyl-2-guanidine
A solution was prepared of 0.1 mol1,2- dihydro-2,2,4-trimethyl-6-methoxyquinoline hydrochloride in 100 mL of water, and was treated with 0.1 mol of dicyandiamide. The resulting mixture was refluxed for approximately four hours until the evolution of gas ceased. The hot reaction mixture was decanted from oils, cooled and treated with concentrated KOH with constant stirring until a pH of between 10 and 11 was obtained. The precipitate was isolated by filtration, washed several times with isopropanol, recrystallized from DMSO, then washed with isopropanol and dried under vacuum. The desired product of 4-methyl-6-methoxy quinazolyl-2-guanidine was obtained at a yield of 84%, a melting point of 298–300 °C, Anal. (calcd/found): 57.14/57.12, H: 57.14/57.12, and N: 30.30/30.32.
General procedure for the synthesis of substituted 4-methylquinazolyl-2-guanidines
Following the procedure described above for 4-methyl-6-methoxy quinazolyl-2-guanidine and substituting the following substituted quinoline hydrochlorides (Formula II, Scheme 1) for 1,2-dihydro-2,2,4-trimethyl-6-methoxyquinoline hydrochloride, the corresponding substituted quinazolyl-2-guanidine compounds (Formula III) were obtained.
4-Hydroxyquinazolyl-2-guanidine (7)
1H NMR: δ 7.15 (dd, 1H, J = 8.0, 8.0 Hz), 7.35 (d, 1H, J = 8.0 Hz), 7.5 (brs, 4H), 7.55 (dd, 1H, J = 8.0, 8.0 Hz), 7.9 (d, 1H, J = 8.0 Hz), 10.95 (brs, 1H). Mp = 286–287 °C. Anal. calcd for C9H9N5O·H2O: C, 53.20; H, 4.46; N, 34.46. Found: C, 52.76; H, 4.42; N, 33.40.
4,6,8-Trimethylquinazolyl-2-guanidine (21)
1H NMR: δ 1.08 (t, 6H, J = 7.2 Hz), 2.43 (s, 3H), 2.45 (s, 3H), 2.59 (br, 4H), 2.82 (s, 3H), 7.57 (s, 1H), 7.95 (s, 1H), 11.6–12.0 (br, 2H). Mp = 228–230 °C.
4-Ethyl-7-methylquinazolyl-2-guanidine (24)
1H NMR: δ 1.35 (t, 3H, J = 7.2 Hz), 2.55 (s, 3H), 3,29 (q, 2H, J = 7.2 Hz), 7.46 (dd, 1H, J = 8.4, 2.0 Hz), 7.74 (brd, 1H, J = 2.0 Hz), 8.16 (d, 1H, J = 8.4 Hz), 8.5 (br, 4H). Mp = 274–276 °C.
4-Methyl-8-methylquinazolyl-2-guanidine (29)
1H NMR: δ 2.48 (s, 3H), 2.71 (s, 3H), 7.15 (br, ~4H), 7.17 (dd, 1H, J = 8.1, 6.9 Hz), 7.56 (d, 1H, J = 6.9 Hz), 7.81 (d, 1H, J = 8.1 Hz). Mp = 252–253 °C.
4-Methyl-8-methoxyquinazolyl-2-guanidine (30)
1H NMR: δ 2.69 (s, 3H), 3.91 (s, 3H), 7.17 (dd, 1H, J = 7.5, 2.5 Hz), 7.19 (dd, 1H, J = 7.5, 6.8 Hz), 7.21 (br, 4H), 7.50 (dd, 1H, J = 6.8, 2.5 Hz). Mp = 179–181 °C.
4-Methyl-8-ethoxyquinazolyl-2-guanidine (31)
1H NMR: δ 1.42 (t, 3H, J = 7.0 Hz), 2.71 (s, 3H), 4,16 (q, 2H, J = 7.0 Hz), 7.17 (dd, 1H, J = 7.5, 2.7 Hz), 7.20 (dd, 1H, J = 7.5, 6.4 Hz), 7.33 (br, ~4H), 7.52 (dd, 1H, J = 6.4, 2.7 Hz). Mp = 218–219 °C.
General procedure for the synthesis of 4-methyl-6-methoxyquinazolyl-2-N-acylguanidines
A suspension of 4-methyl-6-methoxyquinazolyl-2-guanidine (0.01 mol) was prepared in 30 mL DMSO at ambient temperature. The suspension was treated with the corresponding anhydride (0.005 mol) and allowed to stir for 2 h. The reaction mixture was filtered and the residue recrystallized from dioxane yielding the desired 4-methyl-6-methoxyquinazolyl-2-N-acylguanidine.
General procedure for the preparation of substituted 4-methylquinazolyl-2-N-acylguanidines
Following the procedures described for 4-methyl-6-methoxyquinazolyl-2-N-acylguanidine above and substituting the following substituted quinazolyl-2-guanidines (Formula III, Scheme 1) for 4-methyl-6-methoxyquinazolyl-2-guanidine, the corresponding substituted 4-methylquinazolyl-2-N-acylguanidines (Formula V) were obtained with the listed physical and spectral properties.
4-Methylquinazolyl-2-N-propionylguanidine (4)
1H NMR: two forms: δ 1.07 (t, 3H, J = 7,5 Hz), 2.56 (br, 2H), 2.87 and 2.92 (s, 3H), 7.58 and 7.64 (dd, 1H, J = 8.2, 7.5 Hz), 7.75–8.0 (m, 2H), 8.20 and 8.25 (d, 1H, J = 8.2 Hz), 8.5 (br, ~2H), 11.1–11.6 (br, ~1H). Mp = 153–155 °C.
4-Methylquinazolyl-2-N-benzoylguanidine (5)
1H NMR: δ 2.92 (s, 3H), 7.43–7.46 (m, 2H), 7.51 (dd, 1H, J = 8.5, 7.5 Hz), 7.60 (dd, 1H, J = 8.0, 7.5 Hz), 7.91–7.97 (m, 2H), 8.18–8.24 (m, 3H), 9.3 (bs, 1H), 10.0 (bs, 1H), 12.1 (bs, 1H). Mp = 170–172 °C. Anal. calcd for C17H15N5O: C, 66.87; H, 4.95; N, 22.94. Found: C, 66.37; H, 5.09; N, 22.60.
4,6-Dimethylquinazolyl-2-N-propionylguanidine (11)
1H NMR: δ 1.06 (t, 3H, J = 7,5 Hz), 2.35 (q, 2H, J = 7.5 Hz), 2.49 (s, 3H), 2.81 (s, 3H), 7.64–7.69 (m, 2H), 7.86 (s, 1H), 9.9–11.5 (br, ~3H). Mp = 179–180 °C. Anal. calcd for C14H17N5O: C, 61.98; H, 6.32; N, 25.81. Found: C, 61.86; H, 6.01; N, 25.56.
4,6,7-Trimethylquinazolyl-2-N-acetylguanidine (19)
1H NMR, two forms: δ 2.28 (bs, 3H), 2.44 (s, 3H), 2.46 (s, 3H), 2.83 and 2.87 (s, 3H), 7.59 and 7.71 (s, 1H), 7.94 and 7.99 (s, 1H), 8.4 (bs, ~2H). MS (EI) m/z 271 [M]+. Mp = 248–249 °C.
4,7-Dimethylquinazolyl-2-N-propionylguanidine (22)
1H NMR: δ 1.06 (t, 3H, J = 7.2 Hz), 2.35 (q, 2H, J = 7.2 Hz), 2.49 (s, 3H), 2.79 (s, 3H), 7.33 (d, 1H, J = 8.4 Hz), 7.58 (s, 1H), 8.00 (d, 1H, J = 8.4 Hz), 9.9–11.1 (br, ~3H). MS (EI) m/z 271 [M]+. Mp = 159–161 °C.
4 - Methyl - 7 - methoxyquinazolyl - 2 -N - benzoylguanidine (26)
1H NMR: δ 2.91 (s, 3H), 3.95 (s, 3H), 7.51 (d, 1H, J = 2.5 Hz), 7.64 (dd, 1H, J = 9.0, 2.5 Hz), 7.7 (bs, 1H), 7.89 (d, 1H, J = 9.0 Hz), 8.1–8.35 (brm, 4H), 10.9–13.4 (br, ~2H). MS (EI) m/z 335 [M]+. Mp = 236–237 °C.
4-Methyl-7-ethoxyquinazolyl-2-N-acetylguanidine (28)
1H NMR: δ 1.40 (t, 3H, J = 7.2 Hz), 2.05 (s, 3H), 2.80 (s, 3H), 4.19 (q, 2H, J = 7.2 Hz), 7.39 (d, 1H, J = 2.5 Hz), 7.50 (dd, 1H, J = 9.0, 2.5 Hz), 7.73 (d, 1H, J = 9.0 Hz), 9.8 (br, ~3H). Mp = 154–155 °C. Anal. calcd for C14H17N5O2•1/3H2O: C, 57.33; H, 6.07; N, 23.88. Found: C, 57.17; H, 5.60; N, 23.51.
General procedure for the preparation of substituted 4-methylquinazolyl-2-N,N-diacylguanidines
Following the procedures described for 4-methylquinazolyl-2-N-acylguanidines except 0.002 mol of the anhydride was used, and substituting the following substituted quinazolyl-2-guanidines (Formula III, Scheme 1) for 4-methyl-6-methoxyquinazolyl-2-guanidine, the corresponding substituted 4-methylquinazolyl-2-N,N-diacylguanidines (Formula V) were obtained with the listed spectral and physical properties.
4-Methylquinazolyl-2-N,N-diacetylguanidine (6)
1H NMR: δ 2.26 (bs, 6H), 2.88 (s, 3H), 7.60 (dd, 1H, J = 8.0, 7.5 Hz), 7.81 (d, 1H, J = 8.6 Hz), 7.92 (dd, 1H, J = 8.6, 7.5 Hz), 8.21 (d, 1H, J = 8.0 Hz), 11.4–11.9 (br, ~2H). Mp = 196–197 °C. Anal. calcd for C14H15N5O2: C, 58.94; H, 5.30; N, 24.55. Found: C, 58.59; H, 5.23; N, 24.43.
6-Methoxy-4-methylquinazolyl-2-N,N-dipropionylguanidine (15)
1H NMR: δ 1.10 (t, 6H, J = 7.2 Hz), 2.62 (bs, 4H), 2.70 (s, 3H), 3.97 (s, 3H), 7.12–8.15 (m, 3H), 11.6–12.0 (br, 2H). Mp = 156–158 °C.
4,6,7 - Trimethylquinazolyl - 2 -N,N - dipropionylguanidine (20)
1H NMR: δ 1.08 (t, 6H, J = 7,2 Hz), 2.43 (s, 3H), 2.45 (s, 3H), 2.59 (br, 4H), 2.82 (s, 3H), 7.57 (s, 1H), 7.95 (s, 1H), 11.6–12.0 (br, 2H). 1H NMR (DMSO-d6): 2.25 (bs, 6H), 2.49 (s, 3H), 2.84 (s, 3H), 7.43 (d, 1H, J = 8.4 Hz), 7.60 (s, 1H), 8.09 (d, 1H, J = 8.4 Hz), 11.6 (br, ~2H). MS (EI) m/z 299 [M]+. Mp = 169–171 °C. Anal. calcd for C18H23N5O2: C, 63.32; H, 6.79; N, 20.51. Found C, 63.20; H, 6.59; N, 20.46.
4,7-Dimethylquinazolyl-2-N,N-diacetylguanidine (23)
1H NMR: δ 2.25 (bs, 6H), 2.49 (s, 3H), 2.84 (s, 3H), 7.43 (d, 1H, J = 8.4 Hz), 7.60 (s, 1H), 8.09 (d, 1H, J = 8.4 Hz), 11.6 (br, ~2H). MS (EI) m/z 299 [M]+. Mp = 180–182 °C. Anal. calcd for C15H17N5O2: C, 60.19; H, 5.72; N, 23.40. Found: C, 59.80; H, 5.61; N, 23.13.
4-Methyl-7-methoxyquinazolyl-2-N,N-diacetylguanidine (27)
1H NMR: δ 2.28 (bs, 6H), 2.92 (s, 3H), 3.96 (s, 3H), 7.13 (s, 1H), 7.17 (d, 1H, J = 8.4 Hz), 8.09 (d, 1H, J = 8.4 Hz), 11.5–12.1 (br, ~2H). Mp = 176–178 °C.
Preparation of 4-methyl-6-methoxyquinazolyl-2-(2′-amino-4′-imidazolinone) (35)
A mixture of 4-methyl-6-methoxyquinazolyl-2-guanidine (0.01 mole) and ethyl 2-bromoacetate (0.006 mole) in 50 mL of dimethylformamide was refluxed for 4 to 6 h, allowed to cool and poured into water. The resulting precipitate was filtered and the residue recrystallized from dioxane yielding the desired 4-methyl-6-methoxyquinazolyl - 2 - (2′ - amino - 4′ - imidazolinone) with the reported physical and spectroscopic properties.17
General procedure for the preparation of substituted 4-methylquinazolyl-2-(2′-amino-4′-imidazolinones)
Following the procedures described above for 35 and substituting the following substituted quinazolyl-2-guanidines (Formula III, Scheme 1) for 4-methyl-6-methoxyquinazolyl-2-guanidine, the corresponding substituted 4-methylquinazolyl-2-(2′-amino-4′-imidazolinone) (32, 33, 36, 37, and 38) were obtained with the reported spectral and physical properties.10
4-Methyl-6-methoxyquinazolyl-2-N-methylguanidine (13)
A mixture of 4-methyl-6-methoxyquinazolyl-2-guanidine (0.01 mol) and iodomethane (0.015 mol) in 30 mL of dry dimethylformamide was heated under reflux for 10 h. The mixture was allowed to cool, the precipitate was isolated by filtration and washed with acetone yielding the desired product.11
6-Methyl-4-methoxyquinazolyl-2-guanidine
A suspension of 4-hydroxy-6-methylquinazolyl-2-guanidine (5 g) in 100 mL of methanol was treated with 100 mg of p-toluenesulfonic acid and refluxed for 15 h. The precipitate was isolated by filtration, washed with water, dried and recrystallized from dimethylformamide yielding 4 g (80% yield) of the desired 6-methyl-4-methoxyqunolyl-2-guanidine. Mp 265–266 °C.
Preparation of substituted quinolines
N-(7-Methoxy-4-methyl-quinolin-2-yl)-acetamide (49)
To a solution of 2-amino-4-methyl-7-methoxyquinoline (9.4 mg, 0.04 mmol) in CH2Cl2 (1 mL) was sequentially added triethylamine (14 µL, 0.10 mmol) and acetic anhydride (6.6 µL, 0.07 mmol) and DMAP (1 mg, 0.008 mmol). The reaction mixture was stirred at room temperature for 1 h, and the solvent was removed under a stream of nitrogen. The residue was purified by preparative thin layer chromatography (ethyl acetate/hexanes/methanol = 10/10/1) to give 49 (9.8 mg, 84%) as a white solid. 1H NMR (CDCl3) δ 2.25 (s, 3H), 2.68 (s, 3H), 3.94 (s, 3H), 7.13 (s, 1H), 7.82 (dd, 1H, J = 1.1, 8.4 Hz), 8.17 (s, 1H), 9.16 (bs, 1H). MS (CI/NH3) m/z 231 (M + H+)+. FAB+ exact mass: calcd for C13H15N2O2 231.1134. Found: 231.1138.
N-(7-Methoxy-4-methyl-quinolin-2-yl)-carbamic acid methyl ester (50)
To a solution of 2-amino-4-methyl-7-methoxyquinoline (10 mg, 0.053 mmol) in CH2Cl2 (1 mL) was added triethylamine (14 µL, 0.1 mmol) and methylc hloroformate (6 µL, 0.08 mmol). The reaction mixture was stirred at room temperature for 1 h, and the solvent was removed by a nitrogen stream. The residue was purified by preparative thin layer chromatography (ethyl acetate:hexanes:methanol = 10:15:1) to give 50 (10.5 mg, 81%) as a white solid. 1HNMR (CDCl3) δ 2.67 (d, 3H, J = 0.8 Hz), 3.83 (s, 3H), 3.93 (s, 3H), 7.09 (dd, 1H, J = 2.6, 9.1 Hz), 7.14 (d, 1H, J = 2.5 Hz), 7.59 (bs, 1H), 7.81 (d, 1H, J = 9.1 Hz), 7.94 (s, 1H). MS (CI/NH3) m/z 247 (M + H+)+. FAB+ exact mass: calcd for C13H15N2O3 247.1083. Found: 247.1072.
N,N′-Di-tert-butyloxycarbonyl-N″-(7-Methoxy-4-methylquinolin-2-yl)-guanidine (51)
2-Amino-4-methyl-7-methoxyquinoline (19 mg, 0.1 mmol) was added to a solution of di-BOC-protected-triflylguanidine (36 mg, 0.1 mmol) and triethylamine (16 µL, 0.12 mmol) in CH2Cl2 (1 mL), and the mixture was stirred at refluxing temperature until all triflylguanidine was consumed (the reaction was monitored by TLC). After completion of the reaction, the mixture was diluted with CH2Cl2 (2 mL) and the organic layer was washed with 2M sodium bisulfate, saturated sodium bicarbonate, brine, and dried over sodium sulfate, filtered, concentrated in vacuo. The residue was purified by preparative thin layer chromatography (ethyl acetate/hexanes/methanol = 10:30:1) to give 51 (16 mg, 40%) as a white solid. 1H NMR (DMSO-d6) δ 1.52 (s, 9H), 2.63 (s, 3H), 3.89 (s, 3H), 7.15–7.19 (m, 1H), 7.19 (s, 1H), 7.93 (s, 1H), 7.94 (d, 1H, J = 9.6 Hz), 10.81 (s, 1H), 11.95 (s, 1H). MS (CI/NH3) m/z 431 (M+H+)+.
N - (7 - Methoxy - 4- methyl - quinolin - 2 - yl) - guanidine (52) and N-tert-butyloxycarbonyl-N’-(7-Methoxy-4-methylquinolin-2-yl)-guanidine (53)
A solution of 51 (8 mg, 0.019 mmol) in 5% trifluoroacetic acid in CH2Cl2 (1 mL) was stirred for 2 h at room temperature. The solvent was evaporated under reduced pressure and the residue was purified by preparative thin-layer chromatography (ethyl acetate/hexanes/methanol = 10:10:1) to give 52 (0.8 mg, 30%) and 53 (1.6 mg, 57%) as a white solid.
52. 1H NMR (CD3OD) δ 2.68 (s, 3H), 3.95 (s, 3H), 6.82 (d, 1H, J = 0.8 Hz), 7.19 (dd, 1H, J = 2.6, 9.2 Hz), 7.38 (d, 1H, J = 2.6 Hz), 7.95 (d, 1H, J = 9.2 Hz). MS (CI/NH3) m/z 231 (M+H+)+. FAB+ exact mass: calcd for C12H15N4O 231.1246. Found: 231.1238.
53. 1H NMR (CD3OD) δ 1.55 (s, 9H), 2.66 (s, 3H), 3.97 (s, 3H), 6.85 (s, 1H), 7.15 (dd, 1H, J = 2.6, 9.2 Hz), 7.32 (d, 1H, J = 2.6 Hz), 7.91 (d, 1H, J = 9.2 Hz). MS (CI/NH3) m/z 331 (M+H+)+. FAB+ exact mass: calcd for C17H23N4O3 331.1770. Found: 331.1761.
Pharmacology
R-PIA, and 2-chloroadenosine were purchased from Sigma-RBI (St. Louis, MO, USA). Other synthetic reagents were purchased from Aldrich (Milwaukee, WI, USA).
Ki values of quinazoline derivatives were determined in displacement of binding of the non-selective radioligand [3H]ZM241385 (4 - (2 - [7 - amino - 2 - {furyl}{1,2,4}-triazolo{2,3 - a}{1,3,5}triazin - 5 - ylaminoethyl) - phenol (Tocris, Ballwin, MO, USA) at the human A2BAR stably expressed in HEK-293 cell membranes.7 In order to determine selectivity, the compounds were evaluated using standard binding assays at A1, A2A, and A3ARs. The screening2 utilized rat brainA1/A2AARs (with radioligands [3H]R-PIA, Amersham, Arlington Heights, IL, USA and [3H]CGS21680, Perkin–Elmer, Boston, MA, USA8
The functional effects in inhibiting the rise in intracellular calcium elicited by the non-selective agonist NECA in HEK cells expressing the human AAR were examined, by methods described.6 The test compound was incubated with cells at 37° for 2 min. Then the cells (1 million in 2 mL) were transferred to a stirred cuvette maintained at 37 °C within an Aminco SLM 8000 spectrofluorometer (SML instruments, Urbana, IL, USA). The ratios of indo-1 fluorescence obtained at 400 and 485 nm (excitation, 332 nm) was recorded using a slit width of 4 nm. NECA was added after a 100 s equilibration period.
Table 2.
Affinities of 4-methylquinazoline imidazolinone derivatives in radioligand binding assays A1, A2A, and A2B receptorsa–c
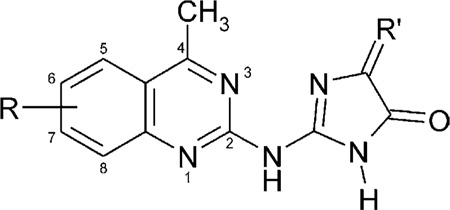 | ||||||
|---|---|---|---|---|---|---|
| Compd | R | R′ | rA1a | rA2Ab | hA2Bc | rA1/hA2B |
| 32 | — | H, H | 10.2 ± 2.3 | 23.8 ± 4.0 | 45% (10−5) | — |
| 33 | 6-CH3 | H, H | 1.44 ± 0.31 | 5.74 ± 1.05 | 0.446 ± 0.096 | 3.2 |
| 34 | 6-CH3 | =CHCOOCH3 | 2.91 ± 0.38 | 1.49 ± 0.40 | 38 ± 8% (10−5) | <1 |
| 35 | 6-OCH3 | H, H | 12.7 ± 1.7 | 17.7 ± 11.9 | 9.44 ± 0.11 | 1.3 |
| 36 | 6-OCH2CH3 | H, H | 5.58 ± 1.95 | 24 ± 2% (10−4) | 14 ± 3% (10−5) | <1 |
| 37 | 7-CH3 | H, H | 1.35 ± 0.26 | 12.9 ± 4.1 | 0.292 ± 0.070 | 4.6 |
| 38 | 7-OCH3 | H, H | 1.29 ± 0.43 | 2.4 ± 0.3 | 0.112 ± 0.015 | 12 |
Displacement of specific [3H]R-PIA binding in rat brain membranes, expressed as Ki ± SEM in µM (n = 3–5).
Displacement of specific [3H]CGS 21680 binding in rat striatal membranes. Expressed a Ki ± SEM in µM (n = 3–6), or as a percentage of specific binding displaced at the indicated concentration (M).
Displacement of specific [3H]ZM241,385 binding at human A2B receptors expressed in HEK cells, in membranes, expressed as Ki ± SEM in µM (n = 3–4), or as a percentage of specific binding displaced at the indicated concentration (M).
Table 3.
Affinities of 4-methylquinazoline dihydropyrimidine derivatives in radioligand binding assays A1,A2A, and A2B receptorsa–c
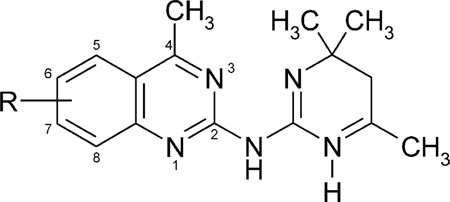 | |||||
|---|---|---|---|---|---|
| Compd | R | rA1a | rA2Ab | hA2Bc | rA1/hA2B |
| 39 | — | 1.89 ± 0.12 | 5.17 ± 1.89 | 38 ± 3% (10−5) | <1 |
| 40 | 6-CH3,7-CH3 | 3.89 ± 1.15 | 8.4 ± 0.4 | 31 ± 1% (10−5) | <1 |
| 41 | 7-OCH3 | 0.648 ± 0.050 | 19.3 ± 8.3 | 4.04 ± 0.05 | 0.16 |
Displacement of specific [3H]R-PIA binding in rat brain membranes, expressed as Ki ± SEM in µM (n = 3–5).
Displacement of specific [3H]CGS 21680 binding in rat striatal membranes. Expressed a Ki ± SEM in µM (n = 3–6).
Displacement of specific [3H]ZM241,385 binding at human A2B receptors expressed in HEK cells, in membranes, expressed as Ki ± SEM in µM (n = 3–4), or as a percentage of specific binding displaced at the indicated concentration (M).
Table 4.
Affinities of 4-methylquinazoline benzoheptanone derivatives (R′ = methyl) in radioligand binding assays A1, A2A, and A2B receptorsa—c
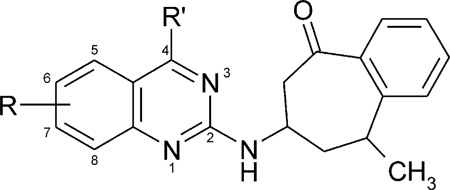 | |||||
|---|---|---|---|---|---|
| Compd | R | rA1a | rA2Ab | hA2Bc | rA1/hA2B |
| 42 | — | 2.68 ± 0.13 | 3.1 ± 1.0 | 44 ± 2% (10−5) | <1 |
| 43 | 6-CH3 | 1.65 ± 0.24 | 1.50 ± 0.79 | 46 ± 4% (10−5) | <1 |
| 44 | 6-OCH3 | 3.41 ± 0.82 | 8.4 ± 2.9 | 25 ± 4% (10−5) | <1 |
| 45 | 6-OCH2CH3 | 2.88 ± 0.06 | 5.71 ± 2.35 | <10% (10−5) | <1 |
| 46 | 6-CH3,7-CH3 | 1.95 ± 0.19 | 2.08 ± 0.27 | 42 ± 8% (10−5) | <1 |
| 47 | 7-CH3 | 0.334 ± 0.038 | 1.44 ± 0.07 | 4.3 ± 2.4 | 0.23 |
| 48 | 7-OCH3 | 1.04 ± 0.04 | 1.57 ± 0.35 | 1.66 ± 0.18 | 0.63 |
Displacement of specific [3H]R-PIA binding in rat brain membranes, expressed as Ki ± SEM in µM (n = 3–5).
Displacement of specific [3H]CGS 21680 binding in rat striatal membranes. Expressed a Ki ± SEM in µM (n = 3–6).
Displacement of specific [3H]ZM241,385 binding at human A2B receptors expressed in HEK cells, in membranes, expressed as Ki ± SEM in µM (n = 3–4), or as a percentage of specific binding displaced at the indicated concentration (M).
Acknowledgements
We are grateful for the help of Sergey Shorshnev of ChemBridge Moscow for his help with NMR spectral interpretation.
References and Notes
- 1.Jacobson KA, Knutsen LJS. P1 and P2 Purine and Pyrimidine Receptors. In: Abbracchio MP, Williams M, editors. Handbook of Experimental Pharmacology, Volume 151/I: Purinergic and Pyrimidinergic Signaling I. Berlin, Germany: Springer; 2001. p. 129. [Google Scholar]
- 2.Kim Y-C, Ji X-D, Melman N, Linden J, Jacobson KA. J. Med. Chem. 2000;43:1165. doi: 10.1021/jm990421v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Webb TR, Melman N, Lvovskiy D, Ji X-D, Jacobson KA. Bioorg. Med. Chem. Lett. 2000;10:31. doi: 10.1016/s0960-894x(99)00583-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feoktistov I, Polosa R, Holgate ST, Biaggioni I. Trends Pharmacol. Sci. 1998;19:148. doi: 10.1016/s0165-6147(98)01179-1. [DOI] [PubMed] [Google Scholar]
- 5.Kirshenbaum AS, Hettinger B, Day Y-J, Gilfillan AM, Metcalfe DD, Kim Y-C, Linden J, Jacobson KA. Amer. Acad. Allergy, Asthma and Immunol; 57th Annual Meeting; 2001; 2001. (abstr 2000803). [Google Scholar]
- 6.Auchampach JA, Jin J, Wan TC, Caughey GH, Linden J. Mol. Pharmacol. 1998;52:846. doi: 10.1124/mol.52.5.846. [DOI] [PubMed] [Google Scholar]
- 7.Ji X-D, Jacobson KA. Drug Des. Discov. 1999;16:217. [PMC free article] [PubMed] [Google Scholar]
- 8.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. Mol. Pharmacol. 1994;45:978. [PMC free article] [PubMed] [Google Scholar]
- 9.Cliffe WH. J. Chem. Soc. 1933;35:1327. [Google Scholar]
- 10.Knövenagel E. Ber. 1921;54B:1722. [Google Scholar]
- 11.Shikhaliev KhS, Falaleyev AV, Ermolova GI, Solov’ev AS. Chem. Heterocycl. Comp. 1999;35:818. [Google Scholar]
- 12.Shikhaliev KhS, Falaleyev AV, Ermolova GI, Solov’ev AS. Khim. Khim. Tekhnol. 1998;41:116. [Google Scholar]
- 13.Fozard JR, Hannon JP. Pulm. Pharmacol. Ther. 1999;12:111. doi: 10.1006/pupt.1999.0191. [DOI] [PubMed] [Google Scholar]
- 14.Salvatore CA, Tilley SL, Latour AM, Fletcher DS, Koller BH, Jacobson MA. J. Biol. Chem. 2000;275:4429. doi: 10.1074/jbc.275.6.4429. [DOI] [PubMed] [Google Scholar]
- 15.Feoktistov I, Polosa R, Holgate ST, Biaggioni I. Trends Pharmacol. Sci. 1998;19:148. doi: 10.1016/s0165-6147(98)01179-1. [DOI] [PubMed] [Google Scholar]
- 16.Jacobson KA, IJzerman AP, Linden J. Drug Devel. Res. 1999;47:45. doi: 10.1002/(sici)1098-2299(199905)47:1<45::aid-ddr6>3.0.co;2-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arya VP, David J, Grewal RS, Marathe SB, Patil SD. Indian J. Chem. 1977;15B:1129. [Google Scholar]
- 18.Rosowsky A, Nadel ME, Modest EJ. J. Heterocycl. Chem. 1972;9:625. [Google Scholar]
- 19.van Muijlwijk-Koezen JE, Timmerman H, Link R, van der Goot H, Ijerman AP. J. Med. Chem. 1998;41:3994. doi: 10.1021/jm980037i. [DOI] [PubMed] [Google Scholar]
- 20.Feichtinger K, Zapf C, Sings HL, Goodman M. J. Org. Chem. 1998;63:3804. [Google Scholar]