

Abstract
Reactive oxygen species (ROS) are essential components of the innate immune response against intracellular bacteria, and it is thought that professional phagocytes generate ROS primarily via the phagosomal NADPH oxidase (Phox) machinery1. However, recent studies have suggested that mitochondrial ROS (mROS) also contribute to macrophage bactericidal activity, although the mechanisms linking innate immune signaling to mitochondria for mROS generation remain unclear2-4. Here we demonstrate that engagement of a subset of Toll-like receptors (TLR1, TLR2 and TLR4) results in the recruitment of mitochondria to macrophage phagosomes and augments mROS production. This response involves translocation of the TLR signaling adapter tumor necrosis factor receptor-associated factor 6 (TRAF6) to mitochondria where it engages evolutionarily conserved signaling intermediate in Toll pathways (ECSIT), a protein implicated in mitochondrial respiratory chain assembly5. Interaction with TRAF6 leads to ECSIT ubiquitination and enrichment at the mitochondrial periphery, resulting in increased mitochondrial and cellular ROS generation. ECSIT and TRAF6 depleted macrophages exhibit decreased levels of TLR-induced ROS and are significantly impaired in their ability to kill intracellular bacteria. Additionally, reducing macrophage mROS by expressing catalase in mitochondria results in defective bacterial killing, confirming the role of mROS in bactericidal activity. These results therefore reveal a novel pathway linking innate immune signaling to mitochondria, implicate mROS as important components of antibacterial responses, and further establish mitochondria as hubs for innate immune signaling.
The phagocytic response of the innate immune system involves the production of ROS via the Phox-dependent respiratory burst, a necessary effector response for the destruction of intracellular microbes1,6. In addition to Phox, the mitochondrial oxidative phosphorylation (OXPHOS) machinery generates ROS when electrons prematurely escape OXPHOS Complexes I and III and react with molecular oxygen to generate superoxide7,8. Mitochondria are major sites of ROS production in most cells; however, mROS have traditionally been regarded as byproducts of oxidative respiration, and therefore their synthesis was believed to be unregulated7,9. To examine whether TLR signaling could enhance mROS production we stimulated RAW macrophages with lipopolysaccharide (LPS; TLR4 agonist), synthetic lipopeptide Pam3CSK4 (TLR1/2 agonist), lipotechoic acid (LTA; TLR2 agonist), Poly(I:C) (TLR3 agonist), R848 (TLR7/8 agonist) and CpG DNA (TLR9 agonist) (Fig. 1a). The production of mROS was triggered only upon signaling from the cell surface TLRs (TLR1/2/4), whereas stimulation of endosomal TLRs (TLR3/7/8/9) failed to augment mROS (Fig. 1a). Exposure of cells to rotenone and antimycin A, compounds known to increase mitochondrial superoxide generation, did augment mROS, but TNFα treatment did not (Fig. 1a)7. We observed similar increases in mROS when bone marrow-derived macrophages (BMM) were stimulated with TLR1/2/4 agonists, but were again unable to detect significant induction of mROS upon ligation of TLR9 (Fig. 1b). We also detected increased cellular hydrogen peroxide (H2O2) generation upon TLR2/4 ligation, but not following TLR9 ligation (Fig. 1b)10-12. As ROS are critical for antibacterial responses, it is not surprising that signaling from cell surface TLRs, which predominantly recognize ligands derived from bacteria, induces ROS generation13. In contrast, ROS are not utilized as direct antiviral effectors, and hence endosomal TLRs, which function primarily in sensing viral infection, do not appear to augment ROS production.
Figure 1. TLR1/2/4 signaling induces mROS generation and mitochondrial recruitment to phagosomes.
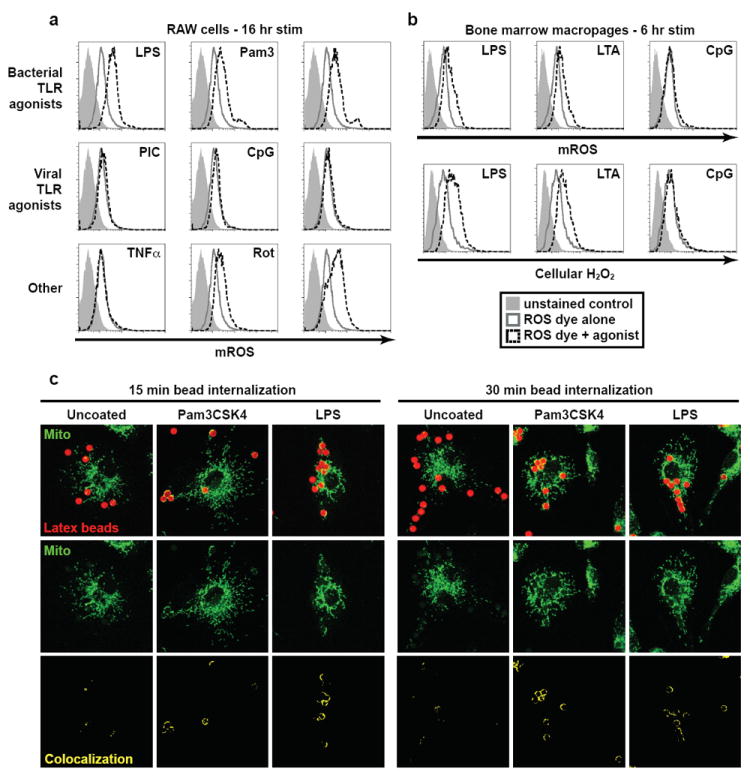
a, RAW cells stimulated as indicated, stained with MitoSOX [mROS], and analyzed by FACS. b, BMM were stimulated as indicated, stained with MitoSOX (top panels) or CM-H2DCFDA [cellular H2O2] (bottom panels), and analyzed by FACS. c, BMM were incubated with uncoated, Pam3CSK4-, or LPS-coated latex beads and mitochondrial networks were immunostained with HSP70 antibodies [Mito]. Confocal Z-stacks were acquired and colocalized beads (red pixels) and mitochondria (green pixels) are displayed in yellow (bottom). Images shown are representative of approximately 100 cells analyzed.
Several reports have indicated that mitochondria are recruited to vacuoles containing intracellular pathogens14-17. To investigate whether recruitment of mitochondria to phagosomes might be an active process mediated by innate immune signaling, we examined mitochondrial localization in cells loaded with latex beads coated with pathogen-associated molecular patterns (PAMPs). Such coated beads have been used previously to investigate signaling in phagocytic cells and have been shown to recruit innate immune signaling components, analogous to phagocytosed bacteria18,19. Interestingly, we observed mitochondrial recruitment and cupping around Pam3CSK4 and LPS coated beads in BMM (Fig. 1c). Uncoated beads, despite being taken up by BMM to a similar extent, did not colocalize efficiently with mitochondrial networks and displayed markedly lower mitochondrial cupping per bead (Fig. 1c and Supplementary Fig. 2).
Based on the above findings, we hypothesized that the inducible juxtaposition of phagosomes and mitochondria should be accompanied by the concomitant translocation of TLR signaling components. A key intermediate in TLR1/2/4 signaling is TRAF6, and immunoblotting of highly purified cellular extracts from LPS-stimulated macrophages revealed that TRAF6 was enriched in mitochondrial fractions (Fig. 2a). This recruitment was specific to TRAF6, as other cytosolic proteins that interact transiently with TLR signaling complexes, such as MyD88, IRAK4 (not shown), IRAK1, TAK1, and IκBα, were not detected in mitochondrial fractions. Furthermore, Pam3CSK4 and LTA stimulation induced TRAF6 recruitment to mitochondria with similar kinetics to that triggered by LPS (Fig. 2b). Consistent with the results on mROS generation, we were unable to detect TRAF6 in the mitochondrial fractions of macrophages stimulated with Poly(I:C) or CpG (Fig. 2c).
Figure 2. TRAF6 is recruited to mitochondria upon TLR1/2/4, but not TLR3/9, signaling to engage ECSIT on the mitochondrial surface.
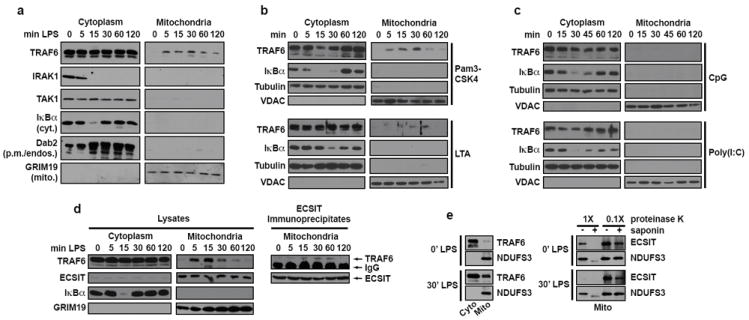
a-e, RAW cells were stimulated with TLR agonists for the indicated times, cells were fractionated, and extracts were blotted. Purified mitochondrial lysates were immunoprecipitated with ECSIT antibody overnight (d). Heavy chain IgG is indicated by an asterisk (d). Equal amounts of extracts were treated with the indicated amount of proteinase K on ice with or without 0.2% saponin to gently permeabilize mitochondrial membranes (e).
The induction of mROS and the recruitment of TRAF6 to mitochondria upon TLR1/2/4 stimulation suggested that TRAF6 potentially interfaces with mitochondrial proteins to control mROS production. Recent studies have shown that ECSIT, a previously characterized TRAF6 interacting protein, localizes to mitochondria and plays a role in OXPHOS Complex I assembly5,20,21. Mass spectrometry analysis of purified ECSIT protein complexes confirmed that ECSIT associates with OXPHOS Complex I components (Supplementary Table 1). Immunofluorescence microscopy and biochemical fractionation experiments revealed that ECSIT localizes predominantly to mitochondria in both fibroblasts and BMM (Supplementary Fig. 3a-c)5,21. Additional analysis confirmed ECSIT localizes to the inner mitochondrial membrane (IMM) consistent with its role in Complex I assembly. However, we also observed some ECSIT molecules proximal to outer mitochondrial membranes (OMM), suggesting that OMM-associated ECSIT might interact with TRAF6 recruited from phagosomal TLR signaling complexes. (Supplementary Figs. 3d-f). Accordingly, we detected inducible interactions between ECSIT and TRAF6 in purified mitochondrial extracts from macrophages stimulated with LPS (Fig. 2d).
TRAF6 possesses E3-ubiquitin ligase activity; therefore, we explored whether ECSIT is ubiquitinated by TRAF622,23. ECSIT was polyubiquitinated when co-transfected with TRAF6 in 293 cells (Supplementary Fig. 4a). In addition, a dominant-negative (ΔN) form of ECSIT lacking the TRAF6 interaction domain was significantly less ubiquinated by TRAF6 (Supplementary Fig. 4b)21. We also detected increasing ECSIT polyubiquitination in macrophages after exposure to LPS, which mirrored the mitochondrial recruitment kinetics of TRAF6 (Supplementary Fig. 4c). In addition, total LPS-induced ECSIT ubiquitination was decreased in TRAF6 knockdown macrophages, indicating a requirement for TRAF6 in the ubiquitination of ECSIT during TLR4 signaling (Supplementary Fig. 4c).
We next investigated the dynamics of ECSIT localization within mitochondria following LPS stimulation. Remarkably, after 30 minutes of LPS treatment, ECSIT became more sensitive to proteinase K in the absence of OMM permeabilization by saponin (Fig. 2e). In contrast, the IMM protein NDUFS3 remained largely proteinase K insensitive without saponin. This suggests that ECSIT becomes enriched at the mitochondrial periphery, and thus more sensitive to protease digestion, upon LPS signaling. Electron microscopy analysis further confirmed these data, as more ECSIT was localized peripheral to the OMM after LPS treatment (Supplementary Fig. 5). Protease sensitivity assays on mitochondria from TRAF6 knockdown RAW cells indicated that TRAF6 is required for LPS-induced ECSIT enrichment on OMMs (Supplementary Fig. 6, compare lanes 5 and 7 with 9 and 11). In addition to influencing the mitochondrial localization of ECSIT, TRAF6/ECSIT signaling also appears to regulate the recruitment of mitochondria around PAMP-coated latex beads. Both TRAF6 knockout and ECSIT knockdown BMM displayed less mitochondrial enrichment around phagosomes containing LPS and Pam3CSK4 coated beads (Supplementary Fig. 7).
The observed link between ECSIT and Complex I led us to hypothesize that ECSIT might modulate mROS derived from this complex5,7. To establish the role of ECSIT in TLR1/2/4-dependent upregulation of mROS, we sought to examine macrophages lacking ECSIT. Although ECSIT knockout mice have been generated, they are very early embryonic lethals24. Heterozygous ECSIT (+/-) animals are viable but display ~40% less ECSIT protein levels (Supplementary Fig. 8a). Interestingly, BMM from ECSIT +/- mice generated modestly lower mROS and cellular H2O2 when stimulated with LPS and LTA (Supplementary Fig. 8b-c). To confirm the importance of TLR1/2/4-induced TRAF6/ECSIT signaling in mROS responses, we analyzed TRAF6 and ECSIT knockdown BMM (Fig. 3a and Supplementary Fig. 11a). Upon LPS (Fig. 3b) or LTA (Supplementary Fig. 9) stimulation, we observed marked reduction in mROS production in both ECSIT and TRAF6 depleted macrophages at all time points tested. Although TLR-generated mROS responses were impaired in ECSIT knockdown cells, mROS production elicited by rotenone or antimycin A was unaffected (Supplementary Fig. 10). Additionally, LPS- (Fig. 3c and Supplementary Fig. 11b), LTA- (Supplementary Fig. 9), and Pam3CSK4-induced (Supplementary Fig. 11c) cellular H2O2 levels were markedly reduced upon ECSIT and TRAF6 knockdown. Thus, induction of mROS and cellular H2O2 by bacterial PAMPs is critically dependent on both TRAF6 and ECSIT. To determine whether the E3-ubiquitin ligase activity of TRAF6 is required for ROS generation, we examined TRAF6 knockout BMM reconstituted with either WT or RING mutant TRAF6 constructs (Fig. 3d)25. In agreement with the knockdown results, TRAF6 null BMM generated strikingly lower mROS and cellular H2O2 in response to both LPS and LTA (Fig. 3e-f). Null macrophages reconstituted with WT, but not RING mutant, TRAF6 regained the ability to generate ROS in response to TLR2/4 agonists (Fig. 3e-f). Therefore, these data suggest a functional RING domain is required for TRAF6 signaling to ROS generation, most likely by mediating ECSIT ubiquitination (Supplementary Figure 4).
Figure 3. TRAF6-ECSIT signaling regulates generation of mitochondrial and cellular ROS, which requires TRAF6 E3-ubiquitin ligase activity.
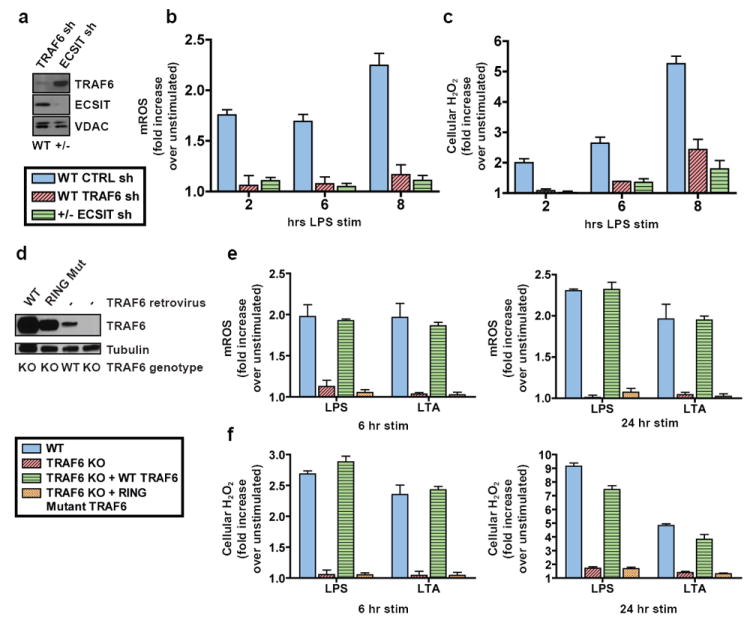
a-c, WT or ECSIT +/- BMM were transduced with shRNAs and left untreated or stimulated with LPS. Untreated BMM were lysed and extracts blotted for TRAF6 and ECSIT (a). Cells were stained with MitoSOX (b) or CM-H2DCFDA (c) and analyzed by FACS. d-f, WT or TRAF6 null BMM were left untreated or transduced with TRAF6 expressing retroviruses, then stimulated with for the indicated times. Unstimulated BMM were lysed and extracts blotted for TRAF6 expression (d). Cells were stained with MitoSOX (e) and CM-H2DCFDA (f) and analyzed by FACS. Error bars represent s.d. of the mean from triplicate samples.
To test the functional significance of these findings, we assessed the responses of ECSIT and TRAF6 deficient macrophages to Salmonella typhimurium, a Gram-negative, facultative, intracellular pathogen that is sensitive to ROS-dependent killing26-28. Consistent with the results obtained using purified PAMPs, we observed decreased mitochondrial and cellular ROS in ECSIT and TRAF6 knockdown BMM when exposed to Salmonella (Supplementary Fig. 12). To determine whether mROS is important in macrophage bactericidal responses, control or ECSIT knockdown BMM were infected with GFP-Salmonella and analyzed by immunofluorescence microscopy and Western blotting. Strikingly, ECSIT depleted BMM harbored significantly more GFP-Salmonella when compared with control knockdown cells (Fig. 4a-b). Direct measurement of intracellular bacterial colony forming units (CFU) demonstrated significantly increased levels of bacteria in ECSIT deficient cells at all time points examined, as compared to control cells (Fig. 4c). The reduced ability of ECSIT deficient BMM to control intracellular bacteria was not the result of a non-specific impairment in Phox activity, as PMA-stimulated respiratory burst was unaffected in ECSIT knockdowns (Supplementary Fig. 13). Additionally, nitric oxide and proinflammatory cytokine production was similar between control and knockdown BMM, collectively indicating that ECSIT depletion does not result in systemic innate immune deficiency (Supplementary Fig. 14).
Figure 4. ECSIT depleted and MCAT transgenic macrophages are less effective at clearing Salmonella.
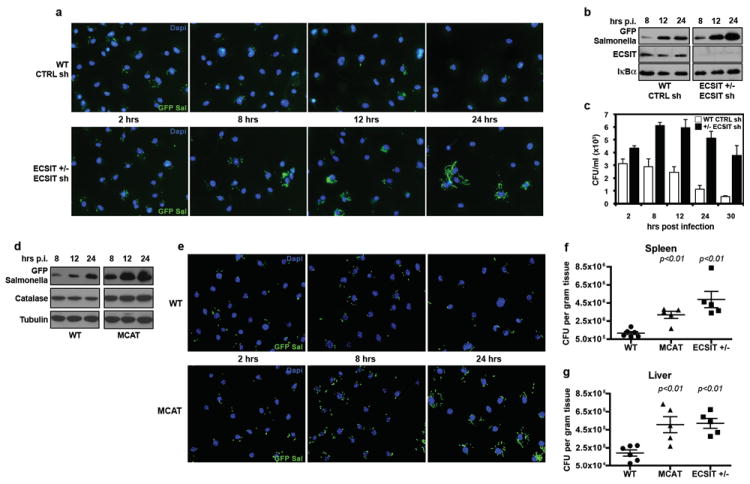
a-c, BMM from WT or ECSIT +/- were transduced with shRNAs and infected with GFP-Salmonella. Cells were fixed and stained with Dapi (a), solubilized in SDS (b), or lysed and plated (c). d-e, WT or MCAT BMM were infected with GFP-Salmonella. Cells were solubilized in SDS (d) or fixed and Dapi stained (e). Triplicate wells were pooled and blotted (b, d), and error bars (c) represent s.d. from triplicate samples. f-g, WT (n=6), MCAT (n=5), and ECSIT +/- (n=5) mice were infected with Salmonella i.p. Five days post infection, spleens (f) and livers (g) were homogenized and CFUs per gram of tissue were determined. Error bars indicate s.e.m., and p values are relative to WT.
Mitochondria are regarded as a significant source of H2O2 in most cell types, and peroxisomal catalase converts H2O2 into water and oxygen and functions to reduce oxidative damage caused by these ROS7. Overexpressing catalase in mitochondria using transgenic approaches (MCAT mice) leads to significantly lower mitochondrial H2O2 levels, and these transgenic mice exhibit lower age-related oxidative damage and extended lifespan29. Consistent with our findings that mROS contribute to total cellular ROS levels, MCAT BMM generated significantly less LPS-induced cellular H2O2 than WT cells (Supplementary Fig. 15). To test the specific role of mitochondrial–derived H2O2 in controlling intracellular bacterial replication, WT or MCAT BMM were challenged with GFP-Salmonella. Similar to ECSIT knockdowns, MCAT BMM exhibited significantly higher bacterial loads between 8 and 24 hours post infection (Fig. 4d-e), relative to WT cells. Finally, to confirm the role of mROS in control of bacterial infection in vivo, we challenged WT, MCAT, and ECSIT +/- mice by intraperitoneal infection with Salmonella and measured bacterial burdens in the spleen and liver five days post infection. In agreement with data from isolated BMM, both MCAT and ECSIT +/- mice harbored roughly 2 to 3 fold more bacteria per gram of tissue when compared to WT littermates, further substantiating the notion that mROS play an integral part in antibacterial innate immunity (Fig. 4f-g).
In conclusion, we have discovered a novel pathway by which macrophages generate ROS in response to bacteria by coupling TLR1/2/4 signaling to mitochondrial Complex I via TRAF6 and ECSIT (Supplementary Fig. 1). Our study demonstrates that in addition to Phox-derived ROS, mROS play an important role in macrophage innate immunity, and to our knowledge, provides the first evidence of direct communication between TLRs and mitochondria. This study also highlights a remarkable symmetry between mitochondrial antiviral signaling protein (MAVS) and ECSIT in innate immune responses. As the clearance of intracellular bacteria requires ROS, TRAF6-ECSIT signaling is engaged downstream of bacterial PAMP-sensing TLRs for robust ROS production; likewise, MAVS signaling is activated by virus-sensing RIG-I-like receptors for type I interferon production and effective antiviral immunity. Our current findings therefore further solidify the emerging idea that mitochondria serve as hubs for innate immune signaling and the generation of effector responses.
Methods
Animal Strains
ECSIT heterozygous and MCAT were previously described and maintained on a C57BL/6 background24,29.
Cell Lines and Reagents
RAW 264.7, J774A.1, and NOR10 cells were obtained from the ATCC and maintained in DMEM (Invitrogen) containing 5% FBS (Atlanta Biological). E. coli LPS (L6529), antimycin A, rotenone, and all other chemicals were obtained from Sigma-Aldrich unless otherwise noted. TNFα was from R&D Systems. CpG 1826 oligonucleotides were synthesized by IDT. S. aureus LTA, R848, and Pam3CSK4 were purchased from Invivogen. Rabbit polyclonal antibodies against ECSIT were previously described24. The following additional antibodies were also utilized: mouse monoclonal NDUFS3, VDAC, GRIM19, cytochrome c (Mitosciences); goat polyclonal HSP60, HSP70, rabbit polyclonal IRAK1, IκBα, mouse monoclonal ubiquitin (Santa Cruz Biotechnology); mouse monoclonal COX1 (Molecular Probes); mouse monoclonal catalase, β-tubulin, FLAG M2, HA (Sigma-Aldrich); rabbit monoclonal TRAF6 (Abcam); rabbit polyclonal MAVS, TAK1 (Cell Signaling Technology); mouse monoclonal Dab2, GFP (BD Biosciences).
ROS Measurements
Cells (RAW or BMM) were plated in non-tissue culture treated 6 well dishes and treated with various agonists or stimulated with TLR ligands as indicated. Concentrations of stimulatants were as follows: 500 ng/ml LPS, 1 μg/ml Pam3CSK4, 2 μg/ml LTA, 10 μg/ml Poly(I:C), 1 μM CpG, 1 μg/ml R848, 10 ng/ml TNFα, 500 nM rotenone, or 5 μM antimycin A. Culture medium was removed, cells washed with PBS, then incubated with MitoSOX (to measure the mROS superoxide) and/or CM-H2DCFDA (to measure total cellular H2O2) (Invitrogen) at 2.5 μM final concentration in serum-free DMEM media (Invitrogen) for 15 to 30 min at 37C. Cells were washed with warmed PBS, removed from plates with cold PBS containing 1mM EDTA by pipeting, pelleted at 1500 rpm for 3 min, immediately resuspended in cold PBS containing 1% FBS, and subjected to FACS analysis. Unstained controls were treated similarly, except that treatments and dyes were omitted. To control for baseline dye fluorescence, samples from each experiment were left unstimulated but stained according to the above procedure. FACS mean fluorescence intensity values (Figs. 3b, c, e, f, Supplementary Fig. 11b-c, Supplementary Fig. 15) were calculated by dividing TLR stimulated by unstimulated values. Error bars were generated by calculating standard deviation (s.d.) of the mean from triplicate samples. All ROS experiments shown are representative of three independent experiments.
Latex Bead Phagocytosis Assays
Yellow orange 3 micron Fluoresbrite carboxy latex microparticles (Polysciences) were left untreated or coated with 30 μg/ml LPS or Pam3CSK4 overnight at 4C in PBS. Beads were washed 10 times in large volumes of PBS-1% FBS to remove unbound TLR agonists. BMM were cultured on coverslips in 12 well dishes and incubated on ice for 10 min. Microparticles were added at a concentration of approximately 4-8 beads per cell and allowed to settle on the cells for an additional 10 min on ice. Warm media was added to the cells and plates were moved to 37C to allow bead phagocytosis for the indicated times. Cells were then fixed for 20 min at room temperature with paraformaldehyde, permeabilized with 0.1% Triton X-100, and stained with the indicated primary and appropriate labeled secondary antibodies for confocal microscopy. Colocalization and quantification analysis was performed using ImageJ software. Three large fields of view were collected for each condition (each field of view contained roughly 30 cells and approximately 120 yellow orange beads), and the integrated density of the colocalized beads (red pixels) and mitochondria (green pixels) was determined for each image. This value was divided by the total number of beads in each field to determine colocalized pixels per bead, and the mean and s.d. were calculated by averaging the three fields of view for each condition. Percent beads with mitochondrial cupping (defined as colocalized mitochondria surrounding ~20 percent or more of the bead periphery) was determined by analyzing the same fields of view described above and calculating the mean and s.d. of the percentage values obtained.
Cellular Fractionation and Mitochondrial Isolation
PBS washed cell pellets were resuspended in 10 pellet volumes of RSB buffer (10 mM NaCl, 1.5 mM CaCl2, 10 mM Tris-HCl pH 7.5), swelled on ice for 10 min, homogenized with a motorized Teflon pestle, then 2.5X MS buffer (525 mM mannitol, 175 mM sucrose, 12.5 mM Tris-HCl pH 7.5, 12.5 mM EDTA) was added to 1X. The homogenate was centrifuged at 980 g for 10 min three times to pellet nuclei, which were saved for western blot analysis. The supernatant was transferred to a fresh tube and spun at 17000 g for 30 min to pellet mitochondria, and the resulting cytosolic supernatant was saved for further analysis. The mitochondrial pellet was washed three times with 1X MS buffer, resuspended in Sucrose-TE (20% sucrose, 50 mM Tris-HCl, pH 7.5, 10 mM EDTA), then purified additionally by centrifugation through a 1.0–1.5 M sucrose step gradient at 100000 g for 60 min. Highly purified heavy mitochondria were removed from the interface of the two sucrose solutions by pipetting and transferred to a fresh tube. Mitochondria were washed with 1 ml of 1X MS buffer three times and pelleted by centrifugation at 13000 rpm. for 10 min. SDS was then added to nuclear and cytosolic fractions to a final concentration of 1% and mitochondrial pellets were resuspended in SDS lysis buffer (20 mM Tris-HCl, 1% SDS, pH 7.5) and boiled for 5 min before normalizing protein concentration and Western blotting. Fraction purity was confirmed by blotting for IκBα and tubulin [cytoplasm, cyt.], Dab2 [plasma membrane and endosomes, p.m./endos.], and GRIM19 and VDAC [mitochondria, mito.].
Mitochondrial Sub-fractionation and Triton X-100 Extraction
RAW cell mitochondria were highly purified as above and sub-fractionated as described31. For Triton X-100 solubility assay, equal amounts of RAW mitochondria were resuspended in 1X MS buffer containing the indicated concentrations of Triton X-100 on ice for 20 min. The samples were then spun at 8000 g to pellet unsolubilized mitochondria from extracted proteins in the supernatant and the resulting pellets were completely solubilized in 1% Triton X-100. Triton concentrations between 0.01 and 0.05% solubilize outer mitochondrial membrane and intermembrane space proteins and those above 0.1% solubilize both outer and inner membrane proteins. Samples were then blotted accordingly.
TRAF6 Recruitment and Protease Sensitivity Assays
For the TRAF6 recruitment assay, RAW cells were left untreated or stimulated with TLR agonists (1 μg/ml LPS, 1 μg/ml Pam3CSK4, 2 μg/ml LTA, 1 μM CpG, or 10 μg/ml Poly(I:C)), subjected to mitochondrial isolation as above, and fractions blotted as indicated. For mito-IP, mitochondria were lysed in TNT buffer (25 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM EDTA, protease/phosphatase inhibitors) and incubated with ECSIT antibody and anti-rabbit IgG agarose overnight at 4C. The samples were washed extensively and blotted accordingly. For the proteinase K sensitivity assay, RAW cells or RAW CTRL GFP sh and TRAF6 sh cells were left untreated or stimulated with LPS, subjected to mitochondrial fractionation, and purified mitochondria were resuspended in digest buffer (25 mM Tris-HCl, pH 7.5, 125 mM sucrose, 1 mM CaCl2). Samples were incubated with proteinase K (1X, 33 ng/μl; 0.1X, 3.3 ng/μl; ++, 12 ng/μl; +, 6 ng/μl) with or without 0.1 % saponin on ice for 30 min. Saponin was used to gently permeabilize mitochondrial membranes to allow proteinase K to enter mitochondria. Proteinase K activity was quenched with PMSF, SDS added to 1% to solubilize mitochondrial proteins, and samples were blotted as indicated.
Ubiquitination Assays
For endogenous ubiqitination assays, one 10 cm dish of J774 CTRL GFP sh or TRAF6 sh cells per time point was stimulated as described and solubilized in boiling SDS lysis buffer and boiled for an additional 10 min. After shearing DNA, the samples were diluted in 2X TNT buffer to 1X and immunoprecipitated overnight with ECSIT antibody and anti-rabbit IgG beads (eBiosceince). Beads were washed in 1X TNT buffer and samples were blotted and probed with antibodies as indicated. All other ubiquitination assays were performed using 6 cm dishes of 293T cells transfected with 0.25-1 μg each of the indicated constructs. After overnight incubation (and 2 hour incubation in MG132 if indicated), cells were lysed as described above, diluted in 2X TNT buffer, and immunoprecipitated with anti-FLAG resin (Sigma-Aldrich) overnight. Beads were washed extensively and samples blotted and probed as indicated. TRAF6, ECSIT-FLAG, and ΔN-ECSIT-FLAG were previously described21.
Bacterial Infection and Gentamicin Protection Assay
Gentamicin protection assays were performed using S. typhimurium SL134432 transformed with a GFP expression plasmid essentially as described30,33. Briefly, BMM were plated at 2 × 105 cells per well in 24 well dishes, incubated with opsonized (using fresh mouse serum) GFP-Salmonella at a MOI of 20 for 1 hour, and further incubated with 100 μg/ml gentamicin for an hour. Wells were washed several times and 2-hour timepoints harvested or cells further incubated in media containing 10 μg/ml gentamicin. Similar results were obtained when the initial 100 μg/ml gentamicin incubation was omitted and cells were only exposed to 10 μg/ml gentamicin (not shown). For colony count analysis, triplicate samples were lysed in PBS containing 1% Triton X-100, serially diluted in PBS, and plated on LB agar plates containing streptomycin. For western blot analysis of GFP-Salmonella, triplicate BMM samples were infected as above and cells were lysed at indicated timepoints in SDS lysis buffer. After boiling and shearing DNA, samples were pooled and blotted as described. For immunofluorescence analysis of GFP-Salmonella, 2 × 105 BMM were grown on coverslips in 12 well dishes and treated as above. At indicated timepoints, cells were fixed with 4% paraformaldehyde for 20 min at room temperature, nuclei stained with Dapi, and mounted and imaged by confocal microscopy. Experiments shown are reperesentative of at least three independent experiments.
In vivo Salmonella Infection Assay
8-10 week old WT, MCAT, or ECSIT +/- littermates were injected interperitoneally with 200 S. typhimurium SL1344 bacteria in 100 μl PBS. Five days post infection, mice were sacrificed, livers and spleens were isolated, and tissues were weighed and homogenized in 1 ml PBS. Lysates were serially diluted in PBS and 100 μl of each dilution was plated on LB streptomycin plates. Colonies were counted the next morning with a BioRad VersaDoc imager using the colony counting feature of the Quantity One software. Error bars represent standard error of the mean (s.e.m.).
Tandem Affinity Purification and Mass Spectrometry
293 or RAW cells were transiently or stably transfected with pCTAP-ECSIT (primers utilized: F, tacgagtccgaattcccaccatgagctgggtgcaggtcaac; R, tacgagtccctcgagactttgcccctgctgctgctc) respectively, and TAP complexes purified using the Interplay Mammalian TAP System (Stratagene) as described. Purified complexes were precipitated in 10% trichloroacetic acid, washed with cold acetone, air dried, and resuspended in 4X Laemmli sample buffer. Samples were electrophoresed on SDS-PAGE gels until just entering the resolving gel and slices were removed and subjected to in-gel trypsin digestion and mass spectrometry analysis in the laboratory of Paul Tempst at Memorial Sloan-Kettering Cancer Center as described34,35.
Immuno-Electron Microscopy
J774 cells were left untreated or stimulated with LPS, washed with PBS and fixed (2% paraformaldehyde, 0.1% gluteraldehyde, 3% sucrose, 0.25M Hepes, pH 7.4) for 30 min at 4C. Samples were then rinsed in PBS and resuspended in 10% gelatin, chilled and trimmed to smaller blocks, and placed in cryoprotectant (2.3M sucrose) rotating overnight at 4C. Small pieces were transferred to aluminum pins and frozen rapidly in liquid nitrogen. The frozen block was trimmed on a Leica Cryo-EMUC6 UltraCut and 75nm thick sections were collected using the Tokoyasu method (1973), placed on a nickel formvar/carbon coated grid, and floated in a dish of PBS ready for immunolabeling. Grids were placed section side down on drops of 0.1M ammonium chloride for 10 minutes to quench untreated aldehyde groups, then blocked for nonspecific binding on 1% fish skin gelatin in PBS for 20 min. Single labeled grids were incubated in primary rabbit anti-ECSIT antibody at 1:20 dilution for 30 min. After rinsing, the grids were placed on protein A gold (UtrechtUMC) for 30 min. All grids were rinsed in PBS, fixed with 1% gluteraldehyde for 5 min, rinsed in water, and transferred to a 0.5% uranyl acetate/1.8% methylcellulose drop for 10 min, then collected and dried. Grids were viewed with a FEI Tencai Biotwin TEM at 80KV. Images were taken using a Morada CCD and item software (Olympus).
Bone Marrow Macrophage Generation and Lentiviral shRNA/Retroviral Transduction
Bone marrow was collected from littermate WT, ECSIT +/-, or MCAT mice and cultured on Petri plates for a period of 7 days in DMEM containing 10% FBS plus 30% L929 conditioned media. Media was replenished on day 4 of culture. Lentiviral shRNA constructs were purchased from Open Biosystems (pLKO.1 vector control ID RHS4080; GFP targeting control clone ID RHS4459; ECSIT clone ID TRCN0000113957 or TRCN0000113958; TRAF6 clone ID TRCN0000040733 or TRCN0000040735) and were packaged as described in the Broad Institute RNAi Consortium protocols (http://www.broadinstitute.org/genome_bio/trc/publicProtocols.html) using 239FT cells (Invitrogen). Macrophages were transduced with shRNA lentiviruses overnight on day 3 of culture and media was changed on day 4 as above, except that 3 μg/ml puromycin was added for the remainder of culture to select transduced cells. On day 7, cells were lifted from plates by incubating in cold TEN buffer (40 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA), re-plated in fresh media without puromycin containing 10% L929 conditioned media, and allowed to rest for a period of 24-48 hours before experimentation. RAW cells stably expressing control, GFP, TRAF6, or ECSIT shRNAs were selected by puromycin resistance and maintained in 3-4 μg/ml.
ECSIT knockdown experiments in the primary and supplementary figures utilized shRNA clone ID TRCN0000113958 (termed ECSIT sh) except Supplementary Fig. 11, which utilized clone ID TRCN0000113957 (termed ECSIT sh4). All TRAF6 knockdown experiments utilized shRNA clone ID TRCN0000040735 (termed TRAF6 sh) with the exception of Supplementary Fig. 11, which utilized clone ID TRCN0000040733 (termed TRAF6 sh1). We observed similar knockdowns and deficiencies in both mitochondrial and cellular ROS production compared to control when either TRAF6 or ECSIT shRNA clone was utilized, but due to space limitations, we have not included both data sets. Furthermore, we detected no significant differences in ROS production or bacterial killing between macrophages transduced with vector control (termed CTRL) or GFP control (termed GFP sh) shRNAs; as such, most of the control samples were transduced with vector control lentiviruses. Finally, we utilized WT BMM for control and TRAF6 knockdown experiments, but made use of ECSIT +/- BMM for ECSIT knockdowns. Heterozygous BMM possess a ~40% reduction in total ECSIT protein abundance by Western blot (Supplementary Fig. 11a); therefore, we utilized these cells to allow for more robust reduction in ECSIT levels upon shRNA silencing. We used age and sex matched littermate controls for the WT CTRL and TRAF6 knockdowns, thus minimizing animal to animal variation when comparing WT and +/- samples to one another. TRAF6 null BMM generation and retroviral reconstitution with WT and RING mutant constructs was performed as previously described36.
Confocal Microscopy
For all microscopy images, cells were grown on coverslips and transfected, stimulated, or infected as described. After washing, cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 in PBS for 5 min, blocked with PBS-10% FBS for 30 min, stained with primary antibodies for 60 min, and stained with secondary antibodies for 60 min. Cells were washed with PBS between each step. Nuclei were stained with TOPRO3 (Invitrogen) or Dapi (Sigma) and coverslips mounted with Prolong Gold anti-fade reagent (Molecular Probes). Cells were imaged on a Zeiss LSM 510 META with a 63X water corrected objective.
Amplex Red Assay
1 × 105 WT or ECSIT +/- BMM infected with control or ECSIT shRNA expressing lentiviruses were plated in 96-well fluorescence microplates (Costar). After two PBS washes to remove residual medium, cells were resuspended in 100 μl pre-warmed, sterile HBSS containing 50 μM Amplex Red (Invitrogen) and 1 U/ml HRP (Sigma) with or without PMA or serum opsonized Salmonella SL1344 for the indicated times. Oxidation of Amplex Red into red-fluorescent resorufin by H2O2 was measured in a fluorescence plate reader using excitation at 530 nm and fluorescence detection at 590 nm. Molar concentrations of extracellular peroxide were determined by comparing fluorescence values from each sample with those generated from a standard curve of H2O2. Average background fluorescence readings from unstimulated triplicate wells were subtracted from each triplicate sample that was stimulated with PMA or infected with Salmonella. This allowed for the measurement of H2O2 induced over unstimulated samples during the same time interval and normalized for cell plating errors.
Nitric Oxide and ELISA Assays
WT or ECSIT +/- BMM were infected with control, TRAF6, or ECSIT shRNA expressing lentiviruses, plated in 24 well dishes, and left untreated or stimulated with 1 μg/ml LPS for the indicated times. Supernatant was collected, serially diluted, and measured for nitrite by the Griess Reagent Kit (Molecular Probes) or TNFα and IL-12p40 by ELISA (eBioscience) per the manufacturers’ instructions.
Supplementary Material
Acknowledgments
We would like to thank Chris Schindler, Boris Reizis, and Laura Ciaccia for comments on the manuscript. We also thank Peter Rabinovitch for MCAT mice, Justin Cotney for technical assistance, Zimei Zhang for animal maintenance, and Morven Graham and Kimberly Zichichi for assistance with immuno-electron microscopy. This work was supported by grants from the NIH to SG (R37-AI33443) and GS (ES-011163).
Footnotes
Author Contributions APW designed and performed experiments and wrote the paper; IEB generated GFP-Salmonella, helped design and perform bacterial challenge experiments, and edited the paper; CR assisted with immuno-electron microscopy; DKW provided MCAT tissues for generating BMM; HEB and PT performed mass spectrometry analysis; MCW and YC provided reagents and technical advice for experiments involving TRAF6 knockout cells; GSS designed experiments and edited the paper; and SG designed experiments and wrote the paper.
Supplementary Information Supplementary information is linked to the online version of the paper at www.nature.com/nature.
The authors declare no competing financial interests.
References
- 1.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 2.Arsenijevic D, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. 2000;26:435–439. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- 3.Rousset S, et al. The uncoupling protein 2 modulates the cytokine balance in innate immunity. Cytokine. 2006;35:135–142. doi: 10.1016/j.cyto.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 4.Sonoda J, et al. Nuclear receptor ERR alpha and coactivator PGC-1 beta are effectors of IFN-gamma-induced host defense. Genes Dev. 2007;21:1909–1920. doi: 10.1101/gad.1553007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogel RO, et al. Cytosolic signaling protein Ecsit also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly. Genes Dev. 2007;21:615–624. doi: 10.1101/gad.408407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu Rev Immunol. 2002;20:825–852. doi: 10.1146/annurev.immunol.20.103001.114744. [DOI] [PubMed] [Google Scholar]
- 7.Koopman W, et al. Mammalian mitochondrial complex I: Biogenesis, Regulation and Reactive Oxygen Species generation. Antioxid Redox Signal. 2009 doi: 10.1089/ars.2009.2743. [DOI] [PubMed] [Google Scholar]
- 8.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnoult D, Carneiro L, Tattoli I, Girardin SE. The role of mitochondria in cellular defense against microbial infection. Semin Immunol. 2009;21:223–232. doi: 10.1016/j.smim.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Adachi Y, et al. IFN-gamma primes RAW264 macrophages and human monocytes for enhanced oxidant production in response to CpG DNA via metabolic signaling: roles of TLR9 and myeloperoxidase trafficking. J Immunol. 2006;176:5033–5040. doi: 10.4049/jimmunol.176.8.5033. [DOI] [PubMed] [Google Scholar]
- 11.Remer KA, Reimer T, Brcic M, Jungi TW. Evidence for involvement of peptidoglycan in the triggering of an oxidative burst by Listeria monocytogenes in phagocytes. Clin Exp Immunol. 2005;140:73–80. doi: 10.1111/j.1365-2249.2005.02740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Werling D, Hope JC, Howard CJ, Jungi TW. Differential production of cytokines, reactive oxygen and nitrogen by bovine macrophages and dendritic cells stimulated with Toll-like receptor agonists. Immunology. 2004;111:41–52. doi: 10.1111/j.1365-2567.2003.01781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-like receptors. Annu Rev Cell Dev Biol. 2006;22:409–437. doi: 10.1146/annurev.cellbio.21.122303.115827. [DOI] [PubMed] [Google Scholar]
- 14.Chong A, Lima CA, Allan DS, Nasrallah GK, Garduño RA. The purified and recombinant Legionella pneumophila chaperonin alters mitochondrial trafficking and microfilament organization. Infect Immun. 2009;77:4724–4739. doi: 10.1128/IAI.00150-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horwitz MA. Formation of a novel phagosome by the Legionnaires’ disease bacterium (Legionella pneumophila) in human monocytes. J Exp Med. 1983;158:1319–1331. doi: 10.1084/jem.158.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsumoto A, Bessho H, Uehira K, Suda T. Morphological studies of the association of mitochondria with chlamydial inclusions and the fusion of chlamydial inclusions. Journal of electron microscopy. 1991;40:356–363. [PubMed] [Google Scholar]
- 17.Sinai AP, Webster P, Joiner KA. Association of host cell endoplasmic reticulum and mitochondria with the Toxoplasma gondii parasitophorous vacuole membrane: a high affinity interaction. J Cell Sci. 1997;110(Pt 17):2117–2128. doi: 10.1242/jcs.110.17.2117. [DOI] [PubMed] [Google Scholar]
- 18.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 19.Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol. 2006;7:1029–1035. doi: 10.1038/ni1006-1029. [DOI] [PubMed] [Google Scholar]
- 20.Calvo S, et al. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat Genet. 2006;38:576–582. doi: 10.1038/ng1776. [DOI] [PubMed] [Google Scholar]
- 21.Kopp E, et al. ECSIT is an evolutionarily conserved intermediate in the Toll/IL-1 signal transduction pathway. Genes Dev. 1999;13:2059–2071. doi: 10.1101/gad.13.16.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature. 2009;458:430–437. doi: 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- 24.Xiao C, et al. Ecsit is required for Bmp signaling and mesoderm formation during mouse embryogenesis. Genes Dev. 2003;17:2933–2949. doi: 10.1101/gad.1145603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deng L, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 26.Shiloh MU, et al. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity. 1999;10:29–38. doi: 10.1016/s1074-7613(00)80004-7. [DOI] [PubMed] [Google Scholar]
- 27.Vazquez-Torres A, Fang FC. Oxygen-dependent anti-Salmonella activity of macrophages. Trends Microbiol. 2001;9:29–33. doi: 10.1016/s0966-842x(00)01897-7. [DOI] [PubMed] [Google Scholar]
- 28.Vazquez-Torres A, Fang FC. Salmonella evasion of the NADPH phagocyte oxidase. Microbes Infect. 2001;3:1313–1320. doi: 10.1016/s1286-4579(01)01492-7. [DOI] [PubMed] [Google Scholar]
- 29.Schriner SE, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 30.Brodsky IE, Ghori N, Falkow S, Monack D. Mig-14 is an inner membrane-associated protein that promotes Salmonella typhimurium resistance to CRAMP, survival within activated macrophages and persistent infection. Mol Microbiol. 2005;55:954–972. doi: 10.1111/j.1365-2958.2004.04444.x. [DOI] [PubMed] [Google Scholar]
- 31.Kang BH, et al. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131:257–270. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 32.Hoiseth SK, Stocker BA. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature. 1981;291:238–239. doi: 10.1038/291238a0. [DOI] [PubMed] [Google Scholar]
- 33.Valdivia RH, Falkow S. Bacterial genetics by flow cytometry: rapid isolation of Salmonella typhimurium acid-inducible promoters by differential fluorescence induction. Mol Microbiol. 1996;22:367–378. doi: 10.1046/j.1365-2958.1996.00120.x. [DOI] [PubMed] [Google Scholar]
- 34.Cooper MP, et al. Defects in energy homeostasis in Leigh syndrome French Canadian variant through PGC-1alpha/LRP130 complex. Genes Dev. 2006;20:2996–3009. doi: 10.1101/gad.1483906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sebastiaan Winkler G, et al. Isolation and mass spectrometry of transcription factor complexes. Methods. 2002;26:260–269. doi: 10.1016/S1046-2023(02)00030-0. [DOI] [PubMed] [Google Scholar]
- 36.Walsh MC, Kim GK, Maurizio PL, Molnar EE, Choi Y. TRAF6 autoubiquitination-independent activation of the NFkappaB and MAPK pathways in response to IL-1 and RANKL. PLoS ONE. 2008;3:e4064. doi: 10.1371/journal.pone.0004064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.