

Abstract
Epigenetics remains a rapidly developing field that studies how the chromatin state contributes to differential gene expression in distinct cell types at different developmental stages. Epigenetic regulation contributes to a broad spectrum of biological processes, including cellular differentiation during embryonic development and homeostasis in adulthood. A critical strategy in epigenetic studies is to examine how various histone modifications and chromatin factors regulate gene expression. To address this, Chromatin Immunoprecipitation (ChIP) is used widely to obtain a snapshot of the association of particular factors with DNA in the cells of interest.
ChIP technique commonly uses cultured cells as starting material, which can be obtained in abundance and homogeneity to generate reproducible data. However, there are several caveats: First, the environment to grow cells in Petri dish is different from that in vivo, thus may not reflect the endogenous chromatin state of cells in a living organism. Second, not all types of cells can be cultured ex vivo. There are only a limited number of cell lines, from which people can obtain enough material for ChIP assay.
Here we describe a method to do ChIP experiment using Drosophila tissues. The starting material is dissected tissue from a living animal, thus can accurately reflect the endogenous chromatin state. The adaptability of this method with many different types of tissue will allow researchers to address a lot more biologically relevant questions regarding epigenetic regulation in vivo1, 2. Combining this method with high-throughput sequencing (ChIP-seq) will further allow researchers to obtain an epigenomic landscape.
Keywords: Genetics, Issue 61, ChIP, Drosophila, testes, q-PCR, high throughput sequencing, epi-genetics
Protocol
(The entire ChIP procedure takes approximately two days. Preparation of ChIP libraries for high-throughput sequencing takes another 2-3 days.)
1. Dissect and Prepare Tissue for ChIP Experiment (~1 million cells)
Dissect tissue of interest (e.g. 200 pairs of Drosophila testes) in cold PBS + 1x protease inhibitor (dissolve 1 pellet of protease inhibitor cocktail in 1.5 mL 1x PBS to obtain 7x stock solution) + PMSF (final concentration 100 μg/mL). Rinse tissue twice and resuspend tissue in 200 μL of the same PBS solution with inhibitors.
To fix the sample, add 5.5 μL 37% formaldehyde (warm formaldehyde in 37 °C water bath for 1 min before using). Incubate at 37 °C for 15 minutes, vortex every 5 minutes in between.
Place the sample on ice for the tissue to settle for 2 minutes. Rinse 2x with 450 μL PBS (with protease inhibitors and PMSF). Sample can now be stored at -20 °C. Repeat dissection several times to obtain enough material for ChIP.
2. Prepare Supernatant with Protein-DNA Conjugation for ChIP Assay
Combine enough sample from step 1.3 in a 1.75 mL eppendorf tube.
Remove PBS from tissue sample. Add 200 μL of lysis buffer (50 mM Tris-HCl, pH 7.6, 1 mM CaCl2, 0.2% Triton X-100 or NP-40, 5 mM butyrate, and 1x proteinase inhibitor cocktail). Add fresh PMSF stock solution to lysis buffer (PMSF final concentration of 0.5 mM). Homogenize with blue homogenizer (Fisher Cat# 749521-1590) until tissues dissociate completely without any aggregation, incubate at RT (Room Temperature) for 10 minutes.
Sonication: use microtip sonicator (Misonix HS-XL200) at power 20. Sonicate for 10 sec and rest on ice for 50 sec. Repeat 4-5 times (optimal sonication time should be determined empirically, as longer sonication will yield smaller fragments. For ChIP using antibodies against histone modifications, we normally sonicate chromatin to approximately 200-300 bp; for transcription factor or chromatin regulator (e.g. Polycomb proteins), we normally sonicate chromatin to 200-1000 bp. However, the optimal fragment size should be tested empirically. Note: ALWAYS KEEP THE TUBE ON ICE EVEN DURING SONICATING TO PREVENT SAMPLE FROM HEATING UP!
Dilute sample by adding 1.8 mL RIPA buffer (10 mM Tris-HCl, pH 7.6, 1 mM EDTA, 0.1% SDS, 0.1% Na-Deoxycholate, 1% Triton X-100, with protease inhibitors and PMSF at the same concentration as described previously). Save 40 μL as input control by adding 2 μL 5M NaCl and incubate at 65 °C overnight (O/N) to reverse crosslink.
To conjugate antibody to beads, add 40 μL Protein A beads to a 1.5 ml eppendorf tube, then add 600 μL PBS and rock at 4 °C for 2 minutes, apply beads to magnet and remove supernatant. Add 100 μL of PBS and the antibody of interest (amount of antibody should be determined empirically). Incubate at RT for 1 hour or 4 °C for 4 hours.
Remove supernatant from beads by magnet and add 1mL chromatin extract from step 2.4 to the beads. Rotate at 4 °C O/N.
Apply ChIP sample to magnet and remove supernatant. Wash the beads with the following buffer at 4 °C for 10 minutes each: 2x with 1 mL of RIPA buffer [1.89 mL 'RIPA buffer' + 315 μL 7x protease inhibitors + 20 μL PMSF]; 2x with 1 mL of RIPA buffer + 0.3 M NaCl [1.89 mL RIPA buffer + 220 μL 3 M NaCl]; 2x with 1 mL of LiCl buffer (0.25 M LiCl, 0.5% NP40, 0.5% NaDOC); 1x with 1 mL of 1x TE + 0.2% Triton X-100; 1x with 1 mL of 1x TE.
To reverse crosslink, resuspend the beads in 100 μL TE buffer + 3 μL 10% SDS + 5 μL of proteinase K (20 mg/mL). Incubate at 65 °C O/N.
Apply beads to magnet and transfer supernatant into a new tube. THE SUPERNATANT CONTAINS YOUR DNA SAMPLE. Wash beads with 100 μL TE + 0.5 M NaCl and combine the two supernatant.
Phenol-chloroform extract the DNA sample. Add 200 μL Phenol:Chlorofrom:IAA (25:24:1) mix and vortex. Spin at 14k for 5 minutes at RT. Alternatively, DNA can be extracted using Qiagen PCR purification kit.
Transfer the supernatant/aqueous layer into a new tube, and add linear acrylamide to a final concentration of 20 μg/mL (Alternatively 1 μL of glycogen at 20 mg/mL for every 1 mL of supernatant can also be used. Glycogen is used for subsequent qPCR or PCR analyses while linear acrylamide is used for sequencing assays.) Add 20 μL of 3M NaOAc and 500 μL 100% EtOH. Mix well and incubate at 80 °C for 10 minutes. Spin at maximum speed for 20 minutes at 4 °C.
Remove supernatant and wash with 300 μL 70% EtOH. Air dry and resuspend sample in 50 μL TE buffer. Sample can now be stored at -20 °C. Sample can be used for quantitative PCR (qPCR) assay (Figure 1).
3. Analyze ChIP-ed DNA
3a. Analyze ChIP-ed DNA using qPCR
Dilute genomic DNA at the following concentration to make a standard curve: undiluted, 1/10, 1/100, 1/1000, 1/5000.
For each primer pair, set up qPCR in duplicate in a 96-well plate with genomic DNA from step 3a.1, input control and ChIP-ed DNA: 10 μL 2x SYBR PCR mix (Fermentas, K0222); 1 μL 10 μM forward primer; 1 μL 10 μM reverse primer; x μL ChIP-ed DNA (input control, or genomic DNA); y μL nuclease-free H2O to adjust the reaction volume to 20 μL.
Spin down the 96-well plate with Mini plate spinner (Labnet).
Perform qPCR with ABI 7300 real time PCR system (or other real time PCR systems) using the following condition: Stage 1: 50 °C for 2 min, 1 cycle; Stage 2: 95 °C for 10 min, 1 cycle; Stage 3: 95 °C for 15 s, 60 °C for 1 min, 40 cycles; Stage 4 (dissociation stage): 95 °C for 15 s, 60 °C for 1 min, 95 °C for 15 s.
Check the dissociation curve: One peak at the thermal dissociation plot suggests a single amplicon from the PCR reaction. More than one peak indicates non-specific product with the primer pair, the qPCR data should not be used.
Determine the linear phase of exponential amplification of PCR reaction for each primer set, by calculating the formula that resolves DNA amount according to the Ct (cycle threshold) value, which is based on the standard curve using the series of diluted genomic DNA in 3a.1.
If the Ct value of ChIP-ed DNA and input control are within the linear range of Ct value, calculate the enrichment of ChIP-ed DNA versus input, according to the diluting factor of ChIP-ed DNA and input control in 2.4.
3b. Amplify ChIP-ed DNA for high throughput sequencing.
End-repair of ChIP-ed DNA mix (Use Epicentre DNA END-Repair kit), set up on ice: 1-34 μl genomic DNA from step 2.12 (0.1 to 5 μg) 5 μl 10x End repair buffer 5 μl 2.5 mM dNTP mix 5 μl 10 mM ATP x μl H2O to adjust the reaction volume to 49 μl 1 μl End-Repair Enzyme mix (T4 DNA Polymerase + T4 Polynucleotide Kinase) Incubate for 45 minutes at RT. Purify reaction using Qiagen MinElute Reaction Cleanup kit. Elute in 30 μl Elution Buffer (EB).
Add A-overhangs to the 3' end: 30 μl of eluted DNA product from step 3b.1 5 μl 10x NEB Buffer #2 10 μl 1 mM dATP 2 μl dH2O 3 μl 5 U/ μl Klenow Fragment (3'→5' exo-) Incubate for 30 minutes at 37 °C. Purify reaction using MinElute ReactionCleanup kit. Elute in 10 μl EB.
Solexa linker ligation: 10 μl of eluted DNA from step 3b.2 10 μl ddH2O 2.5 μl 10x T4 DNA ligase buffer 1 μl Adaptor oligo mix from Illumina (1/10-diluted from stock) 2.5 μl T4 DNA ligase (400 units/μl) Incubate for 1 hour at RT. Purify reaction using MinElute ReactionCleanup kit. Elute in 20 μl EB. Sample can now be stored at -20 °C.
Run the eluted DNA sample from step 3b.3 using the E-gel apparatus. Isolate the 300-500 bp band in the gel and purify using the Qiagen Gel Extraction kit (this step eliminates the ~125 bp self-ligated linkers from the adaptor oligo ligation). Elute in 12 μl EB.
Amplify library using paired-end (PE) primers from Illumina: 10.5 μl of eluted DNA from step 3b.4 12.5 μl of master mix (2X Phusion HF, Finnzymes) 1 μl of PE1 PCR primer 1 1 μl of PE2 PCR primer 2 PCR condition: 98 °C for 30 sec (98 °C 10 sec, 65 °C 30 sec, 72 °C 30 sec), repeat 20 cycles 72°C for 5 minutes.
Samples from step 3b.5 can be used for ChIP-seq after selecting the appropriate sized DNA (300-500 bp) using the standard 2% agarose gel. Sample can now be stored at -20 °C. It is best to isolate each ChIP sample on a single gel to prevent contamination. Samples can be submitted for high-throughput sequencing.
3c. Solexa pipeline analysis
The 25-bp sequencing reads were obtained from the Illumina Genome Analyzer (GA) pipeline.
All reads were aligned to the Drosophila genome (dm3) using the ELAND (Efficient Local Alignment of Nucleotide Data) software, allowing up to two mismatches with the reference sequence.
Uniquely mapped reads were retained for downstream analysis. For multiple identical reads, up to three copies were retained to reduce the possibility of biases from PCR amplification.
The output of the GA Pipeline was converted to browser extensible data (BED) files.
To generate the wig files for uploading to the UCSC browser for visualization, we used a python scrip which has been described in a previous publication3 with 4 bp as the window size and 160 bp as the DNA fragment size.
To classify Drosophila genes into silent and expressed genes, we used a digital number called RPKM (reads/per kilobase merged exonic region/per million mapped reads). Genes with RPKM = 0 were classified as silent group and genes with RPKM ≥ 1 were classified into the expressed groups, which can be classified into three subgroups as genes with low, moderate and high expression levels, and each group has about the same number of genes.
To compare the histone modification level and gene expression, we used a python script which has been described in a previous publication3.
4. Representative Results
Examples of ChIP-qPCR results using bam (bag of marbles) mutant testes are shown in Figure 14. In bam testes, there is a failure in the transition from proliferative spermatogonia to differentiating spermatocytes5, 6. We use bam testes as a source for undifferentiated germ cells, which are enriched in this tissue type. Differentiation genes required for sperm differentiation, such as male-specific transcription factor 87 (mst87F), don juan (dj), and fuzzy onion (fzo) are not expressed in bam testes. These genes are highly enriched with the repressive H3K27me3 histone modification7 (Figure 1A), but are devoid of the active H3K4me3 histone modification8 (Figure 1B), a chromatin signature we called 'monovalent'7. Enrichment of either H3K27me3 or H3K4me3 is determined by normalization to a constitutively expressed Cyclin A (CycA) gene4.
ChIP-seq analysis using the same set of antibodies (i.e. repressive H3K27me3 and active H3K4me3) with bam mutant testes (Figure 2) validated the qPCR results shown in Figure 1. For the three tested terminal differentiation genes mst87F, dj and fzo, their genomic regions are highly enriched with H3K27me3 but not H3K4me3 (Figure 2A-2C). In contrast, the constitutively expressed CycA gene has significant H3K4me3 but little H3K27me3 binding near its transcription start site (TSS) (Figure 2D).
Furthermore, the ChIP profiles of H3K27me3 and H3K4me3 near the TSSs of four classes of differentially expressed genes are consistent with the function of each histone modification. As shown in Figure 3A, enrichment of H3K27me3 downstream of the TSSs is inversely correlated with gene expression level. The silent genes have the highest H3K27me3 whereas the highly expressed genes have no H3K27me3 binding. These data is consistent with the repressive role of H3K27me3 on gene expression. In contrast, enrichment of H3K4me3 around the TSSs showed the opposite correlation with gene expression level (Figure 3B), consistent with the active role of H3K4me3 on gene expression.
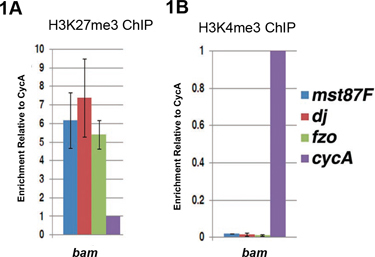 Figure 1. qPCR analysis of ChIP-ed DNA using antibody against either the repressive H3K27me3 histone modification (A) or active H3K4me3 histone modification (B) in undifferentiated cell-enriched bam mutant testes. (A) In bam testes, differentiation genes (Mst87F, dj, fzo) are enriched with the H3K27me3 repressive histone modification. (B) Differentiation genes are depleted with H3K4me3 active mark. The level of ChIP DNA (ChIP DNA/input) at the target gene (Mst87F, dj or fzo) was first normalized to the level of ChIP DNA at the control CycA gene. Error bars indicate standard deviation from three independent biological replicates.
Figure 1. qPCR analysis of ChIP-ed DNA using antibody against either the repressive H3K27me3 histone modification (A) or active H3K4me3 histone modification (B) in undifferentiated cell-enriched bam mutant testes. (A) In bam testes, differentiation genes (Mst87F, dj, fzo) are enriched with the H3K27me3 repressive histone modification. (B) Differentiation genes are depleted with H3K4me3 active mark. The level of ChIP DNA (ChIP DNA/input) at the target gene (Mst87F, dj or fzo) was first normalized to the level of ChIP DNA at the control CycA gene. Error bars indicate standard deviation from three independent biological replicates.
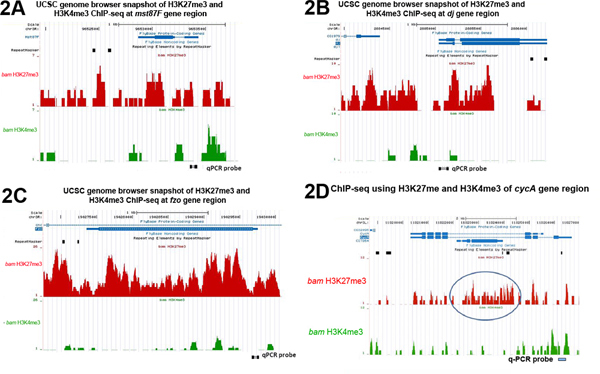 Figure 2. UCSC genome browser snapshots of H3K27me3 and H3K4me3 enrichment across the entire genomic regions of (A) Mst87F (B) dj and (C) fzo, (D) CycA genes. The circled H3K27me3-enriched region in (D) reflects the chromatin status of an overlapping gene CG7264, which is lowly expressed in bam testes (RPKM=1) but highly expressed in wild-type testes (RPKM=130)8 (ChIP-seq data from7). Probes used in quantitative PCR analysis of ChIP results in Figure 1 are labeled at the bottom of each plot. Click here to view larger figure.
Figure 2. UCSC genome browser snapshots of H3K27me3 and H3K4me3 enrichment across the entire genomic regions of (A) Mst87F (B) dj and (C) fzo, (D) CycA genes. The circled H3K27me3-enriched region in (D) reflects the chromatin status of an overlapping gene CG7264, which is lowly expressed in bam testes (RPKM=1) but highly expressed in wild-type testes (RPKM=130)8 (ChIP-seq data from7). Probes used in quantitative PCR analysis of ChIP results in Figure 1 are labeled at the bottom of each plot. Click here to view larger figure.
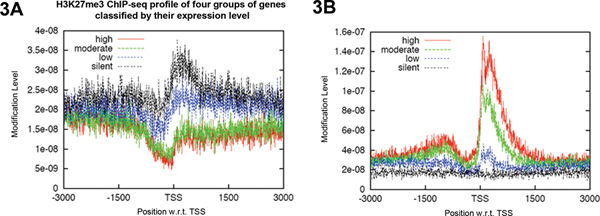 Figure 3. ChIP-seq profiles using H3K27me3 and H3K4me3 in bam testes
7. The four gene groups (7,509 genes) were classified according to their gene expression levels based on RNA-seq results8. The representative classes of genes are plotted for enrichment of a particular histone modification, using sequences from 3kb upstream to 3kb downstream of their transcription start sites (TSSs). This generates a profile of enrichment of (A) H3K27me3 (K27) and (B) H3K4me3 (K4) ChIP-seq analyses in each group. Click here to view larger figure.
Figure 3. ChIP-seq profiles using H3K27me3 and H3K4me3 in bam testes
7. The four gene groups (7,509 genes) were classified according to their gene expression levels based on RNA-seq results8. The representative classes of genes are plotted for enrichment of a particular histone modification, using sequences from 3kb upstream to 3kb downstream of their transcription start sites (TSSs). This generates a profile of enrichment of (A) H3K27me3 (K27) and (B) H3K4me3 (K4) ChIP-seq analyses in each group. Click here to view larger figure.
Discussion
The versatility of ChIP analyses discussed in this protocol can be used on different tissues, which provides an opportunity to study the chromatin state in a biologically relevant system. ChIP experiments using cells from cultured systems are convenient to perform because large quantity of cells can be easily obtained. However, cultured cells do not necessarily reflect cells in a multi-cellular environment. By developing this technique using dissected tissue from live animals, we can address many questions that cultured cells cannot.
Despite the usefulness of this protocol, there are several caveats. For example, the dissected tissue is still heterogeneous with several different cell types. While relatively homogeneous cells can be obtained in Drosophila tissues such as imaginal discs, other tissues like guts, testes, and ovaries which all contain limited adult stem cells are heterogeneous. This complication can be partially addressed using powerful Drosophila genetics. For example, although adult stem cells exist in a small population in tissues discussed above and are extremely difficult to be isolated in a sufficient number for ChIP analysis, stem cell population can be enriched using genetically mutated background. The bam gene is necessary for stem cell differentiation in Drosophila germline lineage5,6. Therefore bam mutant gonads are a good source for enriched undifferentiated germ cells. By performing ChIP using bam mutant, we can obtain an epigenomic landscape in undifferentiated germ cells.
Disclosures
We have nothing to disclose.
Acknowledgments
The authors would like to thank Dr. Keji Zhao's lab (NIH/NHLBI) for their help in providing sequencing results. We would also like to thank the UCSC genome project for the use of Genome Browser to visualize mapped sequencing reads.
This work has been supported by the R00HD055052 NIH Pathway to Independence Award and R01HD065816 from NICHD, the Lucile Parkard Foundation, and the Johns Hopkins University start-up funding to X.C.
References
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, van Steensel B. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 2010;143:212–224. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Chen X, Lu C, Prado JR, Eun SH, Fuller MT. Sequential changes at differentiation gene promoters as they become active in a stem cell lineage. Development. 2011;138:2441–2450. doi: 10.1242/dev.056572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonczy P, Matunis E, DiNardo S. bag-of-marbles and benign gonial cell neoplasm act in the germline to restrict proliferation during Drosophila spermatogenesis. Development. 1997;124:4361–4371. doi: 10.1242/dev.124.21.4361. [DOI] [PubMed] [Google Scholar]
- McKearin DM, Spradling AC. bag-of-marbles: a Drosophila gene required to initiate both male and female gametogenesis. Genes Dev. 1990;4:2242–2251. doi: 10.1101/gad.4.12b.2242. [DOI] [PubMed] [Google Scholar]
- Gan Q, Schones DE, Eun SH, Wei G, Cui K, Zhao K, Chen X. Monovalent and unpoised status of most genes in undifferentiated cell-enriched Drosophila testis. Genome Biol. 2010;11:42–42. doi: 10.1186/gb-2010-11-4-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Q, Chepelev I, Wei G, Tarayrah L, Cui K, Zhao K, Chen X. Dynamic regulation of alternative splicing and chromatin structure in Drosophila gonads revealed by RNA-seq. Cell Res. 2010;7:763–783. doi: 10.1038/cr.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]