

Abstract
Recently, epigenetic regulators have been discovered as key players in many different diseases 1-3. As a result, these enzymes are prime targets for small molecule studies and drug development 4. Many epigenetic regulators have only recently been discovered and are still in the process of being classified. Among these enzymes are lysine demethylases which remove methyl groups from lysines on histones and other proteins. Due to the novel nature of this class of enzymes, few assays have been developed to study their activity. This has been a road block to both the classification and high throughput study of histone demethylases. Currently, very few demethylase assays exist. Those that do exist tend to be qualitative in nature and cannot simultaneously discern between the different lysine methylation states (un-, mono-, di- and tri-). Mass spectrometry is commonly used to determine demethylase activity but current mass spectrometric assays do not address whether differentially methylated peptides ionize differently. Differential ionization of methylated peptides makes comparing methylation states difficult and certainly not quantitative (Figure 1A). Thus available assays are not optimized for the comprehensive analysis of demethylase activity.
Here we describe a method called MassSQUIRM (mass spectrometric quantitation using isotopic reductive methylation) that is based on reductive methylation of amine groups with deuterated formaldehyde to force all lysines to be di-methylated, thus making them essentially the same chemical species and therefore ionize the same (Figure 1B). The only chemical difference following the reductive methylation is hydrogen and deuterium, which does not affect MALDI ionization efficiencies. The MassSQUIRM assay is specific for demethylase reaction products with un-, mono- or di-methylated lysines. The assay is also applicable to lysine methyltransferases giving the same reaction products. Here, we use a combination of reductive methylation chemistry and MALDI mass spectrometry to measure the activity of LSD1, a lysine demethylase capable of removing di- and mono-methyl groups, on a synthetic peptide substrate 5. This assay is simple and easily amenable to any lab with access to a MALDI mass spectrometer in lab or through a proteomics facility. The assay has ~8-fold dynamic range and is readily scalable to plate format 5.
Keywords: Molecular Biology, Issue 61, LSD1, lysine demethylase, mass spectrometry, reductive methylation, demethylase quantification
Protocol
This protocol is modified from Blair et al.6.
1. LSD1 Demethylation Assay
In a final volume of 20 μL, combine 125 ng recombinant LSD1 with 0.25 μg di-methyl histone H3 peptide (ARTKme2QTARKSTGGKAPRKQLYK-biotin) in demethylase buffer (50 mM Tris-Cl pH 8.5, 50 mM KCl, 5 mM MgCl2, 5% glycerol). Additionally, perform a peptide alone control with no enzyme. The control is for section 3 and does not need reductive methylation.
Incubate for 2 hours at 37°C.
To collect the peptides, add 8 μL POROS R2 20 micron beads to each sample and agitate for 15 minutes at room temperature. Prepare POROS beads by suspending 1 volume of beads with 1 volume of methanol, and then add 10 volumes of 5% formic acid / 0.2% TFA.
Condition two C18 ZipTips by aspirating 20 μL of 0.1% TFA into the tip and pushing it back out with a pipettor. Do this twice with 0.1% TFA, four times with 70% acetonitrile / 0.1% TFA, and four times again with 0.1% TFA.
Load the two samples (control and the demethylase reaction) onto the conditioned C18 ZipTips by pipetting the bead slurry behind the C18 resin in the ZipTips and pushing the volume through with a pipettor.
Wash ZipTips twice with 20 μL of 0.1% TFA.
Elute in 40 μL of 70% acetonitrile / 0.1% TFA.
Lyophilize each eluant completely with a SpeedVac concentrator.
2. Reductive Methylation
This portion of the protocol is modified from Blair et al. and Rayment et al.6,7.
Re-suspend the demethylase reaction in 100 μL of 50 mM phosphate buffer, pH 7.4.
To the demethylase reaction, add 8 μL of 15 mg/mL borane dimethylamine. Borane dimethylamine is dissolved in 50 mM phosphate buffer, pH 7.4, to give the 15 mg/mL stock solution. This should be made fresh.
To the demethylase reaction, add 16 μL of 250 mM deuterated formaldehyde. The 250 mM deuterated formaldehyde is prepared by diluting the stock in H20. This should be made fresh.
Incubate at 4°C for 2 hours.
Repeat steps 2.2, 2.3 & 2.4.
Repeat step 2.2.
Incubate at 4°C for ~15 hours.
To the demethylase sample, add 12.5 μL of 1M Tris, pH 7.4 to quench the reductive methylation reaction.
3. MALDI Mass Spectrometry
Add POROS R2 20 micron beads to the demethylase reaction, and collect on ZipTips as described in steps 1.3-1.6.
Note: Re-suspend the control in 100 μL of 50 mM phosphate buffer, pH 7.4 and treat as the other sample starting at step 3.1.
Elute the control and demethylase reaction samples from the ZipTips onto a MALDI sample plate using 2 μL of 33% saturated 2,5-dihydroxybenzoic acid. To prepare the 33% 2,5-dihydroxybenzoic acid, make a saturated solution of 2,5-dihydroxybenzoic acid in 70% acetonitrile / 0.1% TFA and then dilute to 33% saturated in 70% acetonitrile / 0.1% TFA. Allow the eluted samples to dry and crystallize.
Collect mass spectra using a PerkinElmerSciex MALDI-prOTOF mass spectrometer or available high resolution MALDI mass spectrometer 8,9,10.
View spectra and extract peak areas using PerkinElmerSciex TOFworks software or software provided by the mass spectrometer manufacturer.
Verify reaction products by MS2 with an available mass spectrometric system providing tandem mass spectrometric capabilities.
4. Data Analysis
In order to compensate for the isotopic overlap created by using heavy formaldehyde (Figure 1B), determine the peak area (A) ratio of 13C2 and 13C4 isotopes relative to the monoisotopic peak for the control sample, which has not undergone reductive methylation (Fig. 2A). Eq 1: A13C2/A12C = r1 Eq 2: A13C4/A12C = r2 This will give you the ratio of peptide existing in these isotopic states (r1 & r2) specific to your experiment and mass spectrometer.
Use the r1 & r2 values in the following formulas for quantifying the relative amount of peptide existing in each modification state in the demethylase reaction sample (Fig. 2B): Eq 3: H3K4me2= A1 Eq 4: H3K4me = A2 - r1(A1) Eq 5: H3K4 =A3 - r2(A1) - [r1(A2 - r1(A1))]
5. Representative Results
Using LSD1, we show the effectiveness of MassSQUIRM for quantifying lysine demethylation. As described in the Protocol Text section, we performed a demethylase assay with 125 ng of LSD1 and 0.25 μg di-methyl histone H3 peptide (ARTKme2QTARKSTGGKAPRKQLYK-biotin). The control and demethylase reaction samples were subjected to reductive methylation and MALDI mass spectrometry. The control reaction that did not undergo reductive methylation showed the typical isotopic envelope for this peptide and provided peak areas for equations 1 and 2 (Figure 2A). The mass spectrum for the demethylase reaction provided peak areas for equations 3-5 (Figure 2B). Using the peak areas from the control and demethylase reaction, we determined that LSD1 demethylated the peptide to give the following reaction products: 33.7% di-methyl Lys4, 42.3% mono-methyl Lys4, and 24% un-methylated Lys 4. Refer to Blair et al. for the full analysis of LSD1 demethylase activity as well as inhibitor studies 6. Note that changing variables such as reaction time, enzyme concentration and substrate concentration will allow for an in-depth analysis of activity.
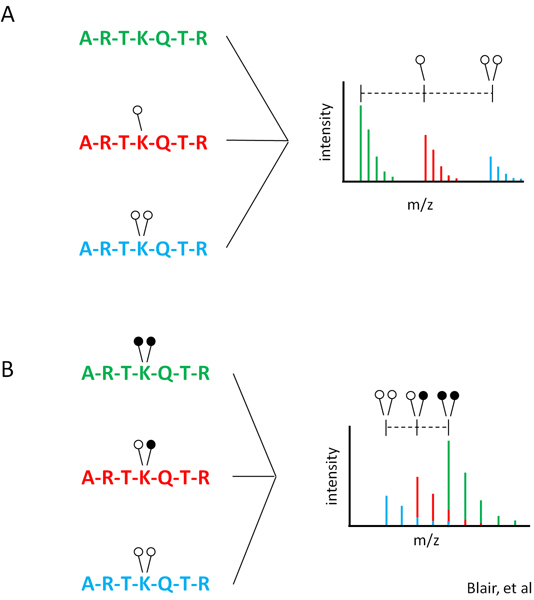 Figure 1. MassSQUIRM overview. (A) An N-terminal peptide from histone H3 is shown as being un- (green), mono- (red), or di- (blue) methylated at lysine 4. Variation in the chemical composition of each peptide leads to differential ionization making quantification complex. (B) Reductive methylation converts all lysine residues to the di-methyl state which causes all peptides to ionize similarly. The use of heavy formaldehyde in the reductive methylation reaction allows retention of the identity of the original methylation. Open circles indicate light methylation while closed circles indicate heavy methylation. Modified from Blair et al. 20116.
Figure 1. MassSQUIRM overview. (A) An N-terminal peptide from histone H3 is shown as being un- (green), mono- (red), or di- (blue) methylated at lysine 4. Variation in the chemical composition of each peptide leads to differential ionization making quantification complex. (B) Reductive methylation converts all lysine residues to the di-methyl state which causes all peptides to ionize similarly. The use of heavy formaldehyde in the reductive methylation reaction allows retention of the identity of the original methylation. Open circles indicate light methylation while closed circles indicate heavy methylation. Modified from Blair et al. 20116.
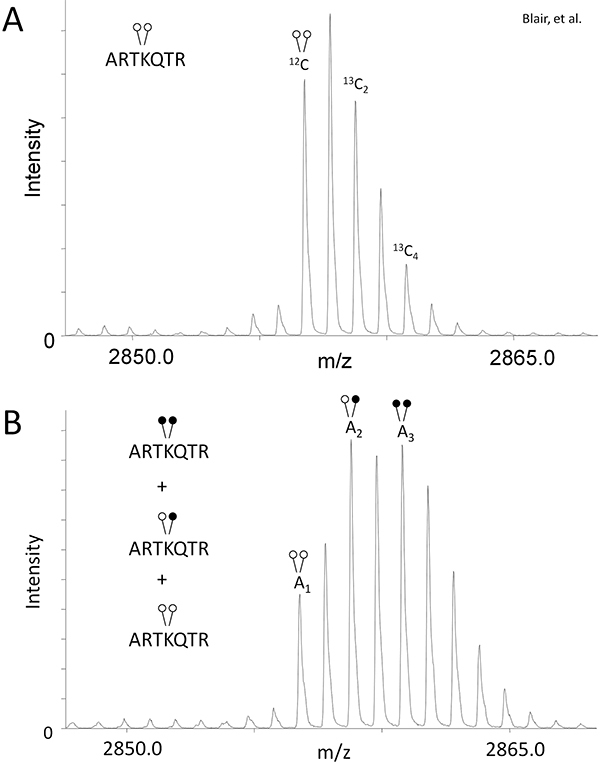 Figure 2. MassSQUIRM can be used to quantify differentially methylated peptides. (A) A di-methylated synthetic histone H3 peptide (ARTKme2QTARKSTGGKAPRKQLYK-biotin) was analyzed using mass spectrometry and peak ratios relative to the monoisotopic peak were noted as r1 and r2 in equations 1 and 2. (B) The same synthetic peptide was incubated with 125 ng LSD1 in demethylase buffer for two hours at 37°C. Samples were then subjected to MassSQUIRM analysis. A mixed population of overlapping peaks represents three different methylation states as seen in Figure 1B. Areas under the monoisotopic peaks were noted as A1, A2, and A3. Peak areas were used in equations 3-5. Open circles indicate light methylation while closed circles indicate heavy methylation. Modified from Blair et al. 20116.
Figure 2. MassSQUIRM can be used to quantify differentially methylated peptides. (A) A di-methylated synthetic histone H3 peptide (ARTKme2QTARKSTGGKAPRKQLYK-biotin) was analyzed using mass spectrometry and peak ratios relative to the monoisotopic peak were noted as r1 and r2 in equations 1 and 2. (B) The same synthetic peptide was incubated with 125 ng LSD1 in demethylase buffer for two hours at 37°C. Samples were then subjected to MassSQUIRM analysis. A mixed population of overlapping peaks represents three different methylation states as seen in Figure 1B. Areas under the monoisotopic peaks were noted as A1, A2, and A3. Peak areas were used in equations 3-5. Open circles indicate light methylation while closed circles indicate heavy methylation. Modified from Blair et al. 20116.
Discussion
MassSQUIRM is an inexpensive and quantitative method for comprehensive analysis of the activity of lysine demethylases involved in mono- and di-methylation. MassSQUIRM offers quantitation not only of the product of the reaction but also for the intermediates. This assay can be used as a powerful tool in studying the mechanism of LSD1 and other histone demethylases. It will also be useful for classifying many newly discovered lysine demethylase enzymes such as PHF8 and could be used for certain methyltransferase enzymes.
Disclosures
We have nothing to disclose.
Acknowledgments
We thank the UAMS Proteomics Facility for mass spectrometric support. Funding for this project was provided by NIH grants P20RR015569, P20RR016460 and R01DA025755.
References
- Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Blair LP, Cao J, Zou MR, Sayegh J, Yan Q. Epigenetic Regulation by Lysine Demethylase 5 (KDM5) Enzymes in Cancer. Cancers (Basel) 2011;3:1383–1404. doi: 10.3390/cancers3011383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole PA. Chemical probes for histone-modifying enzymes. Nat. Chem. Biol. 2008;4:590–597. doi: 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Blair LP. MassSQUIRM: An assay for quantitative measurement of lysine demethylase activity. Epigenetics. 2011;6:490–499. doi: 10.4161/epi.6.4.14531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayment I. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993;261:50–508. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- Collom SL, Jamakhandi AP, Tackett AJ, Radominska-Pandya A, Miller GP. CYP2E1 active site residues in substrate recognition sequence 5 identified by photoaffinity labeling and homology modeling. Arch. Biochem. Biophys. 2007;459:59–69. doi: 10.1016/j.abb.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradolatto A. Saccharomyces cerevisiae Yta7 Regulates Histone Gene Expression. Genetics. 2008;179:291–304. doi: 10.1534/genetics.107.086520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna SD. Yng1 PHD finger binding to histone H3 trimethylated at lysine 4 promotes NuA3 HAT activity at Lysine 14 of H3 and transcripiton at a subset of targeted ORFs. Mol. Cell. 2006;24:785–796. doi: 10.1016/j.molcel.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]