

Abstract
The cystic fibrosis transmembrane conductance regulator (CFTR) is a chloride channel, that when mutated, can give rise to cystic fibrosis in humans.There is therefore considerable interest in this protein, but efforts to study its structure and activity have been hampered by the difficulty of expressing and purifying sufficient amounts of the protein1-3. Like many 'difficult' eukaryotic membrane proteins, expression in a fast-growing organism is desirable, but challenging, and in the yeast S. cerevisiae, so far low amounts were obtained and rapid degradation of the recombinant protein was observed 4-9. Proteins involved in the processing of recombinant CFTR in yeast have been described6-9 .In this report we describe a methodology for expression of CFTR in yeast and its purification in significant amounts. The protocol describes how the earlier proteolysis problems can be overcome and how expression levels of CFTR can be greatly improved by modifying the cell growth conditions and by controlling the induction conditions, in particular the time period prior to cell harvesting. The reagants associated with this protocol (murine CFTR-expressing yeast cells or yeast plasmids) will be distributed via the US Cystic Fibrosis Foundation, which has sponsored the research. An article describing the design and synthesis of the CFTR construct employed in this report will be published separately (Urbatsch, I.; Thibodeau, P. et al., unpublished). In this article we will explain our method beginning with the transformation of the yeast cells with the CFTR construct - containing yeast plasmid (Fig. 1). The construct has a green fluorescent protein (GFP) sequence fused to CFTR at its C-terminus and follows the system developed by Drew et al. (2008)10. The GFP allows the expression and purification of CFTR to be followed relatively easily. The JoVE visualized protocol finishes after the preparation of microsomes from the yeast cells, although we include some suggestions for purification of the protein from the microsomes. Readers may wish to add their own modifications to the microsome purification procedure, dependent on the final experiments to be carried out with the protein and the local equipment available to them. The yeast-expressed CFTR protein can be partially purified using metal ion affinity chromatography, using an intrinsic polyhistidine purification tag. Subsequent size-exclusion chromatography yields a protein that appears to be >90% pure, as judged by SDS-PAGE and Coomassie-staining of the gel.
Keywords: Molecular Biology, Issue 61, Membrane protein, cystic fibrosis, CFTR, protein expression, Cystic Fibrosis Foundation, expression system, green fluorescent protein
Protocol
1. Preparation of Media and Buffers
YNB: For one litre, suspend 6.9 g yeast nitrogen base without amino acids and 0.77 g complete supplement mixture without uracil in 1 l water. Autoclave. Store at 4 °C.
YNBA: For 400 ml, suspend 2.76 g yeast nitrogen base without amino acids, 0.38 g complete supplement mixture without uracil and 8g bacteriological agar in 350 ml water. Autoclave. Mix 8 g glucose with 50 ml water and heat gently until dissolved. Sterilise through a 0.2 μm filter and add to the YNBA whilst the agar is molten. Store at room temperature.
20% glucose medium: For 500 ml, mix 100 g glucose with 500 ml YNB and heat gently until dissolved. Pass through a 0.2 μM filter into a sterile Duran bottle. Store at room temperature.
20% galactose medium: For 2 litres, mix 400 g galactose with 2l YNB and heat gently until dissolved. Pass through a 0.2 μm filter into a sterile Duran bottle. Store at room temperature.
Protease inhibitor stocks: CFTR is highly susceptible to proteolysis4. The authors found the following inhibitors to be effective in limiting proteolysis, though readers may wish to tailor this list to meet their own requirements. Store in 100 μl aliquots at -20 °C to reduce freeze-thaw problems. All inhibitors should be diluted from the stock solutions to the working concentration as below:
| Inhibitor | Stock Concentration | Stock Preparation | Working Concentration |
| AEBSF | 200 mM | Dissolve 48mg in 1ml distilled water | 0.2 mM |
| Benzamidine | 300 mM | Dissolve 36mg in 1ml distilled water | 0.3 mM |
| Chymostatin | 4 mM | Dissolve 2.5mg in 1ml dry DMSO | 4 μM |
| E-64 | 7 mM | Dissolve 2.5mg in 1ml distilled water | 7 μM |
| Leupeptin | 20 mM | Dissolve 10mg in 1ml distilled water | 20 μM |
| Pepstatin A | 15 mM | Dissolve 10mg in 1ml dry DMSO | 15 μM |
| PMSF | 1 M | Dissolve 174mg in 1ml dry DMSO | 1 mM |
DTT (1 M): Dissolve 154 mg dithiothreitol in 1ml distilled water. Store at -20 °C. Use at 1:1000 dilution in buffers indicated.
EDTA (0.5 M): Mix 29.22 g ethylenediaminetetraacetic acid with 100 ml water. Add 10 N NaOH dropwise until all of the EDTA has dissolved and the pH reaches 8. Make up to 200 ml with water and pass through a 0.2 μm filter into a sterile Duran bottle. Store at room temperature.
CRB (300 mM Tris-HCl, pH 7.4, 0.56 M sorbitol, 1 mM EDTA): Dissolve 18.17 g Tris-base in 350 ml water and adjust pH to 7.4 by addition of HCl. Add 51.35 g sorbitol and make volume up to 500 ml with water. Autoclave. Add 1 ml EDTA stock solution and store at 4 °C.
CFTR buffer (50 mM Tris, pH 8.0, 10% v/v glycerol): Dissolve 6.06 g Tris-base in 500 ml water. Adjust pH to 8 with HCl, add 100 ml glycerol and make up to 1 L with water. Autoclave and store at 4 °C.
2x load dye: 50 mM Tris-HCl (pH 7.6), 5% glycerol, 5 mM EDTA (pH 8.0), 0.02% bromophenol blue, 4% SDS, 0.05 M DTT. Make 10 ml, aliquot and store at -20 °C.
2. Screening Transformants
This protocol assumes that the CFTR-GFP-8His fusion gene has been inserted into a yeast plasmid downstream to a GAL1 galactose promoter (Fig.1) and that the plasmid has been transformed into FGY217 cells, a Pep4 deletion mutant of S.cerevisiae10. Cells can be grown on YNBA plates and stored for several weeks at 4 °C. For longer term storage, glycerol stocks should be made and stored at -80 °C. Methods for cloning and transformation are described in detail by Drew et al. (2008)10.
Pick 5-10 well-separated colonies from a transformation plate. Transfer each colony to a separate sterile 50 ml Falcon tube containing 9 ml YNB and 1ml of 20% glucose medium. For this step, it is important to have a final concentration of 2% glucose (w/v) in the culture to maintain cell growth. Grow overnight for 16 hours at 250 rpm, 30 °C in an orbital shaking incubator.
Make glycerol stocks for each of the screened colonies. Aseptically add 0.8 ml of the overnight cultures to 0.2 ml sterile glycerol in labeled screw-top vials, vortex briefly and store at -80 °C.
Dilute the remaining overnight cultures to a final volume of 50 ml in YNB, including 250 μl of 20% glucose medium. For this step, it is important to dilute the glucose concentration to approximately 0.1% (w/w) in the culture because high glucose can repress the GAL1 promoter10. Grow cultures in labeled 250 ml Erlenmeyer baffled flasks to an OD600 of 0.7-0.8 at 250 rpm, 30 °C in an orbital shaking incubator.
Induce the cultures by addition of 5 ml 20% galactose medium to each flask and grow on for 16 hours.
Confirm the expression of CFTR using fluorescence microscopy. Take 100 μl of culture and add 100 μl glycerol to limit cell mobility in solution. Analyse cells on a Delta Vision RT restoration microscope (or similar), using a blue laser under a FITC filter (excitation wavelength of 490 nm and emission wavelength of 528-538 nm). Positive expression of the CFTR-GFP fusion protein should be visible as a ring of fluorescence at the plasma membrane of the yeast cells. Untransformed yeast cells may be used as a control.
Transfer the cultures into 50ml Falcon tubes. Harvest the cells by centrifugation at 3500 x g, 4 °C for 10 minutes in a bench top centrifuge. Whilst the centrifuge is running, prepare 2 ml microfuge tubes with screw tops containing approximately 500 μl acid-washed glass beads and place on ice. Discard the supernatants and resuspend each pellet in 500-800 μl ice-cold CRBwithprotease inhibitors. Transfer the suspensions to the microfuge tubes containing the beads and keep on ice.
Lyse the cells by vigorously shaking/vortexing each microfuge tube for 10 periods of 30 seconds, resting on ice in between periods. A beadbeater can be employed as an alternative, e.g. a BioSpec mini beadbeater operated for 3 min.
Place the tubes into a benchtop microfuge and centrifuge at 3,500 x g, 4 °C for 5 minutes. Transfer the supernatants containing the crude membrane population to clean microfuge tubes and place on ice. Add 500 μl fresh ice-cold CRBwith protease inhibitors to each tube and repeat the process to accumulate the membranes.
Collect the crude membranes by spinning at maximum speed, 4 °C in a benchtop microfuge for 2 hours. Discard the supernatant and resuspend each pellet in 50 μl ice cold CFTR buffer.
In clean microfuge tubes, mix 15 μl of each suspension with 15 μl 2x load dye by pipetting up and down. Incubate at room temperature for a 10 minutes. Do not boil the samples, as this will cause CFTR and other membrane proteins to form SDS-insoluble aggregates and also denature the GFP tag.
Load the samples along with PageRuler Plus prestained protein standards (Fermentas) onto a 4-20% Tris/glycine gradient gel (NuSep) and run at 150 V for 40 minutes or until the dye-front is at the bottom of the gel.
Identify the highest expressing cells by in-gel fluorescence. Place into a fluorescence imaging system such as a Typhoon scanner. Scan the gel using the blue laser at an excitation wavelength of 488 nm and an emission wavelength of 526 nm. The CFTR-GFP fusion should be visible at approximately 220 kDa. There will also be a weak fluorescent band visible at about 70 kDa which is probably an intrinsic yeast FAD-containing membrane protein (such as succinate dehydrogenase subunit A) 11,12.
Stain the gel with Coomassie, destain and scan the gel for comparison to the fluorescence scan using a convenient image viewing software package. The Coomassie-stained gel allows a relative assessment of CFTR-GFP expression levels in different clonal lines after normalization for the amount of total protein loaded onto each track of the gel.
Streak out the highest expressing cell line from its glycerol stock (2.2) onto a fresh YNBAplate and incubate at 30 °C for 2-3 days. This plate may then be stored for up to 2 weeks at 4 °C.
3. Large-scale Fermenter Culture
Prepare pre-cultures for the fermenter. Scrape a 1cm2 area of cells from the YNBAplate (2.13) using a sterile loop and add to 45 ml YNB and 5 ml 20% glucose medium, such that the OD600 is approximately 0.1. Grow in 250 ml Erlenmeyer baffled flasks at 250 rpm, 30 °C in an orbital shaking incubator until the OD600 reaches 1.
Split the culture between two 2 l Erlenmeyer baffled flasks each containing 450 ml YNB and 25 ml 20% glucose medium. Grow these on at 250 rpm, 30 °C in an orbital shaking incubator until the OD600 reaches 1.2.
Whilst these pre-cultures are growing, set up the fermenter. Make 11.2l of YNB as described in 1.1, but dissolve an additional 8.28 g YNB and 0.95 g drop out supplement to compensate for the addition of glycerol at induction. Aseptically add the 11.2 l YNB and 75 ml 20% glucose medium to a sterile 20l fermenter vessel and adjust the running temperature to 30 °C.
Aseptically add the precultures to the fermenter and set the stirring speed to approximately 800 rpm and maintain the temperature at 30 °C. Compressed air should flow at approximately 15 dm3min-1.Once the fermenter culture reaches an OD600 of 1.2, induce by aseptically adding 1.5 l YNB 20% galactose solution and 1.2 l glycerol. Reduce the temperature to 25 °C and grow the culture for 16 hours.
Transfer the fermenter contents into chilled 1l centrifuge pots on ice using a peristaltic pump. Harvest the cells by centrifugation at 3,500 x g, 4 °C for 30 minutes in a large capacity rotor (e.g. 6 litre). Resuspend the cells in 150 ml ice cold CRB with protease inhibitors. From here on, all work should be carried out at 4 °C.
Lyse the cells by passing through a Constant Systems cell disrupter in 4 passes at 25, 30, 32 and 35 kPa, collecting the lysate on ice in each case. Alternatively, use a bead beater (Biospec) with an equal volume of acid-washed 0.5 mm glass beads and agitate for 3 minutes on full power. Transfer the lysate to 50ml Falcon tubes and pellet the cell debris by centrifugation at 3,500 x g, 4 °C for 15 minutes in a benchtop centrifuge.
Transfer the supernatant to chilled centrifuge tubes. Centrifuge at 14,000 x g, 4 °C for 30 minutes in a centrifuge to remove mitochondria.
Transfer supernatant to chilled ultracentrifuge tubes. Centrifuge at 200,000 x g, 4 °C for 90 minutes in an ultracentrifuge to collect microsomes.
Carefully decant and discard the supernatant and add 2ml ice cold CFTR buffer with protease inhibitors and 1mM DTT to each tube. Gently resuspend the pellets using a paintbrush, top up each tube with CFTR buffer and mix using a vortex mixer.
Centrifuge the suspension at 200,000 x g, 4 °C for 60 minutes in an ultracentrifuge, discard the supernatant and resuspend pellets in 2 ml ice cold CFTR buffer with protease inhibitors(no DTT) using a paintbrush.
Pool the resuspended microsomes, adjust the final volume to 50 ml with CFTR buffer and mix well. Reserve a 1 ml aliquot for SDS-PAGE gel analysis, as described in 2.9 - 2.11. The microsomes can now be flash frozen in liquid nitrogen and then stored at -80 °C until needed.
CFTR can be extracted from microsomes using one of the following detergents: lithium perfluorooctonoate acid (LiPFO), tetradecanoly-lyso-phosphatidylglycerol (LPG14), n-dodecyl-β-D- maltoside (DDM). Mix the microsomes with CFTR buffer, protease inhibitors and 5% detergent (w/w). If DDM is used, also add 300 mM NaCl to the buffer. Agitate at 4 °C for 15 minutes on a tube rotator.
Centrifuge the samples at 100,000 x g, 4 °C for 1 hour in an ultracentrifuge. Retain the supernatant and take a small aliquot for SDS-PAGE gel analysis. CFTR may now be purified from the solublised material by immobilised metal affinity chromatography followed by size exclusion chromatography.
4. Representative Results
Transformation of yeast with the CFTR-containing plasmid is not 100% efficient. A representative small-scale screen of CFTR expression in selected colonies from a transformation experiment will yield about 1 in 4 colonies expressing the protein. A typical result from a screen of 5 colonies picked from a plate is shown in panel A of Fig. 3. One of the colonies shows a strong level of expression of the CFTR-GFP fusion protein which typically runs between the 250 kDa and 130 kDa markers, as shown. The CFTR-GFP fluorescence levels will vary considerably between experiments, with colony 4 in Fig. 3 showing at least 10x greater fluorescence than the intrinsic fluorescent band at about 70kDa. If expression levels of CFTR-GFP appear to give less fluorescence than the 70 kDa band, then it is probably worth re-transforming and choosing a colony with higher levels of CFTR-GFP expression. As shown in Fig. 3A, it is unlikely, even with a high expression level of CFTR-GFP, that the CFTR-GFP band will be discernable in the cell extract by Coomassie staining.
Once selected colonies have been grown in larger scale experiments, and microsomes isolated, the presence of CFTR-GFP within the microsomes will need to be assessed, as shown in Fig. 3B. The results of this experiment are important, not only to assess the efficiency of the induction of expression, but also to check that the microsomes have been prepared carefully and that proteolysis has been minimized. The results shown in Fig. 3B imply that in this experiment the CFTR-GFP expression is somewhat lower (as judged relative to the intrinsic 70kDa band) than in the small-scale experiment shown in panel A. However this impression is biased by the overexposure of the fluorescence detector in this measurement. This was because the experimenter was checking for the presence of proteolytic fragments of the CFTR construct. There is some evidence in this experiment for some fluorescent proteolytic fragments between the 130 kDa and 100 kDa markers, but these are very weak compared to the full-length CFTR-GFP band. With the protease inhibitors described here, we find little evidence for proteolytic degradation of CFTR after cell breakage. If significant proteolysis is observed, we recommend making fresh protease inhibitor stock solutions. We have also found that commercial protease inhibitor cocktail tablets are not as effective for this system. Growth of cells beyond 16 hr (post-induction) will give rise to decreased CFTR expression as shown in Figure 4. This is probably due to turnover of the protein, perhaps due to upregulation of the yeast protein quality control machinery6-9,13. It is therefore advisable to monitor CFTR expression levels after induction with galactose, if possible, as the optimal time to harvest the cells may vary from one laboratory to another.
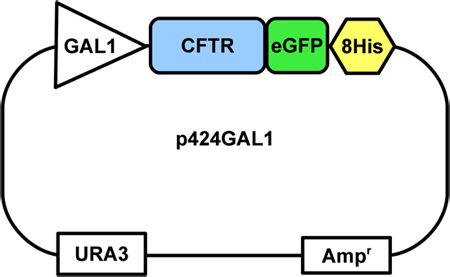 Figure 1. The CFTR construct-containing yeast plasmid. The CFTR-GFP-8His fusion is inserted into the 2μ p424GAL1 expression vector, under the control of a galactose (GAL1) promoter. The vector also contains a uracil selection marker (URA) and an ampicillin resistance gene (Amp).
Figure 1. The CFTR construct-containing yeast plasmid. The CFTR-GFP-8His fusion is inserted into the 2μ p424GAL1 expression vector, under the control of a galactose (GAL1) promoter. The vector also contains a uracil selection marker (URA) and an ampicillin resistance gene (Amp).
 Figure 2. A flowchart summarising the visualised protocol.
Figure 2. A flowchart summarising the visualised protocol.
 Figure 3. Representative SDS-PAGE gels of CFTR expression and purification. Panel A shows five randomly picked transformant colonies (lanes 1-5) that were screened for CFTR expression. Panel B shows microsomes that were isolated from a 15l fermenter culture. Panel C shows purified murine CFTR obtained after two-stage purification using affinity chromatography followed by size exclusion chromatography. All gels are shown under illumination conditions exciting fluorescence from the GFP domain (left) and after Coomassie stain (right). The relative locations of molecular weight standards are listed on the left (kDa).
Figure 3. Representative SDS-PAGE gels of CFTR expression and purification. Panel A shows five randomly picked transformant colonies (lanes 1-5) that were screened for CFTR expression. Panel B shows microsomes that were isolated from a 15l fermenter culture. Panel C shows purified murine CFTR obtained after two-stage purification using affinity chromatography followed by size exclusion chromatography. All gels are shown under illumination conditions exciting fluorescence from the GFP domain (left) and after Coomassie stain (right). The relative locations of molecular weight standards are listed on the left (kDa).
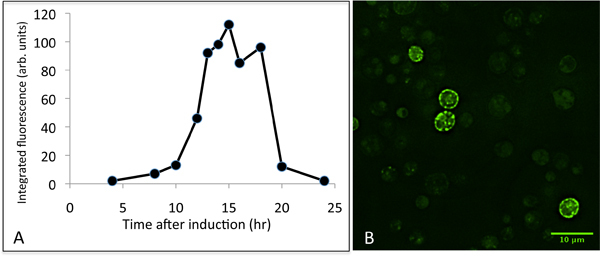 Figure 4. Representative data for the expression of CFTR in yeast. Panel A shows a time-course of CFTR expression after induction with galactose. Cell extracts were analysed by SDS-PAGE, and the GFP fluorescence for the CFTR-GFP protein band was integrated. Panel B shows typical results for fluorescence microscopy of GFP-expressing cells 16 hr post induction. Typically, only a fraction of the cells express CFTR at high levels.
Figure 4. Representative data for the expression of CFTR in yeast. Panel A shows a time-course of CFTR expression after induction with galactose. Cell extracts were analysed by SDS-PAGE, and the GFP fluorescence for the CFTR-GFP protein band was integrated. Panel B shows typical results for fluorescence microscopy of GFP-expressing cells 16 hr post induction. Typically, only a fraction of the cells express CFTR at high levels.
Discussion
This paper provides a method for the expression of murine CFTR protein in yeast cells, which should facilitate research on cystic fibrosis. The aim is to link this paper with the release of the murine CFTR DNA construct, which will be available through the Cystic Fibrosis Foundation (http://www.cff.org/research/CFFT/). Other orthologs should become available later. Transformation of the yeast cells with the CFTR-containing vector is straightforward, but it is important to screen for colonies expressing high levels of CFTR. Variable expression levels may arise from several factors, but the number of copies of the plasmid per cell probably accounts for a significant degree of variation. Critical steps described here should allow production of CFTR-expressing yeast cells and CFTR-containing microsomal membranes. Once the transformation, growth, harvesting and lysis of yeast cells have been mastered, purification of the protein should be possible, and in Figure 3 we have given an example of the purity that should be achievable in this case as a useful benchmark. It is not our intention in this manuscript to provide detailed methodology for purification of the protein. However, there are some critical downstream purification steps that are specific to the S.cerevisae expression system, such as cell lysis and microsome purification, and these have been included in detail in this manuscript. It should be mentioned, however, that apart from the two methods we have used, alternative yeast cell disruption methods can be employed, such as the use of a French pressure cell. The recombinant protein has a TEV-cleavable C-terminal GFP domain that allows the protein to be tracked after induction (Fig. 4). Yeast have an intrinsic 70kDa protein (probably succinate dehydrogenase12) that fluoresces under the same conditions11, and this can provide a useful internal calibration standard for the relative expression levels of CFTR in whole cell extracts or microsomes (Fig. 3). It is clear from the data shown in Figure 4 that the timing of cell harvesting after induction with galactose is crucial. Yields of CFTR drop precipitously after about 16 hr of induction, so that there is barely any detectable CFTR in yeast cells after 24 hr of induction.
The yield of purified protein is about 1-2mg CFTR protein per 15 litre fermenter culture. Recovery can be estimated as about 70% of the total CFTR-GFP protein up to the microsome stage, and about 25% recovery of purified protein. Characterisation of the S. cerevisiae-expressed CFTR is ongoing. As seen in Fig. 4, the protein's location in the cell can be monitored by fluorescence microscopy. Although much of the fluorescence is found around the periphery of the cell as expected10, some of the protein displays a punctate localization, either in, or just inside the plasma membrane which could be due to CFTR recycling through a late Golgi/endosomal pathway14 or perhaps a compartment downstream of the budding of transport vesicles from the ER4. Treatment with PNGaseF, an enzyme that deglycosylates proteins, showed minimal change in the migration of the CFTR protein band on SDS-PAGE, implying that it is unglycosylated, or has minimal glycosylation 15. Experiments on the phosphorylation state of the protein are underway. In some of the detergents tested so far, the purified protein displays ATPase activity (that is inhibited by a CFTR-specific inhibitor16) at rates that are similar to those previously published 2,15 . Measurement of CFTR channel activity will require reconstitution of the purified protein, which would imply a final purification step in a detergent that has a relatively high critical micelle concentration (cmc)17. Yeast microsomes containing CFTR can be solublised with several commonly employed detergents18, including detergents such as dodecyl maltoside2, which are generally considered to be 'mild'. However most high cmc detergents have proven to be inefficient for solubilsation, so far, suggesting that exchange into these detergents should be considered at a late stage in any purification scheme.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank the Cystic Fibrosis Foundation (CFF) for funding this work through its CFTR 3D Structure Consortium (grant number FORD08XX0). We acknowledge the huge contribution of all our colleagues within the Consortium to this work, in particular in the design of the CFTR genes. We also acknowledge the invaluable contributions of Drs. James Birtley (NCSR Demokritos, Greece), Mark Young (University of Cardiff, UK) and David Drew (Imperial College, London, UK) in the early stages of the work. We especially thank Dr. Ina Urbatsch (Texas Tech. University, Lubbock) for critical reading of the manuscript.
References
- Ford RC, Birtley J, Rosenberg MF, Zhang L. CFTR three-dimensional structure. Methods Mol. Biol. 2011;741:329–346. doi: 10.1007/978-1-61779-117-8_22. [DOI] [PubMed] [Google Scholar]
- Rosenberg MF, Kamis AB, Aleksandrov LA, Ford RC, Riordan JR. Purification and crystallization of the cystic fibrosis transmembrane conductance regulator. CFTR). J. Biol. Chem. 2004;279:39051–39051. doi: 10.1074/jbc.M407434200. [DOI] [PubMed] [Google Scholar]
- Zhang L, Aleksandrov LA, Riordan JR, Ford RC. Domain location within the cystic fibrosis transmembrane conductance regulator protein investigated by electron microscopy and gold labelling. Biochim. Biophys. Acta. 2011;1808:399–404. doi: 10.1016/j.bbamem.2010.08.012. [DOI] [PubMed] [Google Scholar]
- Kiser GL. Expression and degradation of the cystic fibrosis transmembrane conductance regulator in Saccharomyces cerevisiae. Arch. Biochem. Biophys. 2001;390:195–205. doi: 10.1006/abbi.2001.2385. [DOI] [PubMed] [Google Scholar]
- Huang P, Stroffekova K, Cuppoletti J, Mahanty SK, Scarborough GA. Functional expression of the cystic fibrosis transmembrane conductance regulator in yeast. Biochim. Biophys. Acta. 1996;1281:80–90. doi: 10.1016/0005-2736(96)00032-6. [DOI] [PubMed] [Google Scholar]
- Ahner A, Nakatsukasa K, Zhang H, Frizzell RA, Brodsky JL. Small heat-shock proteins select deltaF508-CFTR for endoplasmic reticulum-associated degradation. Mol. Biol. Cell. 2007;18:806–814. doi: 10.1091/mbc.E06-05-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Sztul E. ER-associated complexes (ERACs) containing aggregated cystic fibrosis transmembrane conductance regulator (CFTR) are degraded by autophagy. Eur. J. Cell. Biol. 2009;88:215–226. doi: 10.1016/j.ejcb.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F. Derlin-1 promotes the efficient degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) and CFTR folding mutants. J. Biol. Chem. 2006;281:36856–36863. doi: 10.1074/jbc.M607085200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Michaelis S, Brodsky JL. CFTR expression and ER-associated degradation in yeast. Methods Mol. Med. 2002;70:257–2565. doi: 10.1385/1-59259-187-6:257. [DOI] [PubMed] [Google Scholar]
- Drew D. GFP-based optimization scheme for the overexpression and purification of eukaryotic membrane proteins in Saccharomyces cerevisiae. Nat. Protoc. 2008;3:784–798. doi: 10.1038/nprot.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight AW, Billinton N. Seeing the wood through the trees: A review of techniques for distinguishing green fluorescent protein from endogenous autofluorescence. Anal. Biochem. 2001;291:175–197. doi: 10.1006/abio.2000.5006. [DOI] [PubMed] [Google Scholar]
- Robinson KM, Lemire BD. Isolation and nucleotide sequence of the Saccharomyces cerevisiae gene for the succinate dehydrogenase flavoprotein subunit. J. Biol. Chem. 1992;267:10101–10107. [PubMed] [Google Scholar]
- Gnann A, Riordan JR, Wolf DH. Cystic fibrosis transmembrane conductance regulator degradation depends on. the lectins Htm1p/EDEM and the Cdc48 protein complex in. 2004;15:4125–4135. doi: 10.1091/mbc.E04-01-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo JS. Non-conventional trafficking of the cystic fibrosis transmembrane conductance regulator through the early secretory pathway. J. Biol. Chem. 2002;277:11401–11409. doi: 10.1074/jbc.M110263200. [DOI] [PubMed] [Google Scholar]
- Zhang L. Architecture of the cystic fibrosis transmembrane conductance regulator protein and structural changes associated with phosphorylation and nucleotide binding. Journal of Structural Biology. 2009;167:242–251. doi: 10.1016/j.jsb.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Wellhauser L. A small-molecule modulator interacts directly with deltaPhe508-CFTR to modify its ATPase activity and conformational stability. Mol. Pharmacol. 2009;75:1430–148. doi: 10.1124/mol.109.055608. [DOI] [PubMed] [Google Scholar]
- Ramjeesingh M. A monomer is the minimum functional unit required for channel and ATPase activity of the cystic fibrosis transmembrane conductance regulator. Biochemistry. 2001;40:10700–10706. doi: 10.1021/bi0108195. [DOI] [PubMed] [Google Scholar]
- Huang P, Liu Q, Scarborough GA. Lysophosphatidylglycerol: a novel effective detergent for solubilizing and purifying the cystic fibrosis transmembrane conductance regulator. Anal. Biochem. 1998;259:89–97. doi: 10.1006/abio.1998.2633. [DOI] [PubMed] [Google Scholar]