

Abstract
Assignment of glycosylation sites and site microheterogeneity is of both biological and clinical significance. Herein, the detailed N-glycosylation pattern of human serum alpha-2-macroglobulin was studied using an integrative approach, including permethylation of N-glycans, collision induced dissociation (CID) and electron transfer dissociation (ETD) of chymotryptic N-glycopeptides, and partial deglycosylation of chymotryptic N-glycopeptides with endo-β-N-acetylglucosaminidase F3 (Endo F3). Three N-glycosylation sites were found to be occupied by four biantennary complex type N-glycans using N-glycan analysis and the ETD/CID method. Endo F3 assisted mass spectrometric analysis yielded five N-glycosylation sites with and without core fucosylation. In total, six out of eight potential N-glycosylation sites were identified using this approach. This integrative approach was performed using only 10 μL of human serum for both N-glycosylation site assignment and site microheterogeneity determination.
Keywords: Glycopeptide, Site-specific glycosylation, LC-MS/MS, Collision induced dissociation, Electron transfer dissociation, Endoglycosidase, Core fucosylation, Alpha-2-macroglobulin
Introduction
Glycosylation is one of the most prevalent post-translational modifications for proteins and plays an important role in cell recognition, signal transduction and cell proliferation. N-glycosylation is the most common type of glycosylation, where glycans attach to asparagine in the consensus sequence N-X-S/T, where X cannot be proline. Aberrant glycosylation, such as site-specific glycosylation abnormalities, has been found to be associated with various types of cancers or other malignancies [1–3]. Site-specific glycosylation information is of great importance in both clinical research and fundamental biology.
N-glycosylation analysis at the glycopeptide level currently remains challenging for many reasons. Proteins can have multiple glycosylation sites (glycosylation heterogeneity) and each site can be occupied by more than one glycan (glycosylation microheterogeneity), hence the concentrations of individual glycopeptides are usually very low and require highly sensitive methods. Glycopeptides have low ionization efficiency and often suffer from ion suppression from non-glycopeptides during mass spectrometric analysis. Furthermore, collision induced dissociation (CID) of glycopeptides mostly produces fragments from the glycan moiety and no peptide sequence or glycosylation site information is produced [4].
Recently introduced electron transfer dissociation (ETD) overcomes some of the limitations of CID for glycopeptide analysis [5]. ETD uses reagents, such as nitrobenzene or fluoranthene, to produce radical anions that interact with analyte cations (normally with charge +3 or above) to produce fragmentation mainly along the peptide backbone, generating c and z type ions without disrupting the glycan [6]. By combining ETD, which mainly yields information on peptide sequence and glycosylation sites, with CID, which cleaves glycosidic bonds to reveal glycan composition, site-specific glycosylation can be identified in a single LC MS/MS run [5,7].
An alternative method to identify glycosylation sites uses endo-β-N-acetylglucosaminidases to partially deglycosylate glycopeptides [8–10]. Endo H and Endo F1 cleave high-mannose and hybrid type glycans. Endo F2 and Endo F3 are able to cleave complex type glycans with two or three branches [11]. Endo-β-N-acetylglucosaminidase F3 cleaves between the two GlcNAc at the pentasaccharide core, leaving only the innermost GlcNAc and core fucose, if present, attached to the peptides [11]. The fact that fucose remains attached provides endo-β-N-acetylglucosaminidase with the unique ability to identify core fucosylation sites. Core fucosylation occurs during the maturation stage of glycosylation where fucoses are added to the innermost GlcNAc via α (1,6) linkage [12]. Endo-β-N-acetylglucosaminidase leaves a 203 Da (GlcNAc) or 349 Da (GlcNAc-Fuc) modification on the asparagine and such modifications allow for reliable identification of glycosylation sites by CID MS/MS and database searching. False positive identifications are less common because of the large mass increment introduced by GlcNAc or GlcNAc-Fuc attachment.
Alpha-2-macroglobulin is one of the largest and the most abundant proteins in human serum with a molecular weight around 720 kDa. Usually in a tetrameric form, alpha-2-macroglobulin is an acute phase protein that is mainly synthesized in the liver and is known as a proteinase inhibitor [13]. Alpha-2-macroglobulin has eight potential N-glycosylation sites at N55, N70, N247, N396, N410, N869, N991 and N1424, and all the sites have been identified by previous work [14–18]. However, the previous studies were performed on completely deglycosylated peptides where no information on glycans was obtained. In the current work, we have used several complementary approaches to study the site-specific glycosylation pattern of alpha-2-macroglobulin. Three N-glycosylation sites (N70, N396 and N1424) were identified in the CID/ETD MS/MS approach, where heterogeneity of glycans at each site was described. Five N-glycosylation sites (N396, N410, N869, N991 and N1424) were found with the Endo F3 partial deglycosylation method, which uniquely revealed core fucosylation at site N396, N410 and N1424. This integrated approach to studying N-glycosylation was performed with only 10 μL of serum and could serve as a model for studies of other glycoproteins.
Experimental Section
Serum samples
Human normal sera were provided by the University Hospital, Ann Arbor, Michigan according to IRB approval. The samples were aliquoted and stored in a −80°C freezer until further use. All samples were frozen and thawed only once.
Purification of alpha-2-macroglobulin from serum
Alpha-2-macroglobulin was purified from human serum as described in previous work [19]. Briefly, 10 μL of human serum was depleted of immunoglobulin (IgG) with protein A/G agarose beads (Pierce Scientific, Rockford, IL) to avoid IgG interference during immunoprecipitation. Twenty micrograms of alpha-2-macroglobulin antibody (Abcam, Cambridge, MA) was immobilized on protein A/G agarose beads with disuccinmidyl suberate (DSS) crosslinker and then incubated with the depleted serum overnight. Alpha-2-macroglobulin was eluted with 100 mM glycine-HCl at pH 2.8 and desalted with 75 μL Zeba desalting columns (Pierce Scientific, Rockford, IL).
On-plate tryptic digestion and mass spectrometric analysis were performed to verify the success of immunoprecipitation. Desalted alpha-2-macroglobulin was spotted on the MALDI plate and dried in air. Trypsin (Promega, Madison, WI) (0.4 μg) was added to 10 μL of 100 mM ammonium bicarbonate solution with 20% of acetonitrile and added onto the alpha-2-macroglobulin spot and incubated in a humid chamber at 37°C for 10 min. 2,5-dihydroxybenzoic acid (DHB, 10 mg/mL) (Laser Biolabs, France) was dissolved in 50% acetonitrile with 0.1% trifluoroacetic acid and added on top of the dried spot. The peptide peaks were searched against SWISS-PROT Homo sapiens and other mammalia protein database (2012_03) using Mascot, with methionine oxidation set as a variable modification. The tolerance for MS matching was set at 0.2 Da. Mass spectrometric analysis was performed on an Axima MALDI quadrupole ion trap-time of flight mass spectrometer (Shimadzu Biotech, Manchester, UK). A pulsed nitrogen laser (337 nm) at 5 Hz was used for ionization. Helium was used to cool the trapped ions and argon was used for CID fragmentation. All spectra were acquired in the positive ion mode. Spectra were calibrated with an external peptide standard mixture (Bruker Daltonics, Billerica, MA) to a mass accuracy of 30 ppm.
Deglycosylation, purification, permethylation and identification of N-glycans
Alpha-2-macroglobulin was denatured in 10% denaturing solution (0.02% SDS, 10 mM 2-mercaptoethanol) at 60°C for 30 min. Ammonium bicarbonate solution was added to a final concentration of 15 mM. N-glycosidase F (New England Biolabs, Ipswich, MA) was added to release N-glycans at 37°C overnight. Ten microliter porous graphitized carbon tips (Sigma Aldrich, St. Louis, MO) were used to purify N-glycans from proteins and other impurities as described previously [19]. In-solution permethylation was performed on dried purified N-glycans according to published procedures [19]. Permethylated N-glycans were dissolved in 2.5 μL of 20% acetonitrile and 0.5 μL was spotted on a MALDI plate and 0.5 μL sodiated DHB (10 mg/mL in 50% acetonitrile with 100 mM sodium chloride) was spotted on top. N-glycans were analyzed by MALDI-QIT-TOF MS with the same parameters as described before. Glycomod (http://www.expasy.org/tools/glycomod) was utilized to predict the N-glycan compositions. Only N-glycans included in the GlycoSuite database were selected. The glycan compositions were further confirmed with CID MS/MS analysis.
LC-ESI-CID/ETD-MS analysis of chymotryptic glycopeptides
Purified alpha-2-macroglobulin was reduced with 10 mM of dithiothreitol (DTT) at 95°C for 15 min and then alkylated with 22 mM of iodoacetamide (IAA) at room temperature in the dark for 15 min. Alpha-2-macroglobulin was diluted in 50 mM ammonium bicarbonate and incubated with 0.3 U of chymotrypsin (Promega, Madison, WI) at 37°C for 16 h. Chymotrypsin was deactivated by boiling for 3 min. The digested mixture was dried in a SpeedVac and reconstituted in 10 μL 80% acetonitrile with 2% formic acid. ZIC-HILIC ziptips (Protea, Morgantown, WV) were used to enrich glycopeptides. After the tips were equilibrated with 80% acetonitrile with 2% formic acid, the samples were loaded on the tips followed by washing with 80% acetonitrile with 2% formic acid to remove non-specific binding. The glycopeptides were eluted by 98% water with 2% formic acid.
Fused silica PicoTips (New Objectives, Woburn, MA) packed with C18 material (5 μm particle size, 10 cm × 75 μm i.d.) was used for both chromatographic separation and ionization spray. Gradient elution was performed on a Paradigm MG4 micropump system (Michrom Biosciences, Auburn, CA) at 300 nL/min with mobile phase A as 2% acetonitrile with 1% acetic acid in water and mobile phase B as 5% water with 1% acetic acid in acetonitrile. A 70 min gradient was used: (1) 5% B to 60% B in 35 min, (2) 60% B to 95% B in 1 min, (3) isocratic at 95% B for 4 min, (4) decrease from 95% B to 5% B in 0.1 min, (5) isocratic at 5% B for 30 min.
An LTQ-CID/ETD-MS (Thermo Fisher Scientific, San Jose, CA) operated in positive ion mode was used for all LC-MS experiments. The ESI spray voltage was set at 2.2 kV and capillary voltage at 45 V. The mass spectra were generated in a data-dependent manner. After a full scan from m/z 400 to m/z 1800, the three most intense ions were selected for ETD and CID fragmentation. For ETD MS/MS, the reactant temperature was 145°C, the ionization energy was 70V, the emission current was 15 μA, and the ion-ion reaction time with the reagent anion fluoranthene was set at 200 ms. For CID MS/MS, 35% of the normalized collision energy was used for fragmentation.
LC-ESI-CID-MS analysis of endo-β-N-acetylglucosaminidase F3 (Endo F3) treated chymotryptic glycopeptides
Chymotryptic glycopeptides of alpha-2-macroglobulin were purified as described in LC-ESI-CID/ETD-MS analysis of chymotryptic glycopeptides and reconstituted in 50 mM sodium acetate. Ten mU Endo F3 (QAbio, Palm Desert, CA) was added and incubated with chymotryptic glycopeptides at 37°C for 16 h. The resulting peptides were purified with 10 μL C18 ZipTips (Millipore, Billerica, MA). The tips were pre-wetted with 0.1% trifluoroacetic acid in 50% acetonitrile and equilibrated with 0.1% trifluoroacetic acid in water. The peptides were bound to the C18 medium followed by washes to remove non-specific binding. Ten microliters of 50% acetonitrile with 0.1% trifluoroacetic acid were used for elution. The purified two-step digested glycopeptides were analyzed with LC-LTQ-CID-MS using the same LC method and MS parameters described in 2.4. After the MS survey scan, CID MS/MS was performed on the most intense ion, and the most intense fragment ion in MS/MS was selected for further MS3 fragmentation. CID MS/MS and MS3 were also performed on the second to the fourth most intense ions from the survey MS scan.
Data analysis
The MALDI data were acquired and processed in Launchpad software (Kratos, Manchester, U.K.), and the ESI data were acquired with Thermo Xcalibur software (Arlington, VA). The m/z values and intensities were exported as ASCII files and plotted in Sigmaplot (San Jose, CA).
For chymotryptic glycopeptide analysis, the oxonium ion (m/z 366) extracted ion chromatogram (XIC) was re-constructed to locate peptide elution times. CID MS/MS spectra were manually examined and only ions which generated both oxonium ions (m/z 204, 292, 366, 528 and 657) and b, y type glycosidic bond cleavages were considered as glycopeptides. Theoretical N-glycopeptide masses were calculated by adding the masses of theoretical chymotryptic peptides at N-glycosylation sites and the masses of N-glycans obtained from MALDI MS analysis. Theoretical glycopeptide masses were matched against the masses obtained from LTQ-MS experiments. The peptide sequences were confirmed by matching the ETD MS/MS peaks with theoretical c, z type fragments listed in the Protein Prospector database (version 5.10.1) manually.
For glycopeptides resulting from Endo F3 partial deglycosylation, all CID MS/MS spectra were searched against SWISS-PROT Homosapiens database (Release 2010_10, downloaded on Nov 2, 2010) for identification of glycosylation sites. Proteome Discoverer software (version 1.1, Thermo Fisher Scientific, San Jose, CA) incorporated with SEQUEST algorithm was used to perform searches. The following search parameters were used: (1) fixed modification: cysteine carbamidomethylation (+57.0 Da); (2) variable modification: methionine oxidation (+16.0 Da), and addition of N-acetylglucosamine (+203.1 Da) or N-acetylglucosamine-fucose (+349.1 Da) to asparagine; (3) missed cleavages allowed: three; (4) peptide ion tolerance: 1.4 Da; (5) fragmentation ion tolerance: 1.5 Da. All search results containing N-glycosylation sites were validated by manual examination of CID MS/MS spectra.
Results and Discussion
In our work, we sought to develop an integrated mass spectrometry-based workflow to identify site-specific N-glycosylation of human glycoproteins. Alpha-2-macroglobulin was selected for workflow development, because of the overall analytical complexity (8 possible glycosylation sites) and lack of previous studies detailing its glycosylation profile. The developed workflow for this study is outlined in Figure 1. The workflow systematically examines the glycoprotein at the glycan, glycopeptide, and peptide levels to generate data that together provides a detailed description of the glycoprotein’s N-glycosylation profile.
Figure 1.
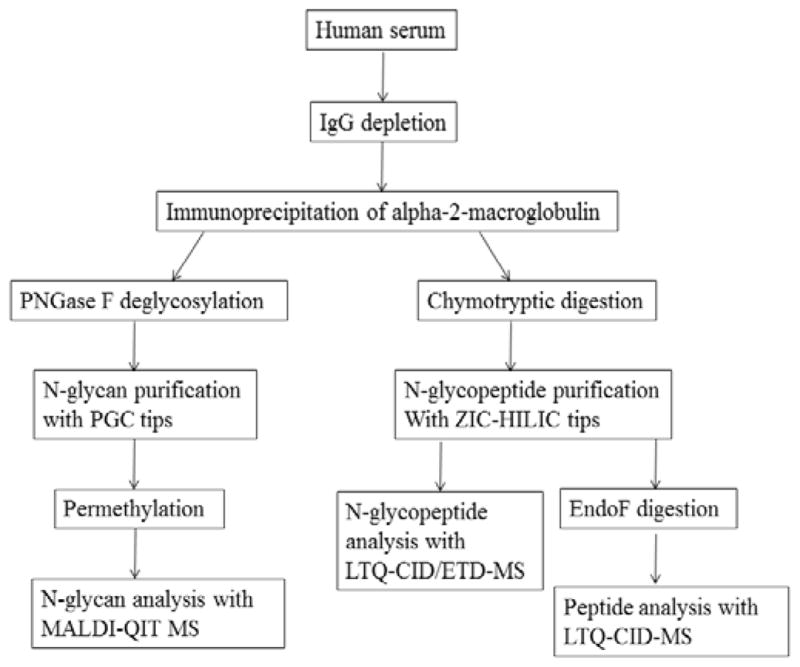
Strategy to characterize site-specific N-glycosylation of alpha-2-macroglobulin from human serum.
Purification of alpha-2-macroglobulin from human serum
In this study, alpha-2-macroglobulin was immuno precipitated from human serum. Ten-minute on-plate tryptic digestion followed by MALDI-QIT-TOF MS analysis was used to confirm the success of immunoprecipitation by peptide mass fingerprinting and MS/MS on the high intensity peaks. The mass spectrum is shown in Supporting Information Figure S1a with all the major peaks corresponding to alpha-2-macroglobulin tryptic peptides. The spectrum was searched against both the Homo sapiens and other mammalia protein database with Mascot, and returned alpha-2-macroglobulin as the only significant protein with 23 matched peptides. Among the 23 peptides, 21 are generated without miscleavages and 2 have 1 miss cleavage, reflecting the reasonable efficiency of on-plate digestion. On-plate fast trypsin digestion has high efficiency because of the high trypsin concentration, and the approximate protein-to-trypsin ratio is 1:1 rather than 50:1 which is normally used in traditional overnight digestion. The purity of alpha-2-macroglobulin was evaluated with SDS-PAGE followed by silver staining (Figure S1b). Alpha-2-macroglobulin monomer (~180 kDa) and two other cleavage fragments (~120 kDa and ~60 kDa) were observed with no other significant bands observed.
N-glycan analysis of alpha-2-macroglobulin
In-solution permethylation was performed on purified N-glycans to improve sensitivity during MS analysis and to stabilize the labile fucose and sialic acids [20]. A typical alpha-2-macroglobulin N-glycan profile by MALDI MS is shown in Figure 2a, which is dominated by the four most abundant N-glycans. These include bi-antennary complex type glycans with one or two sialic acids and with or without fucosylation. CID MS/MS was performed on the four N-glycans to confirm the oligosaccharide compositions. At low energy CID, permethylated glycans typically generate y-ions resulting from cleavage of the labile GlcNAc-Gal bond or NeuAc-Gal bond. Fucoses are usually attached to the innermost GlcNAc via α1-6 linkage or to subterminal GlcNAc via α1-3 or α1-4 linkage. Although CID does not help to determine linkage type, we utilized characteristic CID fragment ions to discriminate between terminal fucosylation and core fucosylation. The CID MS/MS spectrum of the singly sialylated biantennary fucosylated glycan at m/z 2966.44 is shown in Figure 2b. The signals at m/z 2230.03 and 2141.96 are the products after loss of the two terminal NeuAc and terminal GlcNAc-Gal-NeuAc from the parent ion, respectively. The core fucosylation was confirmed by the ion at m/z 1317.38 which corresponds to addition of GlcNAc-Fuc to the trimannosyl-GlcNAc core. The core fucosylation was further verified by glycopeptide analysis by chymotrypsin-Endo F3 two-step digestion.
Figure 2.

(a) Representative mass spectrum of N-glycan profile of alpha-2-macroglobulin of a normal serum sample. Four biantennary complex type glycans were identified. (b) MS/MS spectrum of a fucosylated biantennary glycan at m/z 2966.44.
MS/MS analysis of N-glycopeptides
Trypsin was initially used in our study, but very limited glycopeptide information was obtained, possibly because trypsin generated large glycopeptides which did not ionize well [4]. Chymotrypsin cleaves the amide bonds C-terminal to hydrophobic amino acids, such as Phenylalanine (F), Tryptophan (W) and Tyrosine (Y) and sometimes after Methionine (M) and Lysine (L). Chymotrypsin was found to produce more glycopeptides when compared to trypsin. While the lower cleavage specificity of chymotrypsin, when compared to trypsin, may limit the use of chymotrypsin in strict quantitative studies, the cleavage pattern was found to be very reproducible by precisely controlling the incubation time.
A typical LC-MS base peak chromatogram of a chymotryptic glycopeptide is shown in Figure 3a. The glycan oxonium ion at m/z 366 (GlcNAc-Gal), which is a typical glycopeptide fragment, was used to locate the elution time of the glycopeptides. The extracted ion chromatogram of m/z 366 is shown in Figure 3b, revealing that most of the glycopeptides eluted between 26 min and 42 min. Summed mass spectra within a 1 min elution window around the peak maxima were obtained for all chromatographic peaks (27.15 min, 29.58 min, 37.35 min, 38.27 min and 41.77 min) between 26 min and 42 min in Figure 3a. Most of the mass spectrometric peaks were confirmed to be glycopeptides by manually inspecting the fragment ions from CID and ETD as discussed below. An example of an integrated mass spectrum between 37.60 – 38.60 min (Figure 3a) is shown in Figure 3c. It is noted that some peaks in Figure 3c were not identified as glycopeptides because there were few informative ETD fragments generated for determining peptide sequence and/or glycosylation site.
Figure 3.

(a) Base peak chromatogram of LC-MS analysis of chymotryptic N-glycopeptides purified by ZIC-HILIC. The peaks at 27.30 min, 29.85 min, 37.47 min, 38.22 min and 41.47 min are the primary glycopeptides. (b) Extracted ion chromatogram of oxonium fragment ion at m/z 366 (GlcNAc-Gal) from CID MS/MS spectra. (c) Summed mass spectrum at 37.60–38.60 min revealed some major N-glycopeptides.
All glycopeptides were characterized by combining both CID and ETD fragment information. Since glycosidic bonds are more fragile than amide bonds and CID in the LTQ is a low-energy fragmentation method, the majority of product ions are from glycosidic bond cleavage, leaving the peptide backbone intact. A typical CID MS/MS spectrum of an N-glycopeptide (ESVRGNRSLF) is shown in Figure 4a. The lower mass range is dominated by three oxonium ions (m/z 366: GlcNAc-Gal, 528: Man-GlcNAc-Gal, and 657: GlcNAc-Gal-NeuAc). The higher mass range is dominated by glycan fragments with the intact peptide backbone. The Gal-NeuAc, GlcNAc-Man, Gal-GlcNAc, and Man-Man bonds were broken, resulting in ions at m/z 1397.7, 1357.1, 1130.7 and 1276.1, respectively. It is interesting to note that there are few glycosidic cleavages at the trimannose chitobiose core. Only one Man-Man bond cleavage (m/z 1276.1) was observed, while most of the glycopeptides fragments retain the core structure. Based on the partial glycopeptide fragment information, the glycan composition of glycopeptides was deduced.
Figure 4.

(a) CID MS/MS spectrum of the glycopeptide at m/z 1026.7 (sequence ESVRGNRSLF). Glycosidic bond cleavages were observed, resulting in b,y type ions. The low mass range was dominated by oxonium ions at m/z 366, 528 and 657. The high mass range was dominated by glycopeptides with partial glycan loss and was used to infer glycan composition. (b) ETD MS/MS spectrum of the same glycopeptide. c,z type ions were observed with the intact glycan structure. Peptide sequence and glycosylation site information were obtained from this spectra.
It is well known that ETD generally produces c and z ions resulting from cleavage of the N-Cα bond and retains the post translational modifications such as glycosylation. The ETD spectrum of the same glycopeptide (ESVRGNRSLF) is displayed in Figure 4b. ETD is believed to be less efficient and sensitive than CID [21], and the ETD signals were generally lower than their CID counterparts based on our observation. The most abundant ion is the charge reduced species of the parent ion (m/z 1540.2, charge 2+). The presence of such charge reduced ions can be useful to determine the charge states of glycopeptides when using lower resolution instrumentation, such as the LTQ. In this case, 4 out of 9 z ions and 6 out of 9 c ions predicted were observed, as annotated in Figure 4b. The glycosylation site can be determined by the mass difference between c5 + (m/z 546.3) and c62+ (m/z 1287.5). The mass difference is 2027.7, which is the addition of a glycan mass (1913.7) and the mass of asparagine (114.0). However, the useful signals are often less intense in ETD than in CID. It is reported that ETD is more efficient for charge states over 3+ and m/z lower than 1400 [21]. We also observed that for glycopeptides with a 2+ charge and m/z higher than 1600 (data not shown) that no ETD fragmentation occurred. CID/ETD MS/MS spectra of some other N-glycopeptides are provided in Supporting Information Figure S2–S6.
A summary of all the N-glycopeptides identified for alpha-2-macroglobulin is shown in Table 1. Glycopeptides with the same peptide sequences, but slightly different glycans, eluted at approximately the same time during C18 LC separation [7]. Only glycopeptides that met the following criteria are listed: (1) precursor masses must match with theoretical glycopeptide masses based on identified glycans and theoretical chymotryptic peptide masses, (2) CID spectra must contain both oxonium ions and glycan fragment ions with intact peptide backbones, and (3) ETD spectra must have a total of at least four matching c or z ions. Three sites (N70, N396 and N1424) were identified to be glycosylated with all four N-glycans as shown in Figure 2. However, it should be noted that the other five sites may be glycosylated as well, but were not identified in this ETD/CID approach. This could be because the glycopeptides are of low abundance or these unidentified sites were associated with less abundant N-glycans which were not identified in the glycan analysis. It is also possible that these unidentified glycopeptides have lower ionization efficiencies or low ETD efficiencies and did not provide informative fragments.
Table 1.
N-glycopeptides identified in LC-CID/ETD-MS/MS analysis.
| Site | Peptide sequence | Glycan | m/z | Charge | r.t. (min) |
|---|---|---|---|---|---|
| 70 | ESVRGNRSLF |
|
1026.7 | +3 | 28.77 |
| ESVRGNRSLF |
|
1075.3 | +3 | 29.03 | |
| ESVRGNRSLF |
|
843.4 | +4 | 30.02 | |
| ESVRGNRSLF |
|
1172.2 | +3 | 29.03 | |
| ESVRGNRSLFTDL |
|
852.8 | +4 | 30.51 | |
| 396 | SNATTDEHGLVQF |
|
1111.1 | +3 | 36.60 |
| SNATTDEHGLVQF |
|
1160.8 | +3 | 37.27 | |
| SNATTDEHGLVQF |
|
870.5 | +4 | 37.43 | |
| SNATTDEHGLVQF |
|
1208.5 | +3 | 38.82 | |
| SNATTDEHGLVQF |
|
907.1 | +4 | 37.94 | |
| SNATTDEHGLVQF |
|
1257.3 | +3 | 38.15 | |
| SNATTDEHGLVQF |
|
943.3 | +4 | 38.51 | |
| YSNATTDEHGL |
|
1040.7 | +3 | 38.70 | |
| SNATTDEHGL |
|
1083.8 | +3 | 29.93 | |
| YSNATTDEHGLVQF |
|
947.6 | +4 | 38.90 | |
| 1424 | IYLDKVSNQTL |
|
1069.5 | +3 | 37.09 |
| IYLDKVSNQTL |
|
1118.0 | +3 | 36.91 | |
| IYLDKVSNQTL |
|
1166.9 | +3 | 36.85 | |
| IYLDKVSNQTL |
|
911.8 | +4 | 37.02 | |
| LDKVSNQTL |
|
1123.7 | +3 | 37.58 |
MS/MS analysis of partially deglycosylated N-glycopeptides
Endoglycosidases have been used for the identification of glycosylation sites by partially deglycosylating glycopeptides and was recently used to identify 62 glycosylation sites from 37 serum glycoproteins [9]. Endo-β-N-acetylglucosaminidase hydrolyzes the bonds between the two GlcNAc in chitobiose core linked to asparagine, so that only the innermost GlcNAc or GlcNAc-Fuc is retained on the asparagine side chain. Endo F3 is applicable to complex type biantennary or tri-antennary N-glycans, but is less efficient for tetra-antennary glycans. Compared to the ETD/CID combined approach for glycopeptides analysis, the Endoglycosidases approach does not maintain glycosylation site microheterogeneity information. However, the advantage of this method is that signals from N-glycopeptides sharing the same peptide backbone are merged into two peaks (either core-fucosylated or non-core-fucosylated), so that mass spectral complexity is reduced and overall signal intensity is increased. Furthermore, we found that CID of Endoglycosidases treated peptides produced more peptide sequence ions [22] which could be used for peptide sequencing and glycosylation site determination, as a complement to the ETD/CID approach.
A traditional approach for identification of N-glycosylation sites uses PNGase F to release N-glycans from proteins and depends on the mass shift (+0.98 Da) of deamidation (asparagine to aspartic acid) during PNGase F hydrolysis for site determination [14–17, 23]. However, this method requires high mass resolution mass spectrometers, making linear ion trap instruments less appropriate. Furthermore, deamidation can occur spontaneously as a sample artifact, rather than due to enzymatic action of PNGase F, increasing the chance of false positives [24]. The confidence of glycosylation site assignment can be improved by performing deglycosylation in H2 18O, introducing a mass shift of 2.98 Da [25]. However, partial 18O incorporation in the C-terminus may bring confusion in site identification. With the Endoglycosidases approach, N-glycosylation site assignment is unambiguously confirmed by the residual GlcNAc or GlcNAc-Fuc with greater mass increments, which reduces false positive identification rate.
Based on the N-glycan analysis, most of alpha-2-macroglobulin N-glycans are biantennary complex type, thus we chose Endo F3 for partial deglycosylation. The search results of the acquired CID MS/MS and MS3 spectra revealed N-glycosylation sites as shown in Table 2. Ten partial glycopeptides associated with five N-glycosylation sites were identified with or without core fucosylation. It is interesting to note that the MS/MS spectra of glycopeptides with a GlcNAc-Fuc attached were dominated by the neutral loss of the core fucose [22] where little peptide fragmentation was achieved. Further fragmentation of the most abundant defucosylated ion (MS3) generated fragment ions similar to those from the MS/MS of their non-fucosylated counterparts. An example is shown in Figure 5 where the two glycopeptides selected include fucosylated (m/z 884.6, Figure 5a) and non-fucosylated (m/z 811.6, Figure 5b) with the same peptide backbone. The MS/MS spectrum of the fucosylated peptide at m/z 884.6 in Figure 5a does not provide much peptide backbone fragmentation and the most intense ion at m/z 811.6 results from neutral loss of the core fucose. MS3 of this neutral loss peak at m/z 811.6 in Figure 5a yields extensive peptide backbone fragments (y2–y11 and b2–b11), providing both the peptide sequence and glycosylation site information. The MS3 spectrum of 884.6 ≥ 811.6 has great similarity with the MS/MS spectrum of the non-fucosylated counterpart (m/z 811.6) which is shown in Figure 5c. Thus, by combining Endo F3 partial deglycosylation with MS/MS of non-fucosylated glycopeptides or MS3 of fucosylated glycopeptides, we can obtain the information on peptide sequence, glycosylation sites and attachment sites of core fucosylation. Combined with the ETD analysis of N-glycopeptides, six out of eight N-glycosylation sites were identified (N70, N396, N410, N869, N991 and N1424). However, site N55 and site N247 were not detected with the Endo F3 approach possibly because they were occupied by other N-glycans than the four most abundant complex type glycans identified in this study, and those less abundant glycans were not cleaved due to the substrate specificity of Endo F3. Expected masses of partial glycopeptides associated with site N70 were observed with the Endo F3 method, but Xcorr scores were not sufficient for confident assignment, even though some expected theoretical MS/MS peaks were found by manual inspection.
Table 2.
Glycosylation sites identified by CID MS/MS of Endo F3 treated chymotryptic N-glycopeptides of alpha-2-macroglobulin.
| Site | Sequence | Modification | m/z | r.t. (min) |
|---|---|---|---|---|
| 396 | SNATTDEHGLVQF | GlcNAc | 811.4 (+2) | 25.76 |
| GlcNac-Fuc | 884.6 (+2) | 25.60 | ||
| 410 | SINTTNVMGTSL | GlcNAc | 720.9 (+2) | 28.62 |
| GlcNac-Fuc | 794.0 (+2) | 28.55 | ||
| 869 | AVTPKSLGNVNF | GlcNAc | 725.4 (+2) | 26.89 |
| 991 | DYLNETQQL | GlcNAc | 664.1 (+2) | 26.79 |
| 1424 | LDKVSNQTL | GlcNAc | 611.0 (+2) | 19.12 |
| GlcNac-Fuc | 684.0 (+2) | 19.07 | ||
| 1424 | IYLDKVSNQTL | GlcNAc | 749.0 (+2) | 25.65 |
| GlcNac-Fuc | 822.0 (+2) | 25.70 |
Figure 5.

(a) CID MS/MS of the fucosylated glycopeptide (SN(+GlcNAc-Fuc)ATTDEHGLVQF) at m/z 884.6. The major fragment at m/z 811.6 is the product ion after neutral loss of fucose from the precursor ion. Minimal peptide backbone fragmentation was observed. (b) CID MS3 of m/z 811.6 from (a) showed extensive fragmentation along the peptide backbone, providing both peptide sequence information and the glycosylation site. (c) CID MS/MS of the non-fucosylated glycopeptide (SN(+GlcNAc)ATTDEHGLVQF) at m/z 811.6. (c) has great similarity with (b), revealing presence of both fucosylation and non-fucosylation at the same site.
Conclusion
In this work, an integrated LC-MS/MS strategy was developed for comprehensive identification of both site-specific glycosylation and core fucosylation of glycoproteins. Using this workflow, a volume of only 10 μL of human serum sufficed for two LC-MS/MS analyses on glycopeptides treated with and without Endo F3 treatment. There are three major aspects in this assay including N-glycan analysis, CID/ETD MS/MS analysis of intact glycopeptides, and CID MS/MS analysis of Endo F3 treated glycopeptides.
Glycopeptide CID/ETD MS/MS analysis identified three N-glycosylation sites, N70, N396 and N1424, with four glycoforms found for each site. Endo F3 cleaves the majority of glycan moieties with only the core GlcNAc or GlcNAc-Fuc attached to the peptide backbones, thus reducing the mass spectra complexity and provides the unique ability to identify site-specific core fucosylation. The advantages of Endo F3 assisted mass spectrometric analysis were successfully used to reveal five glycosylation sites at N396, N410, N869, N991 and N1424. With this combined approach, we identified a total of six N-glycosylation sites with site-specific glycosylation or core fucosylation patterns revealed.
Acknowledgments
We acknowledge support of this work from the National Cancer Institute under grant 1 R01 CA154455 01 (DML) and the SPORE program grant 1 P50CA130810 (DML, DMS, MTR) and from the National Institutes of Health under grant R01 GM49500 (DML).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Dube DH, Bertozzi CR. Glycans in cancer and inflammation. Potential for therapeutics and diagnostics. Nat Rev Drug Dicov. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 2.Zhao J, Simeone DM, Heidt D, Anderson MA, Lubman DM. Comparative serum glycoproteomics using lectin selected sialic acid glycoproteins with mass spectrometric analysis: Application to pancreatic cancer serum. J Proteome Res. 2006;5:1792–1802. doi: 10.1021/pr060034r. [DOI] [PubMed] [Google Scholar]
- 3.Nakano M, Nakagawa T, Ito T, Kitada T, Hijioka T, et al. Site-specific analysis of N-glycans on haptoglobin in sera of patients with pancreatic cancer: A novel approach for the development of tumor markers. Int J Cancer. 2008;122:2301–2309. doi: 10.1002/ijc.23364. [DOI] [PubMed] [Google Scholar]
- 4.An HJ, Froehlich JW, Lebrilla CB. Determination of glycosylation sites and site-specific heterogeneity in glycoproteins. Curr Opin Chem Biol. 2009;13:421–426. doi: 10.1016/j.cbpa.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hogan JM, Pitteri SJ, Chrisman PA, McLuckey SA. Complementary structural information from a tryptic N-linked glycopeptide via electron transfer ion/ion reactions and collision-induced dissociation. J Proteome Res. 2005;4:628–632. doi: 10.1021/pr049770q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mikesh LM, Ueberheide B, Chi A, Coon JJ, Syka JE, et al. The utility of ETD mass spectrometry in proteomic analysis. Biochim Biophys Acta. 2006;1764:1811–1822. doi: 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang D, Hincapie M, Rejtar T, Karger BL. Ultrasensitive characterization of site-specific glycosylation of affinity-purified haptoglobin from lung cancer patient plasma using 10 mu m i.d. porous layer open tubular liquid chromatography-linear ion trap collision-induced dissociation/electron transfer dissociation mass spectrometry. Anal Chem. 2011;83:2029–2037. doi: 10.1021/ac102825g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Segu ZM, Hussein A, Novotny MV, Mechref Y. Assigning N-Glycosylation sites of glycoproteins using LC/MSMS in conjunction with Endo-M/exoglycosidase mixture. J Proteome Res. 2010;9:3598–3607. doi: 10.1021/pr100129n. [DOI] [PubMed] [Google Scholar]
- 9.Hagglund P, Bunkenborg J, Elortza F, Jensen ON, Roepstorff P. A new strategy for identification of N-glycosylated proteins and unambiguous assignment of their glycosylation sites using HILIC enrichment and partial deglycosylation. J Proteome Res. 2004;3:556–566. doi: 10.1021/pr034112b. [DOI] [PubMed] [Google Scholar]
- 10.Zhang W, Wang H, Zhang L, Yao J, Yang P. Large-scale assignment of N-glycosylation sites using complementary enzymatic deglycosylation. Talanta. 2011;85:499–505. doi: 10.1016/j.talanta.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, Zhang W, Zhao J, Zhang L, Liu M, et al. N-glycosylation pattern of recombinant human CD82 (KAI1), a tumor-associated membrane protein. J Proteomics. 2012;75:1375–1385. doi: 10.1016/j.jprot.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 12.Miyoshi E, Moriwaki K, Nakagawa T. Biological function of fucosylation in cancer biology. J Biochem. 2008;143:725–729. doi: 10.1093/jb/mvn011. [DOI] [PubMed] [Google Scholar]
- 13.Barrett AJ, Starkey PM. Interaction of alpha-2-macroglobulin with proteinases-characteristics and specificity of reaction, and a hypothesis concerning its molecular mechanism. Biochem J. 1973;133:709–724. doi: 10.1042/bj1330709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu T, Qian WJ, Gritsenko MA, Camp DG, 2nd, Monroe ME, et al. Human plasma N-glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry. J Proteome Res. 2005;4:2070–2080. doi: 10.1021/pr0502065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bunkenborg J, Pilch BJ, Podtelejnikov AV, Wisniewski JR. Screening for N-glycosylated proteins by liquid chromatography mass spectrometry. Proteomics. 2004;4:454–465. doi: 10.1002/pmic.200300556. [DOI] [PubMed] [Google Scholar]
- 16.Chen R, Jiang XN, Sun D, Han G, Wang F, et al. Glycoproteomics analysis of human liver tissue by combination of multiple enzyme digestion and hydrazide chemistry. J Proteome Res. 2009;8:651–661. doi: 10.1021/pr8008012. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 18.Zhao J, Qiu W, Simeone DM, Lubman DM. N-linked glycosylation profiling of pancreatic cancer serum using capillary liquid phase separation coupled with mass spectrometric analysis. J Proteome Res. 2007;6:1126–1138. doi: 10.1021/pr0604458. [DOI] [PubMed] [Google Scholar]
- 19.Lin Z, Simeone DM, Anderson MA, Brand RE, Xie X, et al. Mass spectrometric assay for analysis of haptoglobin fucosylation in pancreatic cancer. J Proteome Res. 2011;10:2602–2611. doi: 10.1021/pr200102h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wada Y, Azadi P, Costello CE, Dell A, Dwek RA, et al. Comparison of the methods for profiling glycoprotein glycans-HUPO Human Disease Glycomics/Proteome Initiative multi-institutional study. Glycobiology. 2007;17:411–422. doi: 10.1093/glycob/cwl086. [DOI] [PubMed] [Google Scholar]
- 21.Alley WR, Jr, Mechref Y, Novotny MV. Characterization of glycopeptides by combining collision-induced dissociation and electron-transfer dissociation mass spectrometry data. Rapid Commun Mass Spectrom. 2009;23:161–170. doi: 10.1002/rcm.3850. [DOI] [PubMed] [Google Scholar]
- 22.Zhao Y, Jia W, Wang JF, Ying W, Zhang Y, et al. Fragmentation and site-specific quantification of core fucosylated glycoprotein by multiple reaction monitoring-mass spectrometry. Anal Chem. 2011;83:8802–8809. doi: 10.1021/ac201676a. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Wu SL, Hancock WS. Approaches to the study of N-linked glycoproteins in human plasma using lectin affinity chromatography and nano-HPLC coupled to electrospray linear ion trap-Fourier transform mass spectrometry. Glycobiology. 2006;16:514–523. doi: 10.1093/glycob/cwj091. [DOI] [PubMed] [Google Scholar]
- 24.Palmisano G, Melo-Braga MN, Engholm-Keller K, Parker BL, Larsen MR. Chemical deamidation: a common pitfall in large-scale N-linked glycoproteomic mass spectrometry-based analyses. J Proteome Res. 2012;11:1949–1957. doi: 10.1021/pr2011268. [DOI] [PubMed] [Google Scholar]
- 25.Kaji H, Saito H, Yamauchi Y, Shinkawa T, Taoka M, et al. Lectin affinity capture, isotope-coded tagging and mass spectrometry to identify N-linked glycoproteins. Nat Biotechnol. 2003;21:667–672. doi: 10.1038/nbt829. [DOI] [PubMed] [Google Scholar]