

Abstract
Stereoselectivities of the dihydroxylations of cis-bicyclo[3.3.0]octene intermediates for a projected total synthesis of chromodorolide A have been explored experimentally. The reaction occurs unexpectedly on the apparently more hindered (concave) face; this result has been explained through computational studies using B3LYP and B3LYP-D3 methods. Torsional effects are largely responsible for the stereoselectivity encountered in the chromodorolide A synthesis. Many literature examples have been reported on related cases. QM calculations show that the stereoselectivities of dihydroxylations of fused cyclopentenes are influenced by the conformational rigidity or flexibility of the substrate. Torsional, electrostatic, and steric effects can all influence stereoselectivity, and the rigidity or flexibility of conformations of reactants provides a predictive guide to stereoselectivity.
Introduction
Chromodorolide A (1) is a structurally unique diterpene.1 We envisaged accomplishing its total synthesis by late-stage intramolecular lactonization of a tetracyclic precursor such as 2 (Figure 1). In our early exploration in a model series of strategies for assembling such cyclization precursors, we examined dihydroxylation of intermediate 3. To our surprise, the reaction of 3 with OsO4 occurred with high selectivity from the concave face, which appears to be more sterically hindered (Figure 1). As osmium tetroxide-mediated dihydroxylation of double bonds is one of the most useful reactions in organic synthesis,2 we felt it worthwhile to explore computationally the origins of the diastereoselectivities of these and related literature reactions. We report the preparation and dihydroxylation of bicyclic alkene 3 and a computational study of the origin of stereoselectivity of the dihydroxylation reaction. We have also reviewed the known cases of osmylations of cis-bicyclo[3.3.0]octenes and hetero-substituted cases; we provide a computational study of these systems and explanation of the origins of selectivities. We also show that the conformations of the reactants provide a good guide to the stereoselectivity of reactions.
Figure 1.
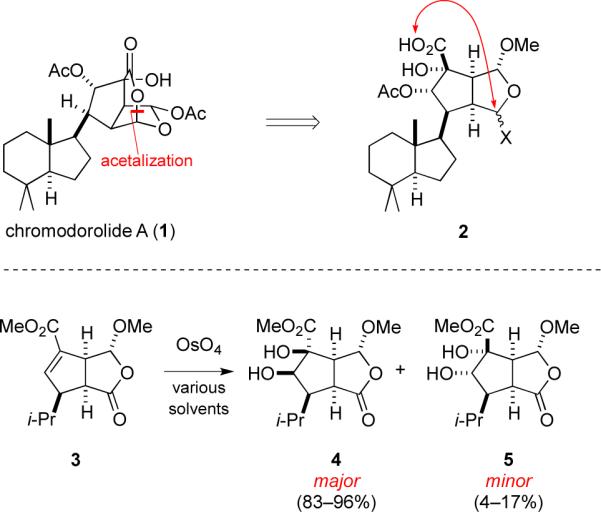
Structure of chromodorolide A (1) and experimental results of dihydroxylation of cis-oxabicyclo[3.3.0]octenone 3.
Results and Discussion
Experimental Studies
Cis-oxabicyclo[3.3.0]octenone 3 was prepared by a two-step sequence; the key step was phosphine-promoted (3+2) annulation3 involving allenoate 64 and butenolide 75 to furnish 8 (Scheme 1). Reductive debromination of 8 with zinc and acetic acid delivered oxabicyclooctanone 3 in 20% overall yield. Dihydroxylation of 3 at room temperature with catalytic OsO4 and NMO in 1:1 tert-butanol/H2O gave a 92:8 mixture of diastereomeric cis-diol products 4 and 5 in 87% yield.6 Recrystallization of this mixture from CH2Cl2 provided the major product, diol 4 (mp 146–147 °C), whose structure was secured by 1H NMR nOe studies and single-crystal X-ray analysis. Note the oxidation on the concave side of 3, syn to the lactone, but anti to the two hydrogens at the ring junction.
Scheme 1.
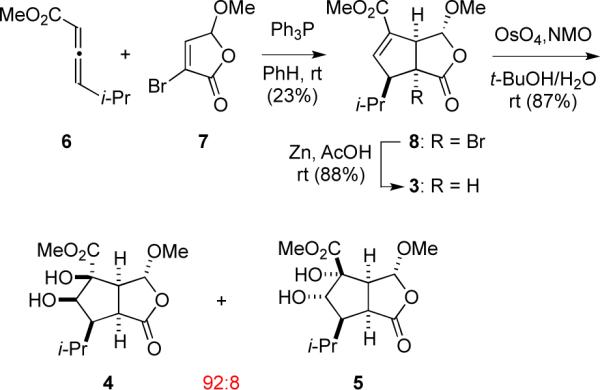
Synthesis of cis-oxabicyclo[3.3.0]octenone 3 and its dihydroxylation with OsO4 in aqueous tert-butanol.
Computational Studies
All calculations were performed with Gaussian09.7 The diastereomeric dihydroxylation transition states of precursor 3 were investigated at the B3LYP8 level using the LANL2DZ9 basis set for osmium and 6–31G(d) for all other atoms. Energies were calculated with both B3LYP and B3LYP-D3; the conclusions are the same with both methods, and the B3LYP-D3 results are discussed in the text. Conformational searches were performed on compounds 3, 9, 11 and 13. For each compound, the two envelope conformations possible for the cyclopentene ring were created with the flap of the envelope either up or down. Each was then minimized. For 3 and 9, the minimizations lead to only one envelope conformation for each compound. For 11 and 13, the minimizations gave two different envelope conformations for each compound.
Only one envelope conformation of the cyclopentene ring of 3 was obtained after transition state optimization, as shown in Figure 2. The isopropyl group can adopt a variety of conformations, but only the two lowest energy transition state conformers are shown in Figure 2. The transition state structures with other isopropyl conformations are higher in energy; they are shown in the Supporting Information. The α-dihydroxylation transition structure TS3α, with the OsO4 approaching from the sterically less hindered face of the cyclopentene ring, is 2.2 kcal/mol higher in energy than the β-dihydroxylation transition structure TS3β at the B3LYP level. With B3LYP-D3, which includes dispersion corrections that may be important for such systems, the energy difference between TS3α and TS3β increases to 4.2 kcal/mol. Although the energy difference is overestimated by both methods as compared to experiment, the results are consistent with the experimental observation that the major product is the β-dihydroxylated compound. A variety of other functionals have been developed, and could be considered for these studies. However, those methods are often computationally much more demanding, and the steric and electronic factors that differ in diastereomeric transition states are known to be adequately differentiated at this level.10
Figure 2.

Optimized β and α-dihydroxylation transition structures of cis-oxabicyclo[3.3.0]octanone 3. The Newman projections of interest shown in the red and green box insets are viewed along the red and green bonds, respectively. Values below each structure are relative energies in kcal/mol calculated by B3LYP. Values enclosed in parentheses are relative energies in kcal/mol calculated by B3LYP-D3.
The energy difference between the transition structures TS3α and TS3β is a result of torsional strain differences. There are two forming C–O bonds in the transition state, one bond is shorter (2.00 Å) and more fully formed than the other (2.17 Å). This asynchronicity is the result of electron-withdrawal and conjugation by the ester group. Inspection of the Newman projections about the bond to the left alkene carbon (shown in red in Figure 2) of two transition structures indicates TS3β is slightly more staggered than TS3α (red box insets); torsional differences at the bond to the right alkene carbon, where bond formation is more advanced, are more striking. As seen from the Newman projections for the bond to the right alkene carbon (shown in green), the α-dihydroxylation transition structure TS3α exhibits substantial eclipsing, in contrast to the substantially more staggered β-dihydroxylation transition structure TS3β. For this reaction, the torsional effects override the steric effects, which makes the more crowded TS3β more stable than the less crowded TS3α. Torsional effects direct OsO4 to attack the cyclopentene ring from the sterically more hindered β-face. Such effects have been reported previously to govern the stereoselectivities in a wide variety of situations.11 The example of epoxidation of an intermediate for a guanacastepene A synthesis11e is especially relevant, and the effect was called “torsional steering”. In this and the other examples in reference 11, attack on an envelope conformation of a cyclopentene occurs on the concave face because of torsional effects.
The origins of the dihydroxylation stereoselectivities can be identified qualitatively from the geometry of the starting substrate. The energy-minimized geometry of compound 3 is shown in Figure 3a.12 Only one envelope conformation was obtained for the cyclopentene ring after minimization, while the isopropyl group can adopt a variety of different conformations. The other two minimized conformers with isopropyl groups in other orientations are 1.1 and 2.5 kcal/mol higher in energy, respectively (Supporting Information). By careful inspection of the substrate structure, in the left C21–C3 bond Newman projections (Figure 3a, red box inset), the dihedral angle between the C6 carbonyl carbon and C7 is 58° and 67° for C6 to the allylic hydrogen H1. This analysis indicates that addition from the β face is more favored torsionally than from the α face, which is consistent with the transition state analysis. The more striking torsional difference on the right alkene carbon shown in the TSs is also reflected in the substrate structure itself. In the right C1–C5 bond Newman projections (Figure 3a, green box inset), the dihedral angle between the vinyl hydrogen H2 and the central carbon of isopropyl C8 is 38° and 80° to the allylic hydrogen H3. The 42° dihedral angle difference makes the β-face attack more favorable due to the torsional effects, which is consistent with the result obtained by TS exploration (Figure 3b). The analysis of the geometries of the substrates again confirms that the stereoselectivities of dihydroxylation result from torsional effects. In this case, the reactant cyclopentene ring is fixed in an envelope conformation, and the analysis of the reactant geometries gives a reliable guide to the direction of attack by osmium tetroxide and, presumably, other electrophiles, radicals and nucleophiles as well. The situation is more complex when two envelope conformers of the cyclopentene are similar in energy. Such examples were found in the literature, as described in the next section.
Figure 3.

(a) Optimized structure of cis-oxabicyclo[3.3.0]octenone 3. (b) The torsional of interest viewed along the green bond. Newman projections shown in the blue and yellow box insets illustrated the resultant torsional effects for the β and α-dihydroxylation transition states.
Literature Examples of Dihydroxylation of Other cis-Bicyclo[3.3.0]octenes
A survey of the chemical literature identified a variety of cis-bicyclo[3.3.0]octene systems that have been subjected to dihydroxylation (see the Supporting Information for full details). Out of a total of 26 relevant transformations, there is rigorous experimental evidence for the structural assignment of the diol product(s) in only seven cases. This striking fact is a result of the difficulty of obtaining solid evidence for the configurational assignments of cis-bicyclo[3.3.0]octane ring systems, as 1H coupling constant analysis and nOe data are often ambiguous.
As shown in the SI, for the 26 cis-bicyclo[3.3.0]octene systems studied, the rings fused to the cyclopentene ring are mainly lactones and cyclic ketones or derivatives with different substituents. After dihydroxylation, most of those dihydroxylated intermediates were incorporated in a total synthesis of natural products or other important target. Despite the general importance of such transformations for cis-bicyclo[3.3.0]octene systems, none of the 26 examples published included any discussion of the factors controlling the stereoselectivities. In order to better understand these transformations and to provide guidelines for synthetic chemists to predict the preferred products of reactions of this type, we explored in detail the three simple examples shown in Figure 4. These are representative of the types of molecules included in the 26 examples in the SI. These do not include substituents, and include one cyclic ketone, 11, and two lactones, 9 and 13.13–15 In example (i), dihydroxylation of cis-oxabicyclo[3.3.0]octenone 9 took place preferentially from the concave face to give the β-diol product 10. In the first report of this transformation,13a the major product was assigned—without experimental evidence—as arising from convex-face dihydroxylation, but this structural assignment was later reversed by means of an X-ray crystal structure.13b In the second example (ii), cis-bicyclo[3.3.0]octenone 11 was dihydroxylated also preferentially from the concave face to give diol 12 with relatively low 4:1 selectivity.14 It was hypothesized in this report that the selectivity may be due to a directing effect of the carbonyl group, as conversion of the ketone into a protected β-alcohol inverts the facial selectivity.
Figure 4.

Selected literature examples of dihydroxylation of cis-bicyclo[3.3.0]octenes using osmium reagents.
In example (iii), high diastereoselectivity for dihydroxylation from the convex face of 13 to give diol 14 is reported; however, no direct experimental evidence was given to support this structural assignment.15 Our computational studies, described below, suggest that the reported assignment is correct.
We first performed a conformational search on 9, 11 and 13. Compounds 11 and 13 have two different energy minima, while 9 has only one low-energy conformation. The cyclopentene ring adopts an envelope conformation in which four of the carbon atoms C1, C2, C3 and C5 are coplanar, while C4 can be above or below the plane. For compounds 11 and 13, two different minimum energy conformations were located having similar energies (Figure 5). However, for compound 9, there is only one minimum energy conformation for the cyclopentene ring, with C4 above the plane (R9, Figure 5). This is the same situation as with compound 3 described earlier.
Figure 5.


Three fused cyclopentene compounds and their optimized geometries. In R9, R11 and R13, the flap of the envelope is up; in R11' and R13', it is down.
This ground state difference is also reflected in the TSs of the osmylation reactions. Compound 9 has only one conformation, and both α and β-dihydroxylation transition structures are based on this conformation. TS9β is calculated to be 1.6 kcal/mol more stable than TS9α using B3LYP-D3 calculations, consistent with the stereoselectivities observed experimentally. From the structures shown in Figure 5, the different stabilities between two transition structures are also most likely due to the torsional strain, as in the TS analysis of dihydroxylation of 3.
For compounds 11 and 13, the situations are more complicated, because there are two possible conformations of the substrates. After TS searches, the lowest energy β-dihydroxylation transition structures TS11β and TS13β were located, and they are related to conformations R11 and R13. Transition structures TS11α and TS13α are structurally related to conformations R11' and R13'. The different conformation preferences of different TSs can be rationalized from the substrate conformation itself. From the substrate geometries shown in Figure 5, the β face is more torsionally favored in R11 and R13, whereas the α face is more torsionally favored in R11' and R13'. The β and α-dihydroxylation transition structures avoid torsional strain and involve the conformations that result in less torsional strain. In both 11 and 13, both α and β attack can occur from a concave face of an envelope cyclopentene, due to the flexibility of the cyclopentene.
For compound 11, TS11α has similar torsional strain as TS11β, but TS11β is still calculated to be 0.8 kcal/mol more stable than TS11α using B3LYP-D3; this result agrees with the 4:1 preference for β attack observed experimentally. The different stabilities of the two TSs are presumably a result of the electrostatic interactions between the carbonyl carbon and OsO4 oxygen inside the concave-face attack TS11β, which makes it a little more stable than the convex-face attack TS11α. This attractive interaction is related to the O…C=O interaction identified by Raines in various protein structures.16
For compound 13, as with 11, the torsional differences are not large between TS13β and TS13α. However, now, TS13α is calculated to be more stable than TS13β by 1.6 kcal/mol by B3LYP-D3, which predicts the correct stereoselectivity in the experiment. The stability difference between these two TSs is likely due to differences in O-O repulsions, which makes the more crowded and electrostatically disfavored TS13β less stable than the less crowded TS13α.
In order to determine the reason for the different conformational properties of different reactants, we performed a conformational analysis. For 3 and 9, we were able to locate only one envelope conformation of the cyclopentene with C4 above the plane. The other envelope conformation of the cyclopentene ring for 3 and 9 was manually created by freezing C4 below the plane.17 The rest skeleton of the molecules was then optimized and the optimized structures R3a', R9' are calculated less stable than R3a and R9 by 2.0, 1.8 kcal/mol, respectively. If no restrictions are given to the systems, R3a', R9' will go back to R3a, R9 after re-optimization. This result indicates that the availability of only one envelope conformation for 3a and 9 is a result of the remarkable instabilities of the other envelope conformations. From the structures of R3a and R3a', all the bond distances of the fused ring in the two conformations are similar; indicating there is little ring strain differences in between. The different stabilities between R3a and R3a' are due to torsional effects. Inspection of the Newman projections of C7-C3 (green bond in Figure 7a) show that R3a is slightly more staggered than R3a'. This is also true for R9 and R9' as shown in the Newman projections (green box insets) in Figure 7b.
Figure 7.

Optimized structures of (a) R3a and R3a' (b) R9 and R9'.
However, for 11 and 13, two envelope conformations with similar energies were located for the substrates. Now, the atom in position 7 is no longer a sp3 carbon, and the torsional differences for bond C7 (or O)-C3 no longer exist according to the Newman projections shown in Figure 8. As a result, two envelope conformations of the reactants are both available and similar in energy.
Figure 8.

Optimized structures of (a) R11 and R11' (b) R13 and R13'.
Conclusions
In dihydroxylations of fused cyclopentene systems, and presumably other additions, when only one envelope conformational minimum is accessible in the reactant, torsional effects always steer attack to the concave face. In cases studied here, the torsional factors override other factors and determine stereoselectivity. However, when the cyclopentene ring is able to adopt two different low-energy conformations, both top and bottom attack can occur on a concave face of the cyclopentene envelope. As a result, torsional factors no longer dominate the stereoselectivity. The stereoselectivity in such cases is then determined by other factors such as steric and electrostatic interactions or both, as demonstrated in the dihydroxylations of compounds 11 and 13.
Supplementary Material
Figure 6.

Optimized β and α-dihydroxylation transition structures of cis-bicyclo[3.3.0]octenes 9, 11 and 13.
Acknowledgement
Work at UCLA and UCI was supported by the National Science Foundation (CHE 0614591) and National Institute of General Medical Sciences, National Institutes of Health (GM36700) and (GM098601). We are grateful for SNSF [Swiss National Science Foundation] postdoctoral fellowship to P.K. Computations were performed on the National Science Foundation Terascale Computing System at the National Center for Supercomputing Applications (NCSA), and on the IDRE Hoffman2 cluster at UCLA.
Footnotes
Supporting Information Detailed experimental procedures and product characterization, optimized geometries and energies of all computed species, summary of literature examples of dihydroxylations of cis-bicyclo[3.3.0]octene systems, CIF files for X-ray structures of 4 and the benzyl ester analog of 3, and full authorship of ref 7. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Dumdei EJ, de Silva ED, Andersen RJ, Choudhary MI, Clardy J. J. Am. Chem. Soc. 1989;111:2712–2713. [Google Scholar]; (b) Morris SA, de Silva ED, Andersen RJ. Can. J. Chem. 1991;69:768–771. [Google Scholar]
- 2.Kolb HC, VanNieuwenzhe MS, Sharpless KB. Chem. Rev. 1994;94:2483–2547. [Google Scholar]
- 3.(a) Ruano JL, Núñez A, Jr., Martín, Fraile A. J. Org. Chem. 2008;73:9366–9371. doi: 10.1021/jo801896a. [DOI] [PubMed] [Google Scholar]; (b) Lu X, Zhang C, Xu Z. Acc. Chem. Res. 2001;34:535–544. doi: 10.1021/ar000253x. [DOI] [PubMed] [Google Scholar]
- 4.(a) De March P, Font J, Gracia A, Qingying Z. J. Org. Chem. 1995;60:1814–1822. [Google Scholar]; (b) Rout L, Harned AM. Chem. Eur. J. 2009;15:12926–12928. doi: 10.1002/chem.200902208. [DOI] [PubMed] [Google Scholar]
- 5.Fariña F, Martín MR, Martín MV. An. Quim. 1979;75:144–149. [Google Scholar]
- 6.VanRheenen V, Kelly RC, Cha RC. Tetrahedron Lett. 1976;17:1973–1976. [Google Scholar]
- 7.Frisch MJ, et al. Gaussian 09. revision B.01 Gaussian, Inc.; Wallingford, CT: 2010. [Google Scholar]
- 8.(a) Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys. Rev. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 9.Hay PJ, Wadt WR. J. Chem. Phys. 1985;82:299–310. [Google Scholar]
- 10.(a) Grimme S. J. Comput. Chem. 2006;27:1787–1799. doi: 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]; (b) Grimme S, Antony J, Ehrlich S, Krieg H. J. Chem. Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]; (c) McMullin CL, Jover J, Harvey JN, Fey N. Dalton Trans. 2010;39:10833–10836. doi: 10.1039/c0dt00778a. [DOI] [PubMed] [Google Scholar]; (d) Antoline JE, Krenske EH, Lohse AG, Houk KN, Hsung RP. J. Am. Chem. Soc. 2011;133:14443–14451. doi: 10.1021/ja205700p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (a) For dispersion correction, see:; For application of dispersion corrected DFT in computational chemistry, see:
- 11.(a) Caramella P, Rondan NG, Paddon MN, Houk KN. J. Am. Chem. Soc. 1981;103:2438–2440. [Google Scholar]; (b) Rondan NG, Paddon MN, Caramella P, Mareda J, Mueller PH, Houk KN. J. Am. Chem. Soc. 1982;104:4974–4976. [Google Scholar]; (c) Paddon MN, Rondan NG, Houk KN. J. Am. Chem. Soc. 1982;104:7162–7166. [Google Scholar]; (d) Lucero MJ, Houk KN. J. Org. Chem. 1998;63:6973–6977. doi: 10.1021/jo980760g. [DOI] [PubMed] [Google Scholar]; (e) Cheong PH, Yun H, Danishefsky SJ, Houk KN. Org. Lett. 2006;8:1513–1516. doi: 10.1021/ol052862g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Martinelli MJ, Peterson BC, Khau VV, Hutchison DR, Leanna MR, Audia JE, Droste JJ, Wu Y-D, Houk KN. J. Org. Chem. 1994;59:2204–2210. [Google Scholar]; (g) Houk KN, Paddon MN, Rondan NG, Wu Y-D, Brown FK, Spellmeyer DC, Metz JT, Li Y, Loncharich RJ. Science. 1986;231:1108–1117. doi: 10.1126/science.3945819. [DOI] [PubMed] [Google Scholar]
- 12.The single-crystal X-ray model of the benzyl analogue of 3 shows no significant differences from the computational model, with torsional angles about the C1–C5 bond being nearly identical (see Supporting Information).
- 13.(a) Broom N, O'Hanlon PG, Simpson TJ, Stephen R, Willis CL. J. Chem. Soc., Perkin Trans. 1. 1995:3067–3072. [Google Scholar]; (b) Fretmanlis J, Gerca L, Turovskis I, Liepins E, Lola D, Mishnev A, Bundule M, Bleidelis J. J. Prakt. Chem. 1987;329:39–48. [Google Scholar]
- 14.Leonard J, Hussain N. J. Chem. Soc., Perkin Trans. 1. 1994:49–60. [Google Scholar]
- 15.(a) Neufellner E, Kapeller H, Griengl H. Tetrahedron. 1998;54:11043–11062. [Google Scholar]; (b) Ernst M, Helmchen G. Angew. Chem., Int. Ed. 2002;41:4054–4056. doi: 10.1002/1521-3773(20021104)41:21<4054::AID-ANIE4054>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]; (c) Marschner C, Baumgartner J, Griengl H. J. Org. Chem. 1995;60:5224–5235. [Google Scholar]
- 16.(a) Choudhary A, Gandla D, Krow GR, Raines RT. J. Am. Chem. Soc. 2009;131:7244–7246. doi: 10.1021/ja901188y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jakobsche CE, Choudhary A, Miller SJ, Raines RT. J. Am. Chem. Soc. 2010;132:6651–6653. doi: 10.1021/ja100931y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.In order to simplify the calculation, all the substituents on 3 were removed, and the truncated substrate is referred to as 3a.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.