

Abstract
Ankyrin repeat domain 1 protein (Ankrd1), also known as cardiac ankyrin repeat protein (CARP), increases dramatically after tissue injury, and its overexpression improves aspects of wound healing. Reports that Ankrd1/CARP protein stability may affect cardiovascular organization, together with our findings that the protein is crucial to stability of the cardiomyocyte sarcomere and increased in wound healing, led us to compare the contribution of Ankrd1/CARP stability to its abundance. We found that the 26S proteasome is the dominant regulator of Ankrd1/CARP degradation, and that Ankrd1/CARP half-life is significantly longer in cardiomyocytes (hrs) than endothelial cells (min). In addition, higher endothelial cell density decreased the abundance of the protein without affecting steady state mRNA levels. Taken together, our data and that of others indicate that Ankrd1/CARP is highly regulated at multiple levels of its expression. The striking difference in protein half-life between a muscle and a non-muscle cell type suggests that post-translational proteolysis is correlated with the predominantly structural versus regulatory role of the protein in the two cell types.
Keywords: cardiomyocytes, endothelial cells, Ankrd1/CARP, 26S proteasome
Introduction
Ankyrin repeat domain 1 (Ankrd1;cardiac ankyrin repeat protein, CARP) is a member of the muscle ankyrin repeat protein (MARP) family [1]. Chu et al. [2] first identified the protein as a cytokine inducible factor (C-193) in human vascular endothelial cells in1995; however, Ankrd1/CARP expression and what little is known about its function, have been best characterized in cardiac and skeletal muscle, where the protein serves roles as a transcriptional co-regulatory protein in the nucleus and a structural/signaling component in the sarcomere [1,3]. Ankrd1/CARP expression is stimulated in many forms of tissue injury, and its overexpression improves aspects of wound healing [4].
In skeletal muscle and cardiomyocytes, nuclear Ankrd1/CARP is distributed between the nucleus, where it interacts with transcription factors such as YB-1 [5], while in the cytoplasm it associates with titin and other sarcoplasmic proteins as part of a putative mechanotransduction system [1]. Ankrd1 expression is prominent during cardiomyogenesis; however, adult expression patterns are largely related to stress. For example, there is a chamber-dependent pattern of cardiac expression that appears to be related to environmental stresses [3]. Pathological stressors such as myocardial infarction and pressure overload often result in increased expression of Ankrd1/CARP. Numerous cardiomyopathies are associated with altered ankrd1 expression, suggesting an adaptive rather than causal role. However, both cardiomyopathies and vascular malformations have also been linked to ankrd1 mutations/polymorphisms[3]. In addition, depletion of Ankrd1/CARP in cardiomyocytes leads to sarcomere disruption [6].
Although its mRNA is present at low levels, Ankrd1/CARP protein is generally undetectable in uninjured, non-muscle tissues. Chemical or mechanical stress induces both Ankrd1/CARP mRNA and protein in the skin, vasculature, and other tissues. Shi et al., reported rapid induction of both the mRNA and protein, which increased to maximum levels by 24h in mouse dermal excisional wounds [4].
Several observations suggest that tight control of Ankrd1/CARP levels may b e important for normal development and function of many tissues. Cinquetti et al. identified two different mutations in ankrd1 in patients with the congenital heart defect, total anomalous pulmonary venous return (TAPVR) [7]: a chromosomal translocation that resulted in >5-fold higher mRNA expression in probands than in normal family members, and a mutation that altered a PEST sequence, resulting in increased stability of the protein. Increased Ankrd1/CARP expression in renal podocytes associates with severity of proteinuria in patients with lupus nephritis [8] and cisplatin resistance in ovarian cancer chemotherapy [9]. Gene expression profiling following crush injury of peripheral and central dorsal root ganglion neurons suggested that Ankrd1/CARP expression was necessary for nerve regeneration [10].
In mice, deletion of ankrd1 or of the other two MARP genes, ankrd2 and ankrd23, either singly or in combination does not appear to have a deleterious effect on development or viability [11]. In the adult, however, Ankrd1/CARP is predominantly induced under stress conditions, and its role as an adaptive factor has not been tested.
Given the foregoing evidence that Ankrd1/CARP protein stability may affect cardiovascular organization, our findings that the protein is crucial to stability of the cardiomyocyte sarcomere, and Ankrd1/CARP upregulation in many cell types including vascular cells following dermal wounding, we compared the contribution of Ankrd1/CARP stability to its accumulation in cardiomyocytes and vascular endothelial cells. While the 26S proteasomal system was dominant in these two cell systems, Ankrd1/CARP half-life differed markedly in muscle and non-muscle cells.
Materials and Methods
Reagents
Epoxomicin was from Peptides International. Clasto-lastacystin ß-lactone, epigallocatechin-3-gallate, NIP-(leu)3-vinyl sulfone, MG-132 and the rabbit anti-cyclophilin antibody were from Enzo Life Sciences (Farmingdale, NY). Medium 131 and microvascular growth supplement (MVGS) were from Invitrogen (Grand Island, NY). ALLN, Z-VAD-fmk, Z-Phe-Gly-NHO-Bz, mouse anti-tubulin antibody and cycloheximide were from Sigma (St. Louis, MO). Donkey anti-rabbit IgG-HRP was from Santa Cruz Biotechnology (Santa Cruz, CA). A rabbit anti-Ankrd1/CARP antibody was prepared by Caprologics (Hardwick, MA) against the N-terminal 104 amino acids of murine Ankrd1/CARP. The antigen was expressed as a GST fusion protein purified from a bacterial lysate by Ankrd1/CARP-GST-affinity chromatography, enzymatically processed to remove the GST moiety, and purified to remove free GST by a second pass over a GST affinity column. The flow through was dialyzed against PBS and concentrated in a dialysis cassette using Spectra/Gel absorbent (Spectrum Labs, Rancho Dominguez, CA). Protein concentration was determined (Pierce BCA assay, Thermo Scientific, Rockford, Il), and the purity of the peptide was confirmed by SDS-PAGE.
Cell Culture
Human microvascular endothelial cells, stably transformed with a perature sensitive SV-40 large T antigen, (HMVEC)[12] were cultured in Medium 131 supplemented with microvascular cell growth supplement and 10 mM glutamine. Cells were maintained at 33°C in 5% CO2. For experiments, cells were seeded into 150 mm tissue culture dishes and placed at 37°C for the duration of the experiment as described in the figure legends. Adult rat ventricular myocytes (ARVM) were isolated as previously described [6]. Long-term primary ARVM were established by culturing cells in DMEM supplemented with 7% FBS, 100 Units/ml penicillin-streptomycin and (+)-5-bromo-2′-deoxyuridine (BrdU) – Sigma Aldrich, St.Louis,MO) at 37°C, 5% CO2 with media changed every 72 h.
Ankrd1/CARP Half-life and Cell Density
To determine the effects of HMVEC density on Ankrd1/CARP expression and half-life (t1/2), cells were plated at 1×, 2× or 3× cell densities as indicated in the figure legends. To determine Ankrd1 t1/2, cells were plated in 150 mm dishes and incubated for 18h, fresh medium was added for 6h, and then cycloheximide was added to a concentration of 10 μg/ml. Cells were harvested at 0, 5, 10, and 15 min after the addition of cycloheximide. Ankrd1/CARP t1/2 was determined in ARVM by the same method except that cells were harvested at 0, 10, 24 and 48 hours after the addition of cycloheximide. For cell density experiments, 24 h before harvest, cells were plated in 150 mm dishes and placed at 37°C. The medium was changed after 18 h and cells were treated with 0.3μM epoxomicin or the equivalent amount of DMSO for 6 h. The Ct values of Ankrd1/CARP RNA were normalized to those of cyclophilin, and the fold changes were expressed as a comparison to the ΔCt of the control value of low density cells.
DNA, RNA and Protein Preparation
Culture media were replaced with cold PBS and cells were detached with a rubber spatula and pelleted by low speed centrifugation. Cell pellets were resuspended in 0.5 ml cold PBS with a 50μl aliquot reserved for DNA determination. The remaining cell suspension was divided to prepare whole cell protein extracts and RNA. Whole cell protein extracts were made by lysing the cells in RIPA buffer containing protease inhibitors (CØmplete Mini Protease Inhibitor Tablets; Roche, Indianapolis, IN) followed by sonication using a Branson 250 Sonifier with a water bath cup horn attachment. Protein concentration in cleared lysates was determined by BCA protein assay, and lysates were stored at −80°C. RNA was isolated using the Illustra RNAspin Mini Isolation Kit (GE Healthcare, Piscataway, NJ) according to manufacturer's instructions. RNA concentration was determined spectrophotometrically (Nanodrop,Thermo Scientific, Wilimington, DE), and samples were stored at −80°C.
DNA and RNA Analysis
The 50 μl cell suspension was diluted with 350 μl TE pH 7.5 and 100 μl 2% SDS in TE pH7.5, and heated at 55°C for 15 min. DNA concentration was determined using the Quant-iT PicoGreen assay (Invitrogen) according to manufacturer's directions using 2-8 μl of the cell lysate. The concentration was converted to cell number, assuming 6 pg of DNA per cell. Taqman qPCR for Ankrd1/CARP (see probe and primer sequences below) and cyclophilin (Applied Biosystems, Foster City, CA) as the housekeeping control, was performed on reverse transcribed RNA (50 ng):
| Ankrd1/CARP: | Forward primer: AGACTCCTTCAGCCAACATGATG |
| Reverse primer: CTCTCCATCTCTGAAATCCTCAGG | |
| Probe: CCCCTGCCTCCCCATTGCCATTCT |
Immunoblotting
Whole-cell protein extracts (30 μg per lane) were separated by electrophoresis using 10% NuPage Bis-Tris gels (Invitrogen) and transferred to PVDF membrane (Immobilon, Millipore, Billerica, MA) using the NuPage SureLock blotting apparatus. After blocking with TBST (10mM Tris-HCL pH 8, 150 mM NaCl, 0.05% tween-20) containing 5% milk powder, membranes were incubated overnight at 4°C with anti-Ankrd1/CARP antibody (1:2000) and anti-cyclophilin antibody (1:100,000) in TBST-5% milk. The membrane was washed and incubated with donkey anti-rabbit IgG-HRP (1:10,000) for 2h at room temperature. After rinsing, the membranes were incubated with Western Lightning Plus Enhanced Chemiluminescent Reagent (Perkin Elmer, Waltham, MA), and protein bands were visualized and quantified using a Kodak Image Station 4000MM Pro with Kodak MI software.
Statistics
Where appropriate, t-test or 2-way ANOVA were performed on data entered into GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA). Data were determined to be significant at p ≤ 0.05.
Results
In light of previous findings on Ankrd1/CARP stability [13] we designed studies to examine degradation and stability of the protein in two physiologically relevant cell types with predominantly cytoplasmic (ARVM) [6] and nuclear (HMVEC) distribution [2].
Inhibition of Ankrd1 degradation by protease inhibitors
We treated ARVM with several protease and 26S proteasome inhibitors at concentrations as recommended by the suppliers. The calpain inhibitor ALLN (2.5 and 5 μM), the caspase 3 inhibitor Z-VAD-fmk (25 and 50 μM) and the broad spectrum cathepsin inhibitor Z-Phe-Gly-NHO-Bz (25 and 50 μM) had no effect on Ankrd1/CARP degradation, excluding these proteolytic pathways (data not shown). In contrast, two of three 26S proteasome inhibitors, MG-132 (0.25 and 0.5 μM), and epoxomicin (50 μM) had effects on Ankrd1/CARP stability. Based on Western blot analysis, epoxomicin was a more potent inhibitor of Ankrd1/CARP degradation than MG-132. Clasto-lactacystin ß-lactone (lactacystin; 0.5 and 1.0 μM) did not prevent degradation at the concentrations tested (Figure 1).
Figure 1. 26S Proteasome inhibitors impede Ankrd1 degradation in ARVM.

ARVM were treated with epoxomicin, lactacystin and MG-132 using the concentrations shown for 24 h, and cell extracts were harvested and subjected to western blotting with anti-Ankrd1/CARP and anti-tubulin antibodies. Epoxomicin was the most effective inhibitor followed by MG-132. Lactacystin did not inhibit degradation in these cells at the concentrations used. Blots are representative of 3 separate experiments.
HMVEC were treated with the 3 proteasome inhibitors as well as epigallocatechin gallate (EGCG), a specific inhibitor of the chymotrypsin-like activity of the 26S proteasome, and NIP-(leu)3-vinyl sulfone (NIP), which inhibits the trypsin-like, chymotrypsin-like and peptidyl-glutamyl peptide hydrolyzing activity of the catalytically-active B subunits of the 26S proteasome. While neither EGCG nor NIP protected Ankrd1/CARP in HMVEC, MG-132, lactacystin and epoxomicin elevated endogenous levels dose-dependently (Table 1). Epoxomicin (0.5 μM) was more potent than either MG-132 (∼10×) or lactacystin (∼100×) in elevating Ankrd1/CARP to equivalent cellular levels as estimated by Western blot analysis.
Table 1. Effects of Proteasomal Inhibitors on Relative Abundance of Ankrd1/CARP in HMVEC.
| Inhibitor | Epoxomicin | Lactacystin | MG-132 | ||||
|---|---|---|---|---|---|---|---|
| Concentration (μM) | 0.5 | 0.5 | 5 | 50 | 0.5 | 5 | 50 |
| Fold Change* | 31 | 1.9 | 14 | 32 | 5.7 | 26 | 56 |
Relative to untreated cells.
Epoxomicin dose-dependently inhibited Ankrd1/CARP protein degradation in both ARVM and HMVEC, but the maximal protective effect occurred at a >20-fold lower dose in cardiomyocytes than in endothelial cells (Figure 2). In the absence of epoxomicin, Ankrd1/CARP protein was easily detectable in ARVM but undetectable in HMVEC. The greater sensitivity to the proteasome inhibitor in ARVM could reflect lower proteasomal activity and/or greater stability of Ankrd1/CARP in the cardiomyocyte where this protein is a prominent structural component of the sarcomere.
Figure 2. Differential dose-dependent epoxomycin inhibition of proteasomal Ankrd1 degradation in cardiac and endothelial cells.

A > 20-fold lower concentration of the proteasome inhibitor, epoxomicin, was required to permit maximal accumulation of Ankrd1/CARP in ARVM (20 nM, Panel B) vs. HMVEC (500 nM, Panel A).
Ankrd1/CARP Half-life
Because of the apparent difference in epoxomicin stabilization of Ankrd1/CARP in ARVM and HMVEC, we determined the protein half-lives (t1/2) in each cell type after cycloheximide treatment, as illustrated in Figure 3. The t1/2 of Ankrd1/CARP in ARVM was 13 ± 2.5 h (Figure 3A). In sharp contrast, Ankrd1/CARP t1/2 in HMVEC, plated at low density (4×105 cell/150 mm dish), was 12.2 ± 3.0 min (Figure 3B). Furthermore, the t1/2 of Ankrd1/CARP in HMVEC plated at a higher density (1.2×106 cells/150 mm dish) was so rapid (<5 min, data not shown) that it was not possible to estimate the rate of decay accurately by this kinetic protocol. In endothelial cells, where, Ankrd1/CARP is undetectable in the steady state, protein abundance was much more highly affected by its rate of degradation than it was in cardiomyocytes, where Ankrd1/CARP is readily detectible. Furthermore, cell density influenced the protein t1/2 in HMVEC.
Figure 3. Ankrd1/CARP protein t1/2 is profoundly different in ARVM (hours) and HMVEC (minutes).
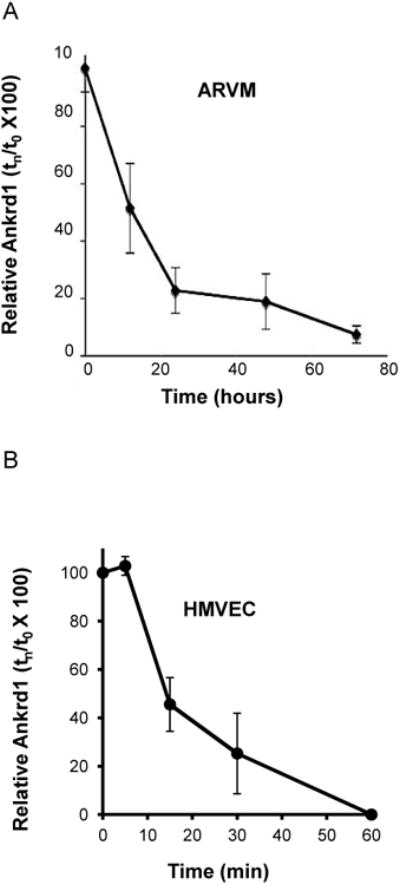
Cycloheximide (10μg/ml) was added and cells were harvested at times indicated. n = 3 for both cell types. (Panel A) Ankrd1/CARP decay in ARVM (t1/2=13 ± 2.5 h) (Panel B) Ankrd1/CARP decay in HMVEC (t1/2=12.2 ± 3.0 min) plated at low density (4 × 105 cells per 150 mm dish).
Cell density affects Ankrd1/CARP expression in HMVEC
To further examine the relationship of Ankrd1/CARP protein to mRNA expression under low- and high-density conditions, both parameters were compared in the presence and absence of epoxomicin. In the absence of epoxomicin (Figure 4, Panel A), HMVEC grown at high density consistently contained lower levels of Ankrd1/CARP than low density cultures (Figure 4A, inset), although the variability of measuring low protein levels prevented the several-fold difference from reaching statistical significance (p = 0.051). The addition of epoxomicin to low-density HMVEC increased Ankrd1/CARP content >30-fold, whereas at high cell density this effect was only 8-fold. The changes in Ankrd1/CARP protein were not reflected at the mRNA level. There was no significant difference in Ankrd1/CARP mRNA levels under either low- or high-density conditions in the absence or presence of epoxomicin, although epoxomicin increased Ankrd1/CARP mRNA by 50-75% (p = 0.023) at both cell densities (Figure 3B). From these data we conclude that the primary regulation of Ankrd1/CARP expression in HMVEC grown at high and low density is through its degradation by the 26S proteasome. Since Ankrd1/CARP protein was not increased by epoxomicin to the same extent in high density cells as in low density cells, despite having comparable mRNA content, density may have an effect at the translational level.
Figure 4. Uncoupling of ankrd1 mRNA and protein levels as a function of HMVEC density.

(Panel A) Fold change in Ankrd1/CARP protein. The values are normalized per cell and compared to the control value at low density. Epoxomycin strongly increased protein levels under low and high density conditions. In the absence of epoxomicin (inset) there was a strong trend for cell density to reduce Ankrd1/CARP (8-fold; p=0.051, paired t-test) and a significant effect (p = 0.04) of cell density (30-fold) in the presence of epoxomicin. (B) There was a small but significant effect (p = 0.023) of epoxomicin on ankrd1 mRNA but no effect of cell density. Values are expressed as the mean ± SEM, n = 3
Discussion
Protein stability has emerged as a major cellular regulatory mechanism [14], and the 26S proteasome is the principal regulator responsible for marking and destroying the majority of cellular proteins. This process affects biological functions including cell proliferation, differentiation, and apoptosis [14,15]. The present study revealed a sharp contrast in the rate of proteasomal degradation of Ankrd1/CARP in the contractile cardiomyocyte and the microvascular endothelial cell. The findings also emphasized the significant uncoupling of ankrd1 transcript and protein levels in the endothelial cell, a discrepancy that was further amplified by cell density.
Ankrd1/CARP has attributes of both a structural and a nuclear regulatory factor [2,16,17], and both the ankrd1 transcript and its product possess regulatory elements. For example, ankrd1 mRNA has degradation motifs in its 3′-UTR, removal of which greatly increased its stability [16]. Ankrd1/CARP protein has potential sites for phosphorylation and glycosylation, and it contains a conserved PEST sequence and other degrons that were shown to mediate protein degradation [13]. Ankrd1/CARP also contains a calpain target sequence [18] and a caspase 3 target sequence [19] within the N-terminal 71 amino acids. Cinquetti et al. [7] confirmed calpain cleavage of in vitro transcribed and translated Ankrd1/CARP, but the same group later demonstrated that the calpain inhibitor, calpeptin, as well as NH4Cl, a lysosomal inhibitor, were unable to prevent degradation of Ankrd1/CARP-3Flag in transfected Hela cells[13]. However, MG-132, a proteasomal inhibitor, inhibited the degradation of the transfected Ankrd1/CARP-3Flag and also endogenous Ankrd1/CARP in SK-MES, a human lung carcinoma cell line.
In our study, failure of another calpain inhibitor, ALLN, confirmed that calpain is not involved in cellular degradation of Ankrd1/CARP in ARVM. However, calpain cleavage of Ankrd1/CARP in skeletal muscle strengthens its association with the N2A region of titin in the sarcomere and alters its translocation rate between the sarcoplasm and the nucleus [18]. Therefore, calpain protease activity, though not important for the degradation of Ankrd1/CARP, appears to influence its function by altering its cellular distribution.
Another study identified a caspase 3 target sequence in Ankrd1/CARP from a human proteomic screen, but the ability of caspase 3 to cleave or degrade Ankrd1/CARP or affect its function was not investigated [19]. We have shown that the caspase 3 inhibitor Z-VAD-fmk had no effect on Ankrd1/CARP degradation in ARVM, nor did the broad-spectrum cathepsin inhibitor, Z-Phe-Gly-NHO-Bz. Because we have not repeated these assays in HMVEC, we cannot state definitively that neither caspase 3 nor cathepsins have an effect on Ankrd1/CARP degradation in this non-muscle cell type.
Epoxomicin acts as a potent, highly specific, and irreversible inhibitor of chymotrypsin-like, trypsin-like, and peptidyl-glutamyl peptide hydrolyzing activities of the 26S proteasome by modifying the proteasomal catalytic subunits LMP-7, MECL1, and Z, but it does not affect the activities of non-proteasomal proteases such as trypsin, cathepsin B, or chymotrypsin [20]. Epoxomicin was the most effective inhibitor of Ankrd1/CARP degradation in our experiments, followed by MG-132, a potent but reversible inhibitor of the 26S proteasome, and then, at higher doses in HMVEC compared to ARVM, lactacystin, an inhibitor of the 20S component of the proteasome.
The differences in the maximally effective dose of epoxomicin between ARVM and HMVEC may be due to the tight association of Ankrd1/CARP within the titin-based sarcoplasmic complex as well as the consequent concentration of the protein in the sarcomere. Our recent report using siRNA-induced loss of Ankrd1/CARP underscores the importance of this protein in sarcomeric integrity of cultured cardiomyocytes [6]. The absence of an analogous cellular compartment in the cytoplasm of HMVEC may render Ankrd1/CARP a more susceptible target for degradation in these cells. The significantly longer Ankrd1/CARP stability in ARVM (hours) versus HMVEC (minutes), supports this hypothesis.
The localization of Ankrd1/CARP in the nucleus of HMVEC is consistent with a primary role as a transcriptional co-factor [21]. Our cell density results suggest that this function of the protein becomes important when cells are stressed by the culture conditions. Low-density plating of HMVEC on a 2-dimensional plastic substrate increased detectable Ankrd1/CARP protein but not mRNA, and In the presence of epoxomicin, the density-dependent differences in the amounts of Ankrd1/CARP were much more evident. The lack of difference in the mRNA levels in cells grown at differing densities in the presence of epoxomicin suggests two mechanisms for the increase in Ankrd1/CARP protein. Our data would suggest that altered proteasomal activity, possibly by stress induced modification of either the 26S proteasome or of Ankrd1/CARP, render the protein less susceptible to degradation. However, presuming that we used the maximally effective dose of inhibitor under both growth conditions, altered regulation of protein translation could play a part in the increased Ankrd1/CARP, since there was 4-5 fold more protein in the cells grown at low density (Figure 4). As cells reached higher density, there appeared to be a reversal of the mechanisms that allow more Ankrd1/CARP to be expressed, until it became undetectable in the absence of epoxomicin and detectable at low levels in its presence.
Because ARVM do not proliferate, cell density experiments were not conducted in these cells. However, evidence that cardiac myocytes may behave similarly comes from a study by Akins et al.[22], in which primary rat ventricular cells were grown in 2D- and 3D-culture under serum free conditions to hold proliferation in check. Cells grown in 2D formed islands of endothelial cells surrounded by primary cardiomyocytes, and Ankrd1/CARP expression progressively declined from days 1-6. In 3D culture, endothelial and cardiomyocyte differentiation was much more efficient, and Ankrd1/CARP expression remained low at all times.
The proteasomal mechanism we have described allows for a rapid modulation of protein abundance independent of transcriptional or translational control. The aberrant overexpression of Ankrd1/CARP may be associated with pathologies, as seen in some patients with TAPVR [7] or cancers that are resistant to chemotherapy [9]. Likewise, disruption of Ankrd1/CARP regulation leading to overexpression is likely involved in pathological heart remodeling [3]. Reduced and/or absence of expression has also been shown to be deleterious as reported by Stam et al. in their identification of Ankrd1/CARP as a transcriptional modulator involved in successful nerve regeneration after injury [10]. Taken together, our data indicate that the expression of Ankrd1/CARP in cultured vascular cells is significantly affected by post-transcriptional regulation. The striking difference in protein half-life between muscle and non-muscle cells and the loss of sarcomeric integrity following depletion of muscle Ankrd1/CARP also suggest cell type-specific regulatory mechanisms.
Research Highlights.
The 26S proteasome regulates Ankrd1 levels in cardiomyocytes and endothelial cells
Ankrd1 protein degrades 60-fold faster in endothelial cells than cardiomyocytes
Differential degradation appears related to nuclear vs. sarcolemmal localization
Endothelial cell density shows uncoupling of Ankrd1 mRNA and protein levels
Acknowledgments
This work was supported by funding by NIH-NIDDK 65656 and the Department of Veterans Affairs to JMD, NIH-NHLBI HL068144 to DBS, and NIH-NHLBI HL095813 and a Vanderbilt University Stahlman Grant to CCL. We wish to thank Susan R. Opalenik, Ph.D. for her critical reading of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Miller MK, Bang ML, Witt CC, Labeit D, Trombitas C, Watanabe K, Granzier H, McElhinny AS, Gregorio CC, Labeit S. The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. Journal of molecular biology. 2003;333:951–964. doi: 10.1016/j.jmb.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 2.Chu W, Burns DK, Swerlick RA, Presky DH. Identification and characterization of a novel cytokine-inducible nuclear protein from human endothelial cells. The Journal of biological chemistry. 1995;270:10236–10245. doi: 10.1074/jbc.270.17.10236. [DOI] [PubMed] [Google Scholar]
- 3.Mikhailov AT, Torrado M. The enigmatic role of the ankyrin repeat domain 1 gene in heart development and disease. The International journal of developmental biology. 2008;52:811–821. doi: 10.1387/ijdb.082655am. [DOI] [PubMed] [Google Scholar]
- 4.Shi Y, Reitmaier B, Regenbogen J, Slowey RM, Opalenik SR, Wolf E, Goppelt A, Davidson JM. CARP, a cardiac ankyrin repeat protein, is up-regulated during wound healing and induces angiogenesis in experimental granulation tissue. The American journal of pathology. 2005;166:303–312. doi: 10.1016/S0002-9440(10)62254-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zou Y, Evans S, Chen J, Kuo HC, Harvey RP, Chien KR. CARP, a cardiac ankyrin repeat protein, is downstream in the Nkx2-5 homeobox gene pathway. Development. 1997;124:793–804. doi: 10.1242/dev.124.4.793. [DOI] [PubMed] [Google Scholar]
- 6.Chen B, Zhong L, Roush SF, Pentassuglia L, Peng X, Samaras S, Davidson JM, Sawyer DB, Lim CC. Disruption of a GATA4/Ankrd1 Signaling Axis in Cardiomyocytes Leads to Sarcomere Disarray: Implications for Anthracycline Cardiomyopathy. PloS one. 2012;7:e35743. doi: 10.1371/journal.pone.0035743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cinquetti R, Badi I, Campione M, Bortoletto E, Chiesa G, Parolini C, Camesasca C, Russo A, Taramelli R, Acquati F. Transcriptional deregulation and a missense mutation define ANKRD1 as a candidate gene for total anomalous pulmonary venous return. Human mutation. 2008;29:468–474. doi: 10.1002/humu.20711. [DOI] [PubMed] [Google Scholar]
- 8.Matsuura K, Uesugi N, Hijiya N, Uchida T, Moriyama M. Upregulated expression of cardiac ankyrin-repeated protein in renal podocytes is associated with proteinuria severity in lupus nephritis. Human pathology. 2007;38:410–419. doi: 10.1016/j.humpath.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Scurr LL, Guminski AD, Chiew YE, Balleine RL, Sharma R, Lei Y, Pryor K, Wain GV, Brand A, Byth K, Kennedy C, Rizos H, Harnett PR, deFazio A. Ankyrin repeat domain 1, ANKRD1, a novel determinant of cisplatin sensitivity expressed in ovarian cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:6924–6932. doi: 10.1158/1078-0432.CCR-07-5189. [DOI] [PubMed] [Google Scholar]
- 10.Stam FJ, MacGillavry HD, Armstrong NJ, de Gunst MC, Zhang Y, van Kesteren RE, Smit AB, Verhaagen J. Identification of candidate transcriptional modulators involved in successful regeneration after nerve injury. The European journal of neuroscience. 2007;25:3629–3637. doi: 10.1111/j.1460-9568.2007.05597.x. [DOI] [PubMed] [Google Scholar]
- 11.Barash IA, Bang ML, Mathew L, Greaser ML, Chen J, Lieber RL. Structural and regulatory roles of muscle ankyrin repeat protein family in skeletal muscle, American journal of physiology. Cell physiology. 2007;293:C218–227. doi: 10.1152/ajpcell.00055.2007. [DOI] [PubMed] [Google Scholar]
- 12.Xu Y, Swerlick RA, Sepp N, Bosse D, Ades EW, Lawley TJ. Characterization of expression and modulation of cell adhesion molecules on an immortalized human dermal microvascular endothelial cell line (HMEC-1) The Journal of investigative dermatology. 1994;102:833–837. doi: 10.1111/1523-1747.ep12382086. [DOI] [PubMed] [Google Scholar]
- 13.Badi I, Cinquetti R, Frascoli M, Parolini C, Chiesa G, Taramelli R, Acquati F. Intracellular ANKRD1 protein levels are regulated by 26S proteasome-mediated degradation. FEBS letters. 2009;583:2486–2492. doi: 10.1016/j.febslet.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 14.Myung J, Kim KB, Crews CM. The ubiquitin-proteasome pathway and proteasome inhibitors. Medicinal research reviews. 2001;21:245–273. doi: 10.1002/med.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nandi D, Tahiliani P, Kumar A, Chandu D. The ubiquitin-proteasome system. Journal of biosciences. 2006;31:137–155. doi: 10.1007/BF02705243. [DOI] [PubMed] [Google Scholar]
- 16.Baumeister A, Arber S, Caroni P. Accumulation of muscle ankyrin repeat protein transcript reveals local activation of primary myotube endcompartments during muscle morphogenesis. The Journal of cell biology. 1997;139:1231–1242. doi: 10.1083/jcb.139.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeyaseelan R, Poizat C, Baker RK, Abdishoo S, Isterabadi LB, Lyons GE, Kedes L. A novel cardiac-restricted target for doxorubicin. CARP, a nuclear modulator of gene expression in cardiac progenitor cells and cardiomyocytes. The Journal of biological chemistry. 1997;272:22800–22808. doi: 10.1074/jbc.272.36.22800. [DOI] [PubMed] [Google Scholar]
- 18.Laure L, Suel L, Roudaut C, Bourg N, Ouali A, Bartoli M, Richard I, Daniele N. Cardiac ankyrin repeat protein is a marker of skeletal muscle pathological remodelling. The FEBS journal. 2009;276:669–684. doi: 10.1111/j.1742-4658.2008.06814.x. [DOI] [PubMed] [Google Scholar]
- 19.Ju W, Valencia CA, Pang H, Ke Y, Gao W, Dong B, Liu R. Proteome-wide identification of family member-specific natural substrate repertoire of caspases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:14294–14299. doi: 10.1073/pnas.0702251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zolk O, Frohme M, Maurer A, Kluxen FW, Hentsch B, Zubakov D, Hoheisel JD, Zucker IH, Pepe S, Eschenhagen T. Cardiac ankyrin repeat protein, a negative regulator of cardiac gene expression, is augmented in human heart failure. Biochemical and biophysical research communications. 2002;293:1377–1382. doi: 10.1016/S0006-291X(02)00387-X. [DOI] [PubMed] [Google Scholar]
- 22.Akins RE, Jr, Rockwood D, Robinson KG, Sandusky D, Rabolt J, Pizarro C. Three-dimensional culture alters primary cardiac cell phenotype. Tissue engineering Part A. 2010;16:629–641. doi: 10.1089/ten.tea.2009.0458. [DOI] [PMC free article] [PubMed] [Google Scholar]